


The basis for each individual's opportunity for good health or risk of common complex disease, e.g. CVD or type 2 diabetes, is established at conception by the consortium of genetic variants acquired from each parent. However, many environmental factors acting throughout development in utero and for the duration of postnatal life influence the expression of those genetic variants as altered phenotype. For example, excellent epidemiological research and studies using animal models have established that being born small, especially if followed by faster than expected growth (early adiposity rebound), is a significant risk factor for several chronic diseases. These ideas are encapsulated in the developmental origins of health and disease hypothesis(Reference Gluckman and Hanson1). ‘High-risk’ lifestyles that are characterised by poor dietary choices, low levels of physical activity and exposure to environmental pollutants, including those in cigarette smoke, when sustained over long time periods, contribute to premature ageing and increase the likelihood that an individual will succumb to CVD, type 2 diabetes or other age-related and obesity-related diseases. These influences on individual susceptibility have been conceptualised in the ‘health pendulum’(Reference Mathers2).
These observations suggest a number of key questions including:
1. how is dietary exposure ‘remembered’ over long time periods. In the case of increased risk of type 2 diabetes in those poorly nourished in utero, the effects of the nutritional insult may not be apparent for decades so the ‘memory’ of the insult has to be retained for a substantial part of the lifespan;
2. how are the effects of dietary exposure ‘revealed’ as altered phenotype. At its most fundamental, this process is likely to involve altered gene expression;
3. what is the mechanistic (molecular) basis for individual differences in response to diet.
Epigenetics
Epigenetics is the science of chromatin modifications resulting in altered regulation of gene expression without involving changes in the primary DNA sequence. Chromatin is much more than just a clever means of packaging a large amount of DNA into the cell nucleus. It seems probable that this ‘smart packaging’ is a highly-sophisticated information store that regulates gene expression in response to environmental signals. The best understood epigenetic marks are (a) patterns of methylation of cytosine bases in DNA and (b) post-translational modifications of the histone tails that protrude from the globular core of eight proteins around which DNA is wrapped(Reference Bernstein, Meissner and Lander3). More than 100 modifications of histones including acetylation, methylation, phosphorylation and ubiquitination have been described(Reference Kouzarides4). Although the functional consequences of most of these ‘marks’ remain obscure, it is possible that individually and collectively they constitute a sophisticated code (the histone code(Reference Jenuwein and Allis5)) that regulates which genes are transcribed in which cells under specific circumstances. Throughout the genome most cytosine residues are methylated at position 5 in the cytosine ring. However, in certain parts of the genome, notably in the 5′ region upstream of genes, there are unusually dense collections of CpG dinucleotides known as CpG islands in which the cytosines are unmethylated. Indeed, a recent detailed study of human chromosomes 6, 20 and 22 has shown that there is little cytosine methylation near transcription start sites(Reference Eckhardt, Lewin and Cortese6). About half human genes have CpG islands in their promoter regions and, for genes that are being transcribed, these islands are normally unmethylated. In association with chromatin modifications, promoter methylation prevents access by the transcription machinery to specific DNA domains and results in gene silencing. In effect, the epigenetic marks act as a switch turning on or off expression of specific genes. Cell differentiation, X chromosome inactivation and genetic imprinting are all consequences of epigenetic processes. The totality of epigenetic marks in a given cell is described as the epigenome.
