


Monozygotic (MZ) twins have an almost identical genetic background, providing us a precious opportunity to determine the genetic contribution underlying diverse human diseases. Particularly in complex disease where multiple genes are involved, studies based on MZ twins are crucial to elucidate the roles of candidate genes. The mechanism underlying the MZ twinning remains unclear, and recently McNamara et al. (Reference McNamara, Kane, Craig, Short and Umstad2016) have summarized the theory models explaining this process. Statistical data shows that the MZ multiple birth incidence rate is about 3% all over the world (Eriksson, Reference Eriksson1962). This rate remains stable in almost all geographic areas, and genetic backgrounds of the inhabitants or special environment factors seem to play limited roles in MZ twins’ occurrence. No confirmative evidence exists for all known theory models, and one hypothesis is that the MZ twins occur randomly at a low frequency.
To date, nearly 20 familial MZ twinning reports have been seen, with 2.5 identical twin pairs in each family on average (Hamamy et al., Reference Hamamy, Ajlouni and Ajlouni2004; Harvey et al., Reference Harvey, Huntley and Smith1977; Machin, Reference Machin2009a; Segreti et al., Reference Segreti, Winter and Nance1978; Shapiro et al., Reference Shapiro, Zemek and Shulman1978). Therefore, genetic factors may underlie a small proportion of MZ twins, providing opportunities to dissect potential mechanisms. Meanwhile, Bamforth et al. (Reference Bamforth, Brown, Senz and Huntsman2003) suggested E-cadherin, a cell adhesion molecule, as a contributory factor in twinning process by analyzing its gene polymorphism in MZ twins. According to familial MZ twinning reports, worldwide constant incidence, and female excess in MZ twinning, Machin (Reference Machin2009b), believed that MZ twinning was not random. Rather, he speculated that genotyping in familial MZ twinning may identify candidate genes related to cell adhesion. Nevertheless, it is still challenging to obtain direct evidence to support each speculation due to ethical issues of human embryo studies or lack of appropriate animal model.
