


I. INTRODUCTION
Currently, magnesium hydride is the subject of many scientific studies as a promising material for hydrogen storage. Obstacles to its widespread use are the high decomposition temperature (~300 °C) and the slow kinetics of hydrogen absorption–desorption. Traditionally, the solution to these problems is found in nanostructuring that can be achieved, for example, by mechano-chemical treatment (Huot et al., Reference Huot, Liang, Boily, Van Neste and Schulz1999; Paskevicius et al., Reference Paskevicius, Sheppard and Buckley2010), use of effective catalysts (Schimmel et al., Reference Schimmel, Huot, Chapon, Tichelaar and Mulder2005; Varin et al., Reference Varin, Zbroniec, Polanski and Bystrzycki2011) or by introducing of additives that alter the thermodynamic properties of the system (Jin et al., Reference Jin, Shim, Ahn, Cho and Yi2007a; Cuevas et al., Reference Cuevas, Korablov and Latroche2012). Nanosized metals may have lower melting point or spontaneous alloying as compared with their bulk form (Hirokazu and Kitagawa, Reference Hirokazu and Kitagawa2012). In addition, nanostructuring by mechano-chemical treatment often improve kinetics but may also lead to surface contamination with material from the milling equipment (Pasquini et al., Reference Pasquini, Callini, Brighi, Boscherini, Montone, Jensen, Maurizio, Antisari and Bonetti2011; Huot et al., Reference Huot, Ravnsbæk, Zhang, Cuevas, Latroche and Jensen2013). A remaining challenge is the stabilization of magnesium nanoparticles and preventing agglomeration, sintering, oxidation, and potential side reactions leading to rapid deterioration of material properties upon cycling (Sato and Ishikawa, Reference Sato and Ishikawa2012). Nanoconfinement of magnesium has been investigated and was shown to increase the kinetics for hydrogen release and uptake and may also provide improved thermodynamic properties (Nielsen et al., Reference Nielsen, Manickam, Hirscher, Besenbacher and Jensen2009, Reference Nielsen, Besenbacher and Jensen2011; Paskevicius et al., Reference Paskevicius, Sheppard and Buckley2010; Vajo, Reference Vajo2011).
A wide range of different types of additives including metals, oxides, halides, and hydrides has been considered for possible prolific effects on hydrogen release and uptake. Nanonickel (n-Ni) is well established as efficient catalyst for the Mg/MgH2 system, which acts via the intermediate Mg2NiH4 (Jensen et al., Reference Jensen, Andreasen, Vegge, Andreasen, Stahl, Pedersen, Nielsen, Molenbroek and Besenbacher2006; Varin et al., Reference Varin, Czujko and Wronski2009, Reference Varin, Zbroniec, Polanski and Bystrzycki2011; Pasquini et al., Reference Pasquini, Callini, Brighi, Boscherini, Montone, Jensen, Maurizio, Antisari and Bonetti2011; Callini et al., Reference Callini, Pasquini, Jensen and Bonetti2013). Other metals, e.g., Al, Ti, Cu, Pd may have both a thermodynamic and kinetic effect by forming magnesium containing alloys (Andreasen et al., Reference Andreasen, Sorensen, Burkarl, Moller, Molenbroek, Pedersen, Andreasen, Nielsen and Jensen2005, Reference Andreasen, Sørensen, Burkarl, Møller, Molenbroek, Pedersen, Vegge and Jensen2006; Callini et al., Reference Callini, Pasquini, Rude, Nielsen, Jensen and Bonetti2010). In particular, titanium is relevant because of low costs, lighter weight, and fast kinetics for hydrogen release and uptake for Mg/MgH2. Moreover, magnesium does not form intermetallic compounds with V and Nb (Massalski, Reference Massalski1990). Therefore, both metals are considered as heterogeneous catalysts (Schimmel et al., Reference Schimmel, Huot, Chapon, Tichelaar and Mulder2005).
Transition metal oxides are also good catalysts for enhancement of hydrogen release and uptake e.g., Nb2O5 (Barkhordarian et al., Reference Barkhordarian, Klassen and Bormann2006) and V2O5 (Oelerich et al., Reference Oelerich, Klassen and Bormann2001a, Reference Oelerich, Klassen and Bormann2001b; Jung et al., Reference Jung, Lee and Lee2006). A drawback may be that some metal oxides have a tendency to reduce during cycling hydrogen release and uptake, which leads to the formation of inert magnesium oxide. The detailed mechanism for metal oxides prolific action may be complex and in some cases is not fully understood. An in situ synchrotron radiation powder X-ray diffraction (SR-PXD) investigation of MgH2 and Nb2O5 (8 mol%) revealed reduction of Nb(V) and formation of Mg1− x Nb x O, which may facilitate release and uptake of hydrogen (Nielsen and Jensen, Reference Nielsen and Jensen2012).
Transition metal halides have also been successfully utilized as additives for improving the kinetics of the absorption/desorption of hydrogen in MgH2, e.g., VCl3 and TiCl3 (Malka et al., Reference Malka, Czujko and Bystrzycki2010). Moreover, halides of Fe, Ni, and Nb have also shown attractive catalytic properties (Ivanov et al., Reference Ivanov, Konstanchuk, Bokhonov and Boldyrev2003; Deledda et al., Reference Deledda, Borissova, Poinsignon, Botta, Dornheim and Klassen2005; Bhat et al., Reference Bhat, Rougier, Aymard, Darok, Nazri and Tarascon2006; Kim et al., Reference Kim, Ahn, Jin, Lee, Chung, Shim, Cho and Oh2008; Ma et al., Reference Ma, Kang, Dai, Liang, Fang, Wang, Wang and Cheng2009).