Many plants flower only after exposure to an extended cold period (winter), which is the process of vernalisation. The suppression of sexual reproduction is caused by the protein flowering locus C, which blocks flowering by repressing those genes needed for flower development. During vernalisation, expression of the flowering locus C gene is down regulated and it is now known that this down-regulation is a result of epigenetic silencing(Reference Bastow, Mylne, Lister, Lippman, Martienssen and Dean7). This process is a clear example in which the environment (in this case low temperature) is a major determinant of phenotype (flowering) by regulating gene expression through an epigenetic mechanism. A second (more nutritional) example of the role of epigenetics in linking environmental exposure with altered phenotype comes from the honeybee (Apis mellifera). To create a few queens among a vast number of workers, female larvae are fed the poorly-characterised ‘royal jelly’, which results in the production of larger adult bees with a very different physiology and lifespan (2 years v. a few weeks) than their worker siblings(Reference Kucharski, Maleszka, Foret and Maleszka8). Very recently, it has been shown that the different nutritional input to the larval queens results in a profoundly different developmental fate because of changes in epigenetic markings (altered DNA methylation(Reference Kucharski, Maleszka, Foret and Maleszka8)). More generally, evidence is accumulating to support the idea that the epigenome plays a key role in orchestrating the complex interplay between the environment and the genome that determines an individual's phenotype (Fig. 1)(Reference Zoghbi, Beaudet, Allis, Jenuwein and Reinberg9). These ideas form the basis for the hypotheses that epigenetic markings (a) enable plasticity of phenotype in a fixed genotype and (b) connect environmental exposure with gene expression and function(Reference Feinberg10). A simple model described as the ‘4 Rs of epigenomics’ has been developed to articulate the key processes underlying these hypotheses (Fig. 2)(Reference Mathers and McKay11). This model proposes that environmental exposures (notably diet) and, of course, time impact on the genome and that at least some of these impacts are ‘Received’ and ‘Recorded’ by epigenetic mechanisms. If such information is to be important in the medium to long term, then it must be ‘Remembered’ though subsequent cell divisions. For DNA methylation, this process of ‘memorisation’ is well understood since DNA methylation marks on the parental strand are copied to the daughter strand by the housekeeping DNA methyltransferase 1. The memorisation of histone modifications is much less well understood, but it is apparent that histones segregate randomly during cell division so that each daughter cell acquires some of the marked proteins, which then spread to the newly-deposited histones(Reference Hatchwell and JM12). Finally, the consequences of earlier environmental exposures may be ‘Revealed’ as altered gene expression leading to changes in cellular function with potential to modify health.
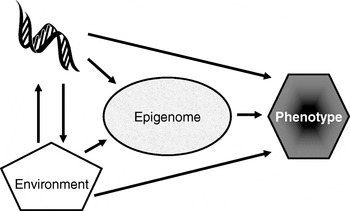
Fig. 1. Role of the epigenome in orchestrating the complex interplay between the environment and the genome that determines an individual's phenotype. (Adapted from Zoghbi & Beaudet(Reference Zoghbi, Beaudet, Allis, Jenuwein and Reinberg9).)
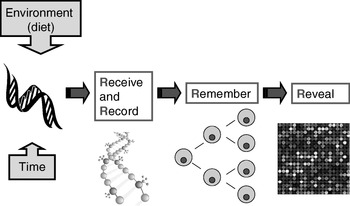
Fig. 2. The four ‘Rs’ of epigenomics. A conceptual model of the key processes through which altered epigenomic markings as a result of environmental (dietary) exposures are ‘Received’, ‘Recorded’, ‘Remembered’ and ‘Revealed’. (For more details, see Mathers & McKay(Reference Mathers and McKay11).)
Nutritional influences on epigenetic marking, gene expression and phenotype in mammals
The viable yellow agouti (A vy) mouse harbours an intracisternal A-particle transposon upstream from the Agouti locus, which acts as an alternative promoter for this gene. When this alternative promoter is active it results in mice with yellow coats and a propensity to obesity and obesity-related diseases(Reference Morgan, Sutherland, Martin and Whitelaw13). However, even among inborn strains, not all littermates have the same phenotype; some are born with greater or lesser amounts of brown fur and the darker-coloured mice are leaner. The latter phenotype is associated with methylation of cytosine residues in the intracisternal A-particle and provides a striking example of how phenotype can vary markedly as a result of epigenetic variability. In this mouse model altering maternal nutrition during pregnancy also affects epigenetic markings and phenotype. Higher intakes of methyl donors (folic acid, vitamin B12, choline and betaine) by the dams increase the proportion of mottled and pseudo-agouti offspring, which is accompanied by greater methylation of the cryptic promoter in the proximal end of the A vy intracisternal A-particle(Reference Waterland and Jirtle14). Such an effect might be anticipated if higher methyl donor supply facilitates more methylation of this locus. However, such a mechanism does not explain the remarkably similar effects on both phenotype and DNA methylation of including genistein (250 mg/kg diet) in the diet of the pregnant dams(Reference Dolinoy, Weidman, Waterland and Jirtle15). Since genistein is not a methyl donor, an alternative mechanistic explanation is required for the epigenetic silencing of the Agouti cryptic promoter and the subsequent changes in coat colour and reduced adiposity(Reference Dolinoy, Weidman, Waterland and Jirtle15). Recent studies in sheep have shown that limiting folate and vitamin B12 supply in the immediate peri-conceptual period only can result in fatter adult offspring with the expected higher blood pressure and greater insulin resistance(Reference Sinclair, Allegrucci and Singh16). Using restriction landmark genomic screening, these authors have subsequently shown that the observed phenotypic changes are associated with altered patterns of methylation at many genomic loci.