Here, we have collected a four-generation pedigree of MZ twinning. Particularly, all twins are female, and gave birth to the twins in the next generation. We performed whole-genome sequencing (WGS) for seven family members to explore the genetic background of this pedigree and to identify candidate variants in MZ twins.
Materials and Methods
Participants and Genomic DNA Extraction
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Shandong Academy of Medical Sciences’ Institutional Review Board. The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008. Whole blood samples were collected from six individuals in this pedigree, including III 4, III 5, III 9, III 10, III 11, IV14, IV16 (Figure 1), among which III 10 and III 11, IV14 and IV16 were two pairs of MZ twins. Blood samples were stored at −20°C before usage. Genomic deoxyribonucleic acid (DNA) was extracted using QIAamp Blood Kit (Qiagen, Hilden, Germany) and quantified on a Qubit Fluorometer Qubit (Thermo Fisher Scientific Inc., Cleveland, OH, USA). The quality of the DNA samples were examined on an agarose gel.
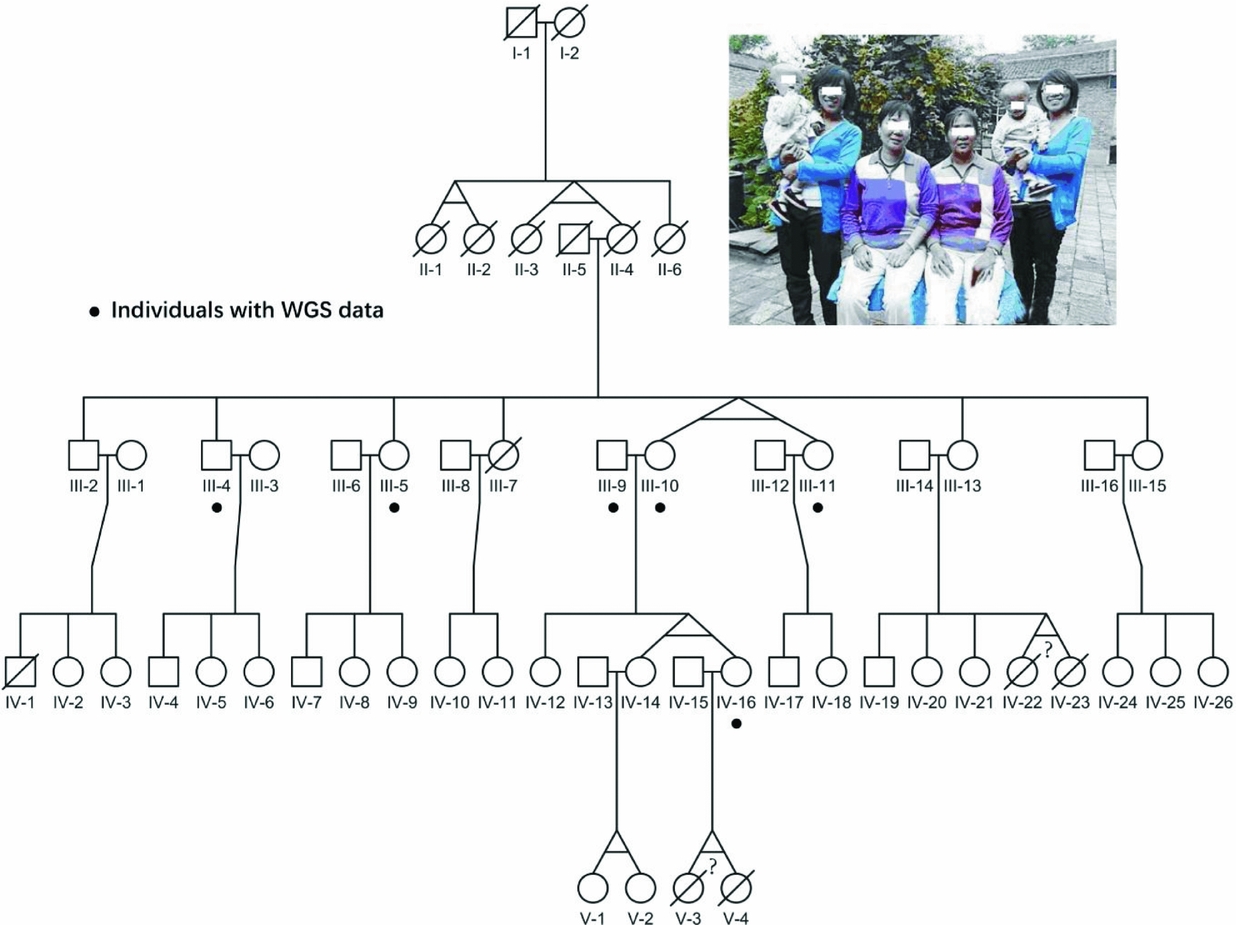
FIGURE 1 Pedigree of the four-generation MZ twins family. The photograph on the top shows all three pairs of alive MZ twins in this pedigree. The black dots point out six individuals with whole-genome sequencing data; the question marks indicate the abortion MZ twins according to verbal questionnaire.
Whole-Genome Sequence (WGS) Experiments
Genomic DNA samples were sent to the Core Genomic Facility (CGF) in Beijing Institute of Genomics for whole-genome sequencing and sequenced using Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA) with 101bp (base pair) pair-end reads or Illumina HiSeq 2500 (Illumina, Inc., San Diego, CA, USA) with 150bp pair-end reads. For each sample, the target coverage was 30X, and two sequencing libraries with target insert size around 300–400 bp were constructed and sequenced in four lanes. The quality of sequencing libraries was evaluated using Agilent 2100 bio-analyzer (Agilent Technologies, Palo Alto, CA).
Reads Alignment
Sequence reads in FASTQ format were evaluated by FastQC (v0.10.1) software, and any adapter segments and low Q-score bases in reads were then removed with cutadapt (v1.9) software (Martin, Reference Martin2011). The trimmed sequence reads were then aligned to the human genome hg19 reference with the BWA (v0.5.9) algorithm (Li & Durbin, Reference Li and Durbin2009). To reduce the false-positive rate in variants detection, only unique mapping reads were used in subsequent analysis. Those mapped reads were sorted by the physical location with Picard (v1.86) software. The raw reads generated from two libraries for one sample were merged into one using Samtools (v1.0) software (Li et al., Reference Li, Handsaker, Wysoker, Fennell, Ruan and Homer2009). Then, PCR duplicate reads were removed with Picard (v1.86) software. Processed bam files were processed via local indel realignment and base-quality recalibration using the Genome Analysis Tool Kit (GATK, v2.7.4) (McKenna et al., Reference McKenna, Hanna, Banks, Sivachenko, Cibulskis, Kernytsky and DePristo2010; Van der Auwera et al., Reference Van der Auwera, Carneiro, Hartl, Poplin, Del Angel, Levy-Moonshine and DePristo2013). Samtools was used to perform the reads depth calculation of output files. In addition, considering the possibility of misalignment in subsequent single nucleotide variant (SNV) or copy number variation (CNV) calling procedures, we only kept the variants covered by strict mask regions according to 1000 Genomes Project phase I for further analysis (1000 Genomes Project Consortium et al., Reference Abecasis, Altshuler, Auton, Brooks, Durbin and McVean2010; 1000 Genomes Project Consortium et al., Reference Abecasis, Auton, Brooks, DePristo, Durbin and McVean2012).
Single Nucleotide Variant (SNV) Detection
Subsequently, sorted bam files were used for SNV calling and the UnifiedGenotyper method based on Bayesian genotype likelihood model was used with GATK (v2.7.4). Then, variants supported by less than 20% alternative allele reads were removed, filtering out the somatic variants or false positive variants. The individual specific variants in each individual of MZ twins were detected by each comparison with MuTect (v1.1.4; Cibulskis et al., Reference Cibulskis, Lawrence, Carter, Sivachenko, Jaffe, Sougnez and Getz2013).
Identity-by-Descent (IBD) Calculation
Identity-by-descent analysis was applied to estimate the inbreeding coefficients in the MZ twin pairs of III10, III11, IV14, and IV16 respectively, using the PLINK toolset (v1.07; Purcell et al., Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira, Bender and Sham2007). The probability of the IBD equal to 0, 1, or 2 was calculated separately, and PI_HAT was calculated based on the probability.
Copy Number Variation Detection
Copy number variations were identified from the read-depth of each sorted bam file with CNVnator (v0.2.7) software (Abyzov et al., Reference Abyzov, Urban, Snyder and Gerstein2011). The output files showed the probability of each candidate copy number changing region with the Gaussian distribution. To reduce the false positive regions, we precluded those with length larger than 100kb, or with the probability lower than 0.01. Genes located in the copy number changing regions were used for pathway analysis.
Annotation of Variants and Pathway Analysis
All detected SNVs were annotated using ANNOVAR (2016Feb01) and SeattleSeq Annotation server (http://gvs.gs.washington.edu/SeattleSeqAnnotation). To preclude common variants, we filtered out the variants observed in 1000 Genome project (2015), ExAC database, and dbSNP147 database.
Pathway analysis was performed using WebGestalt analysis software and Ingenuity® Pathway Analysis (IPA) for candidate genes, according to Gene Ontology (GO) categories, as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways database. The statistical significance was examined by hypergeometric test and adjusted for multiple testing with the Benjamini and Hochberg false discovery rate. The most significantly enriched pathways were summarized based on the report exported from the Ingenuity Knowledge Base.
Mitochondrial DNA (mtDNA) Sequencing
We conducted Sanger sequencing for whole mitochondrial genome from III10, III11, IV14, and IV16. All the primer design and PCR procedures were designed and published by Ramos, et al. (Reference Ramos, Santos, Alvarez, Nogues and Aluja2009), which contained 31 pairs of resequencing primers including nine pairs of primers for mtDNA amplification.
SNV Validation
Sequenom MassARRAY was applied to identify variants specific to MZ twins in this pedigree at the Beijing Institute of Genomics. The primers (Table S3) were designed by AgenaCx online tool (https://agenacx.com/online-tools/) with assay design suite (V2.0). And all the primers were synthesized by Thermo Fisher Scinetific. Finally, 28 of the variants were validated in 12 members and 27 of them were co-separate with twins.
Data Availability
The sequencing data and variants data generated during the current study are available from the corresponding author on reasonable request.
Results
WGS for Seven Individuals in This MZ Twins Pedigree
The pedigree we collected showed an extremely high recurrent rate of MZ twins of around 27% (Figure 1), which was 90-fold of the previous reported 3% recurrent rate around the world. Particularly, consecutive MZ twins occurred in four generations, and we believe there are some genetic factors, especially germline variants. However, the limited offspring for each family in this pedigree makes it difficult to evaluate their ability to conceive MZ twins. Therefore, we assumed that only twins who carried the candidate sites were responsible for having MZ twins. Additionally, shared variants by MZ twins would also have high reliability.