Environmental modulation of epigenetic marking producing altered phenotype is not restricted to development in utero. The adult offspring of rat dams that exhibit ‘better’ maternal behaviour (more licking and grooming of pups and arched-back nursing) during the first week of postnatal life show less fearfulness and have more modest hypothalamic–pituitary–adrenal responses to stress(Reference Liu, Diorio, Tannenbaum, Caldji, Francis, Freedman, Sharma, Pearson, Plotsky and Meaney17). Mechanistic studies have revealed that the better maternal behaviour is associated with demethylation of specific CpG sites in the glucocorticoid receptor gene within the hypothalamus affecting transcription factor binding to the glucocorticoid receptor promoter(Reference Weaver, Cervoni, Champagne, D'Alessio, Sharma, Seckl, Dymov, Szyf and Meaney18). The Canadian team undertaking these studies have speculated that other environmental exposures during the early postnatal period might have profound effects on behaviour in later life through interfering with epigenetic programme driven by maternal care(Reference Szyf, I and M19). There are several other examples of dietary factors influencing DNA methylation but in most cases the consequences, if any, for phenotype have not been established clearly.
Inter- and intra-individual variation in epigenetic markings
Monozygotic twins are a good model for studies of effects of environment v. genetics on phenotype since the members of such pairs are genetically identical. A ground-breaking study of monozygotic twins has reported that young twins are much more alike epigenetically than are older monozygotic pairs(Reference Fraga, E and Paz20). Not only are the older twin pairs epigenetically more distinct but they also have less-similar gene expression profiles(Reference Fraga, E and Paz20). In addition to the likely stochastic effects of age in modulating epigenetic marks(Reference Martin21), there is a priori evidence of the impact of (unknown) environmental factors, since those adult twins who have lived further apart for substantial periods of their lives have more dissimilar epigenetic markings(Reference Fraga, E and Paz20). This study provides evidence of altered DNA methylation patterns associated with altered gene expression despite identical genotypes. It provides a potential mechanistic explanation for divergent phenotype (e.g. disease experience) of monozygotic pairs. Further, it provides encouragement to investigate the role of nutrition as an environmental modifier of individual epigenomic patterns.
Much of the current data on inter-individual variation in DNA methylation profiles is restricted to analyses of nucleated blood cells, but it is apparent that there are also tissue differences in methylation patterns that may be associated with cellular differentiation(Reference Rakyan, Hildmann and Novik22). Indeed, studies of CpG islands in the CYP21A2 and TNF genes have revealed complex inter- and intra-individual variation in patterns of methylation of cytosine residues(Reference Rakyan, Hildmann and Novik22).
To a large extent, the determinants of differential DNA methylation patterns are unknown but it would be reasonable to suppose that both genetic and environmental factors will contribute, with the balance between these two types of factor differing in particular circumstances. Detailed analysis of the extent of methylation of selected domains within the maternally-imprinted gene encoding the insulin-like growth factor II has shown inter-individual variation among human twins(Reference Heijmans, Kremer, Tobi, Boomsma and Slagboom23). However, the estimated heritability of methylation is high (ranging from 57% to 97% for individual CpG sites) and there is no evidence that heritability is less in middle-aged twins than in adolescent twins(Reference Heijmans, Kremer, Tobi, Boomsma and Slagboom23). From these observations it appears that for methylation of the insulin-like growth factor II gene (at the genomic sites investigated), genetic factors rather than age or environmental exposures are the dominant explanatory factors.
In seeking genetic explanations for inter-individual differences in DNA methylation patterns the genes encoding DNA methyltransferases are an obvious target. There are at least five members of this gene family(Reference Bestor24), with DNA methyltransferase 1 being the housekeeping enzyme responsible for maintaining DNA methylation patterns through cell division whilst DNA methyltransferase 3A and 3B play roles in de novo methylation. Several polymorphisms in DNA methyltransferases are known(Reference Cebrian, Pharoah and Ahmed25), which will form the basis for genotype–epigenotype (and phenotype) association studies.
Anticipated developments
Epigenetics has been described by the National Institutes of Health (Bethesda, MD, USA) as an emerging frontier of science(26) and there is ample evidence of burgeoning interest in the field in many biological sciences and especially research on the biological determinants of health. This aspect is best illustrated by the moves to initiate the Human Epigenome Project, which aims to identify, catalogue and interpret genome-wide methylation patterns of all human genes in all major tissues(Reference Brena, Huang and Plass27). Initial work on bisulfite DNA sequencing of human chromosomes 6, 20 and 22 has provided approximately 2×106 CpG methylation values from twelve tissues(Reference Eckhardt, Lewin and Cortese6). Over one-sixth of the 873 genes analysed show differential methylation in the 5′ untranslated region and in one-third of cases, these differentially methylated 5′ untranslated regions are inversely correlated with transcription(Reference Eckhardt, Lewin and Cortese6), which is what would be expected if methylation is acting as a transcription switch.