Here, we sequenced the whole genome of six individuals in this pedigree, as shown in Figure 1, including two MZ twin pairs; one is III10 and III11, and the other is IV14 and IV16. The sequencing coverage of the whole genome was around 21~32-fold, with over 90% of the genome covered by sequence reads in each individual (Material and Methods section, Figure S1). On average, for each individual, the amount of SNVs was 3.2M (Figure S2A), similar to previous studies (Mallick et al., Reference Mallick, Li, Lipson, Mathieson, Gymrek, Racimo and Reich2016). In addition, similar amounts of variants were also observed for each chromosome in all seven individuals (Figure S2B), indicating the even sequencing coverage of all chromosomes. Besides, we applied Sanger sequencing for mtDNA from III10, III11, IV14 and IV16 (Material and Methods section), and verified all novel variants with the observed high alternative allele fractions.
The MZ Twins Shared Almost Identical Genomes
We performed IBD analysis for the MZ twin pairs and they are almost identical (PI_HAT = 0.9974 for III10 and III11, 0.9946 for IV14 and IV16). In all, for SNVs specific to each individual with high confidence, III10 had 64, while III11 had 9, none of which were non-synonymous substitutions according to subsequent annotation (Material and Methods section). Moreover, IV14 had 82 high confidence-specific SNVs, and IV16 had 70. Similarly to III10 and III11, none of these 152 specific variants were non-synonymous substitutions. Additionally, the amount of individual specific polymorphism was similar to Baranzini et al.’s (Reference Baranzini, Mudge, van Velkinburgh, Khankhanian, Khrebtukova, Miller and Kingsmore2010) study; nevertheless, none of these were verified (Baranzini et al., Reference Baranzini, Mudge, van Velkinburgh, Khankhanian, Khrebtukova, Miller and Kingsmore2010). Therefore, the inconsistency between these two MZ twin genomes was likely to have false positive results produced during sequencing. Considering the possibility of incomplete dominance in III11, we considered whether both of them carried the candidate sites responsible for MZ twins. To achieve a high accuracy, we focused on the shared variants in all four individuals — III10, III11, IV14, and IV16 from MZ twins — and defined them as variants specific to MZ twins in our analysis.
A Novel Candidate SNV of ZCCHC12 in the X Chromosome
Since, all four generations of MZ twins were female (Figure 1), we analyzed the genetic characteristics of the X chromosome from the MZ twins, comparing to other family members. On average, ~0.1 million variants in the X chromosome were identified for each individual. Particularly, 212 variants were specific to MZ twins (Table S1). Furthermore, five of these variants were novel (Table 1). Among them, one variant (chrX: 117958597, G > A) is located at the 5′UTR region of ZCCHC12 (zinc finger CCHC-type containing 12), which encodes a downstream effector of bone morphogenetic protein (BMP) signaling. In addition, the remaining 5 variants were located in the intronic or intergenic regions (Table 1). Additionally, all these 5 short nucleotide polymorphisms (SNPs) were validated in this pedigree using a mass spectrometry platform (Method, Figures S4–S15, Table S3).
TABLE 1 Novel Variants in Chromosome X

Consistent mtDNA Composition Among Twins and Non-Twin Siblings
For individuals from the same maternal lineage (III4, III5, III11, III10, IV14, and IV16), we detected 42 candidate sites (Figure 2C). Among them, 34 were novel variations specific to this pedigree with 100% alternative allele fraction in all these 6 individuals; and the remaining 8 sites had novel minor allele with fractions less than 60% in each individual. However, no composition differences were observed either among MZ twins or among all individuals.
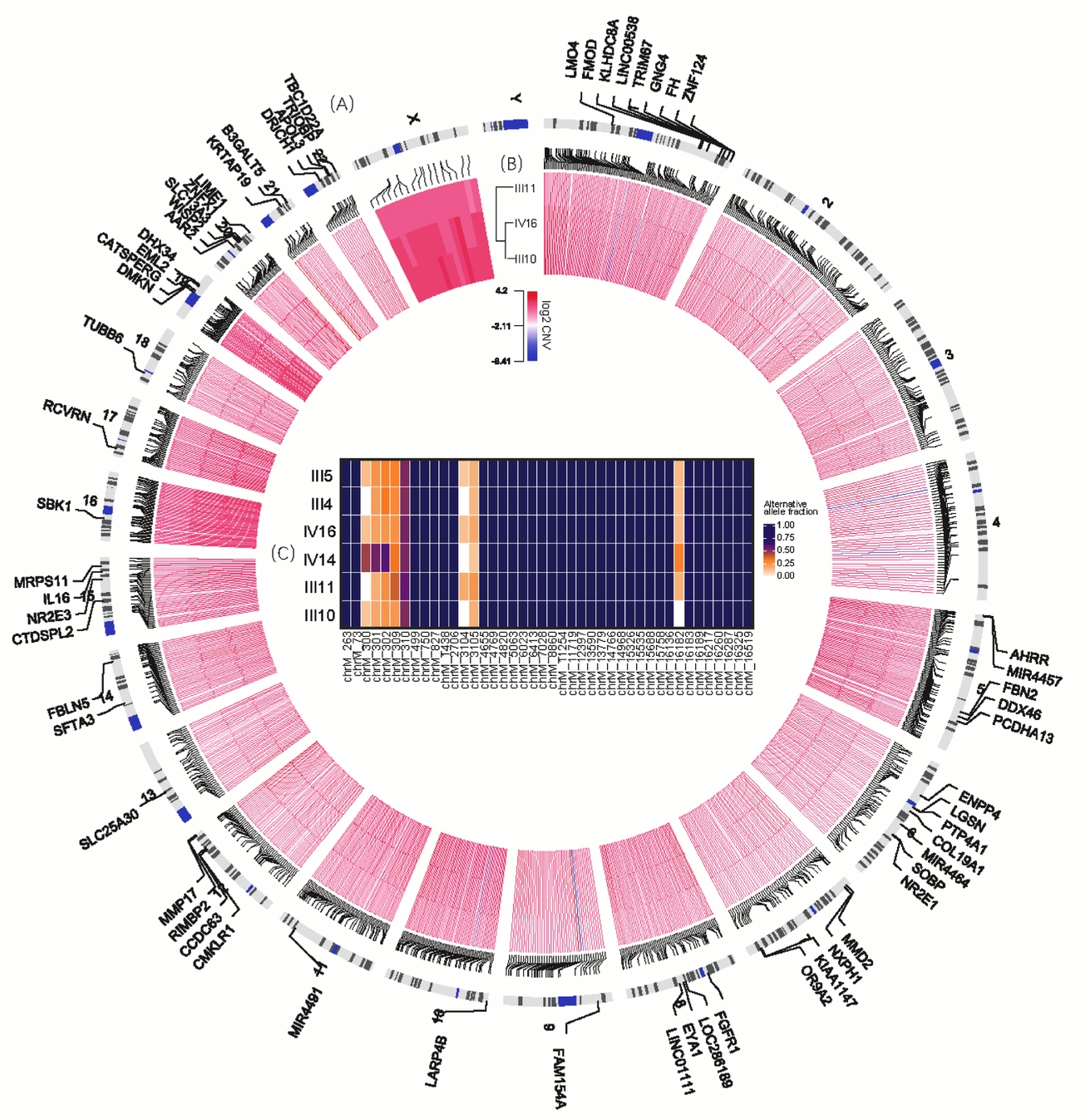
FIGURE 2 Variants shared in MZ twins. (A) The outmost circle shows 71 genes containing 74 functional SNVs specific to MZ twins. Particularly, chromosomes 19 and 20 have more SNVs of this kind observed. Due to limited space, some are not shown here, including genes ALKBH7, ZNF526, MARK4, ZNF812, C19orf40 and LOC102723617 in chromosome 19 and GINS1, DEFB115, SPO11, MYH7B, and CABLES2 in chromosome 20. (B) The inner circle demonstrates the CNVs shared by III10, III11 and IV16. (C) The heatmap in the center illustrates the allele fraction for all 43 sites specific to this family in mtDNA.