This growing research impetus to undertake the Human Epigenome Project has been stimulated by two major factors. The first is the realisation that, despite the explosion of genomic information emerging from the genome mapping projects, these data are of limited utility without information on what genes are expressed in given circumstances. This need drives research to understand the complexities of chromatin structure and how epigenetic information regulates gene expression. The second major factor is technological developments. Until recently, most of the techniques for studying DNA methylation have provided information on either the extent of methylation of the whole-cell DNA (without reference to genomic location) or targeted information on single genes. The earliest approach to genome-wide profiling used restriction landmark genomic scanning, which involves two-dimensional electrophoresis to separate genomic fragments after digestion with methylation-sensitive endonucleases. This approach has been successful in identifying differentially-methylated sequences in sheep exposed to a nutritional intervention peri-conceptually(Reference Sinclair, Allegrucci and Singh16). However, restriction landmark genomic scanning is labour intensive and is associated with considerable bioinformatics problems in identifying the differentially-methylated sequences. Rapid developments in oligonucleotide tiling arrays to provide increasingly comprehensive coverage of the genome at more modest cost make this approach the current state-of-the-art. However, the dominance of array-based methodologies is being challenged by the availability of the next-generation sequencing platforms, which provide astonishingly-high throughput at lower cost. Already, massively parallel bisulfite pyrosequencing has been used to identify tumour-associated methylated DNA loci in blood samples, which may be useful as novel biomarkers of breast cancer(Reference Korshunova, Maloney and Lakey28). It has been predicted that the first sequencing-based mammalian methylome (whole-genome DNA methylation profile) will be reported in 2008(Reference Beck and Rakyan29). Looking a little further ahead (perhaps 2015), it is possible that quite different technologies such as positron emission tomography scanning will be sufficiently powerful and sensitive to provide non-invasive whole-body imaging of the methylome in specific tissues(Reference Beck and Rakyan29).
From the nutrition researcher's perspective, these developments are exciting and will provide the tools with which to discover the impact of nutritional status and/or dietary exposures on the genome and its implications for health. In addition, if, as seems likely, epigenomic marks record a wide variety of environmental exposures there is an opportunity to explore such marks for evidence of long-term dietary choices. Such research could be the basis for the development of radically-new tools to characterise (and quantify) nutritional intake over long time periods that avoid the substantial limitations of current memory and recording-based approaches.
Conclusions
It is now established that epigenetic processes allow plasticity of phenotype in a fixed genotype and that altered epigenetic markings are implicated in the aetiology of several diseases and may be causal for some(Reference Feinberg10). Although there is proof of principle that dietary factors influence epigenetic markings and gene expression and through such mechanisms may modify health, research in nutritional epigenomics is just beginning and there are many unanswered questions. These questions include:
1. which genes, contributing to altered diseases susceptibility, are epigenetically deregulated by dietary and other environmental exposures;
2. which environmental (nutritional) factors, in what doses and over what time scales, affect epigenomic markings;
3. in what periods of the life course (or other circumstances) is the genome especially vulnerable to altered epigenetic markings by dietary factors and are such changes sustained over long time periods;
4. can nutritional interventions modulate the adverse effects of other environmental exposures, e.g. xenobiotics, or of ageing;
5. can epigenomic marks form the basis for novel diet-related biomarkers of disease risk(Reference Jirtle and Skinner30).
Epigenetic heterogeneity among individuals is now established as a functionally-important mechanism responsible for individual phenotypic differences(Reference Brena, Huang and Plass27) and looks likely to be a very fruitful area for research aimed at understanding inter-individual differences in responses to diet.
Acknowledgements
Nutritional epigenomics research in my laboratory is funded by the Biotechnology and Biological Sciences Research Council and the Engineering and Physical Sciences Research Council through the Centre for Integrated Systems Biology of Ageing and Nutrition (BB/C008200/1) and by ‘The European Nutrigenomics Organisation; linking genomics, nutrition and health research’ (NuGO; CT-2004-505944); which is a Network of Excellence funded by the EC's Research Directorate General under Priority Thematic Area 5, Food Quality and Safety Priority, of the Sixth Framework Programme for Research and Technological Development.