Autosomal SNVs of MZ Twins Located at Genes Overrepresented by Epithelial Adherens Junction Signaling Pathway
We have obtained 37,716 SNVs in autosomes and explored their distribution pattern in human genome (Material and Methods section). Among these SNVs, 389 are located at the coding regions, 21,497 at the intergenic areas, while only 707 were in the regulatory regions. After removing SNVs either in repeat regions or recorded in 1000 Genome and ExAC databases, we identified 1,937 novel SNVs and over 92% (2,361) were located at intergenic or intronic regions. Among the remainder, 61 were functional candidates by prediction, including 20 in the gene upstream regions or 5′UTR, 31 were in the 3′UTR or gene downstream regions and 6 non-synonymous substitutions specific to MZ twins in this pedigree (Table S1; Table 2). As shown in Figure 2B, these candidate functional variants are located in 59 genes. Subsequent pathway analysis revealed a significant enrichment of genes (FGFR1, TUBB6, and MYH7B) in the epithelial adherens junction-signaling pathway (p = .011, Table 3). Two of these genes (TRIOBP and TUBB6) were involved in the GTPase family mediated signal pathway. For the SNPs showing functional importance, we further validated 23 of them in the pedigree using the mass spectrometry platform (Method, Table S3). The result showed that all of them were verified (Figures S4–S15).
TABLE 2 Novel Non-Synonymous Variants in Autosomes

TABLE 3 Significant Enriched Pathways

Moreover, we noticed some variants had extremely low frequency (<0.001) in the population, and it is difficult to preclude their contribution in the occurrence of MZ twins, and some of this kinds of variants with the genes involved in GTPase family mediated the signal pathway. For instance, one variant (chr22: 47571026, C > T, frequency = 0.0004) was located in the 3′UTR region of TBC1D22A. The protein TBC1D22A is known to interact with the Rab family of proteins, which are responsible for membrane trafficking and intracellular signaling (Schwartz et al., Reference Schwartz, Cao, Pylypenko, Rak and Wandinger-Ness2007).
Tight Junction-Related Signaling Pathway Enriched for Genes Covered by CNVs Specific to MZ Twins
On average, the length of CNVs for III 10, III 11, IV 14 and IV16 were 17.4 Kb, 9 Kb, 30 KB and 21.3 Kb respectively, and 50 calls ranging from 1M to 30M were precluded in further analysis. About 1,000 common CNVs were finally detected specific to MZ twins (Table S2), and almost all of these CNVs were copy number gain (Figure 2A). Subsequently, gene-set enrichment analysis for 533 genes covered by these CNVs illustrated that the tight junction signaling pathway was the significantly enriched pathway (p < .001, Table 3). In addition, 30 of them were involved in adherens junction, cell adhesion molecules, or related to focal adhesion. For all six genes containing novel autosomal SNVs in the epithelial adherens junction-signaling pathway and GTPase family-mediated signal pathway, only FGFR1 is located in a candidate CNV region (chr8:38266001-38270601), with 1.5–1.9 copies in all three individuals.
Discussion
To our knowledge, this family is the first documented pedigree with consecutive four-generation MZ twins. Interestingly, MZ twinning in this pedigree was a phenotype transmitted among female twins across four generations. Although familial MZ twinning is uncommon, Machin et al. (Reference Machin2009a) reviewed seven pedigrees and believed this phenotype should be an autosomal dominant trait with undetermined penetrance. However, genetic information is not available for these pedigrees. In this study, we first analyzed the polymorphisms of whole-genome sequences from one pedigree. This pedigree demonstrated an X-linked dominant inheritance pattern, but only one out of all twins was passing the trait to the next generation. One explanation could be the incomplete penetrance of the responsible site. We first screened for polymorphisms across the X chromosome, and identified six novel variations, and only one located at the exonic region.
Discordant phenotypes of MZ twins are common and widely discussed in genetic studies (van Dongen et al., Reference van Dongen, Slagboom, Draisma, Martin and Boomsma2012; Zwijnenburg et al., Reference Zwijnenburg, Meijers-Heijboer and Boomsma2010), and mtDNA heteroplasmy is believed to involve in the difference of phenotypes (Detjen et al., Reference Detjen, Tinschert, Kaufmann, Algermissen, Nurnberg and Schuelke2007; Li et al., Reference Li, Bi, Fan, Wu, Tang, Li and Yao2016). To examine whether mtDNA composition contributes to this pedigree, we also compared mtDNA composition in the maternal lineage. WGS datasets also contained the mtDNA sequences with relatively high coverage (>250 × in our data), which provided us an opportunity to explore them. However, there was highly consisted mtDNA compositions in MZ twins and non-twin family members, showing that mtDNA heteroplasmy cannot explain twinning hereditary and seemingly sex bias.
However, there still exists the possibility that the inheritance pattern observed in the family may be formed by coincidence due to limited amount of offspring in each trio family, and it may be an autosomal dominant trait with incomplete penetrance. Therefore, we also screened for the novel variations in autosomes specific to the MZ twins. Our analysis demonstrated a relatively major proportion of featured genetic polymorphisms in MZ twins of this family that came from genes related to cell junctions, either containing novel SNVs possibly having functional effects, or locating at CNVs specific to MZ twins. Previous studies believed that MZ twinning may originate from inner-cell mass separation during different stages of split (McNamara et al., Reference McNamara, Kane, Craig, Short and Umstad2016). Recently, Herranz (Reference Herranz2015) proposed an alternative “fusion” model in addition to the traditional “fission” model, which leads to the debate on evidences sustaining each hypothesis (Denker, Reference Denker2015; Herranz, Reference Herranz2015). However, potential interruption of cell–cell connections exists in both models. Similarly, in our genes containing novel SNVs shared by MZ twins, the FGFR1 gene in mice seems to be an interesting candidate. It is widely expressed in mouse embryo, and can be detected as early as embryonic day 4.5, as illustrated in the MGI database (Goldin & Papaioannou, Reference Goldin and Papaioannou2003). Meanwhile, some FGFR1 mutations are known to lead to diverse syndromes in an autosomal dominant manner. Additionally, mouse embryo also has MYH7B expression detected. It is still challenging to evaluate the functional impacts of those novel SNVs, as well as the novel CNVs detected (Conrad et al., Reference Conrad, Pinto, Redon, Feuk, Gokcumen, Zhang and Hurles2010). In all, 33 genes were involved in cell junctions-related pathways, including 3 genes with novel candidate SNVs and 30 genes covered by novel CNVs. Upstream regulation analysis illustrated that estrogen receptors (ESR1, ESR2) regulate seven of them (SMAD3, FGFR1, COL4A2, CLDN4, CDH4, AKT3, AKT2). Nevertheless, all these candidates may only increase the “susceptibility” of giving birth to MZ twins.
Here, our analysis indicated the novel changes in the X chromosome may be responsible for MZ twinning, and also provided candidates across autosomes, giving clues to increase our understanding about the underlying mechanism. However, due to no appropriate animal models, it is very challenging to evaluate the functional changes caused by them, which needs verification and more evidences from further efforts around the world. Moreover, dissecting the epigenetic factors as the most popular direction nowadays to explain the phenotype discordance between MZ twins (Castillo-Fernandez et al., Reference Castillo-Fernandez, Spector and Bell2014; Fraga et al., Reference Fraga, Ballestar, Paz, Ropero, Setien, Ballestar and Esteller2005) and epigenetic characterization for human germ cells may provide interesting clues (Baumann, Reference Baumann2015; Mill & Heijmans, Reference Mill and Heijmans2013; Zuccala, Reference Zuccala2016). This aspect was not included in our study due to ethical issues around obtaining germ cells.
Acknowledgments
The authors would like to thank all participants and Wubalem Desta Seifu at the Beijing Institute of Genomics for language editing. This work was supported by Innovation Promotion Association CAS (D.Z., 2016098) and National Natural Science Foundation of China (S.L., 31071104; C.Z., 31471199). The sponsor or funding organization had no role in the design or conduct of this research.
Author Contributions
All of the authors were involved in preparing this manuscript. D.Z., D.H., S.L. and C.Z. conceived and designed the experiments; S.L., K.C., L.H. and Y.O. collected the data; Z.X. and K.G. performed the sequencing experiments; Y.H., D.Z., J.G., S.L., K.C. and W.C. analyzed the data; D.Z., D.H. and Y.H. wrote the paper; all authors contributed to manuscript revision.
Disclosure of Interests
The authors declare no conflict of interest.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/thg.2018.41