Transition metal oxides and halides may in some cases react with MgH2 or Mg and form other compounds. Currently, there is no clear picture of their mechanism of action and the active compound may not be known (Barkhordarian et al., Reference Barkhordarian, Klassen and Bormann2006; Jin et al., Reference Jin, Shim, Cho and Yi2007b). This has prompted the present investigation of binary early d-block hydrides, oxides, and chlorides used as additives for the Mg/MgH2 system. We utilize in situ SR-PXD to provide a more detailed picture of mechanism for hydrogen release and uptake as well as identification of active compounds in the mixtures.
II. EXPERIMENTAL
A. Sample preparation
Mixtures of MgH2 (98%, Alfa Aesar) with 5 mol% of additive were ball milled under argon atmosphere using tungsten carbide (WC) hard alloy vial (80 ml) and balls (10 mm diameter) in a Fritsch Pulverisette P4 planetary mill. As additives, commercially available chemicals were used: vanadium pentoxide (V2O5, 98 + %, Aldrich), titanium hydride (TiH2, 98%, Aldrich), zirconium hydride (ZrH2, 99%, Aldrich), vanadium chloride (VCl3, 97%, Aldrich), and scandium chloride (ScCl3, 99.9%, Aldrich). The typical mass of each mixture constituted 3 g with 1 : 41 powder-to-ball weight ratio. The milling was performed in time intervals of 30 min at a rotation speed of 300 rpm (Huot et al., Reference Huot, Ravnsbæk, Zhang, Cuevas, Latroche and Jensen2013).
Experimental details regarding samples composition and preparation can be found in Table I. All handling and manipulation of the chemicals were carried out under argon atmosphere in an MBraun Unilab glove box with a recirculation gas purification system and gas/humidity sensors. Oxygen and water levels were kept well below 1 ppm during all operations.
Table I. Composition of the MgH2 + additives samples prepared mechano-chemically.
B. In situ synchrotron radiation powder X-ray diffraction (SR-PXD)
The detailed reaction mechanisms and kinetics of hydrogen release and uptake in MgH2 with different additives were studied by in situ SR-PXD experiments. Data were collected at beamline I711 at the synchrotron MAX II, in the research laboratory MAX-lab, Lund, Sweden using a Mar165 CCD detector. A LaB6 external standard was used to determine sample-detector distance, detector orientation, and the wavelength. The samples were mounted in sapphire single crystal tubes (Al2O3, o.d. 1.09 mm, i.d. 0.79 mm) in an argon-filled glove box (p(O2, H2O) <1 ppm). The sample holder was specially developed for the study of gas/solid reactions and hydrogen pressures were allowed up to 20 MPa and temperatures from room temperature (RT) to 800 °C (Jensen et al., Reference Jensen, Nielsen, Filinchuk, Jorgensen, Cerenius, Gray and Webb2010). The selected wavelength was 0.94608 Å and the X-ray exposure time was 5 s per PXD pattern. Samples for investigation of mechanism and kinetics of hydrogen release and uptake were heated stepwise to fixed temperatures of 270 and 320 °C (15 °C min−1) in a hydrogen pressure of 100–150 bar. The hydrogen pressure was reduced to p(H2) ~10−2 bar (dynamic vacuum) when the fixed temperature was reached and then again increased to 100–150 bar when the sample was fully dehydrogenated (after 15 to 30 min) measuring one full hydrogen release and uptake cycle at each temperature. Selected samples were additionally heated to 450 °C and two extra H2 desorption/absorption cycles were performed. Finally, the samples were cooled to 270 °C and one dehydrogenation was conducted.
All obtained raw images were transformed to two-dimensional powder patterns using the FIT2D program (Hammersley et al., Reference Hammersley, Svensson, Hanfland, Fitch and Hausermann1996), which was also used for calibration using measurements of the standard NIST LaB6 sample. Crystal structure refinements were performed using the Rietveld method implemented in the Fullprof program (Rodríguez-Carvajal, Reference Rodríguez-Carvajal1993). The backgrounds were described by linear interpolation between selected points, while pseudo-Voigt profile functions were used to fit the diffraction peaks. Sequential refinements provide the weight fractions for the compounds present in the sample as a function of time. The extracted normalized curves represent phase formation or decomposition curves as a function of time denoted α(t). These sigmoidal shaped curves can be used for evaluation of the activation process.
The decomposition curves for MgH2 can be fitted well with the equation of shrinking core model:
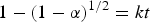 $$1 - \lpar 1 - \alpha \rpar {^{1/2}}=kt$$
$$1 - \lpar 1 - \alpha \rpar {^{1/2}}=kt$$
where α is the phase fraction at time t and k is the rate constant (Liang et al., Reference Liang, Huot, Boily and Schulz2000; Varin et al., Reference Varin, Czujko and Wronski2009).
An apparent activation energy, E A, and a pre-factor, A, for the overall reaction can be calculated using Arrhenius law:
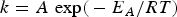 $$k=A\, \, {\rm exp\lpar } - {E_A}/RT\rpar $$
$$k=A\, \, {\rm exp\lpar } - {E_A}/RT\rpar $$
where R is the gas constant and T is the absolute temperature.
III. RESULTS AND DISCUSSION
A. Reactions during mechano-chemical and heat treatment
Dispersion of transition metals in Mg/MgH2 system is difficult because of the ductility of the metals. Therefore, binary salt-like oxides, chlorides, and hydrides based on early d-block metals were selected as additives in the present investigation, i.e., V2O5, VCl3, ScCl3, TiH2, and ZrH2.
Figures 1–5 show in situ SR-PXD data for the ball-milled samples S1–S5 (see Table I), respectively. Bragg diffraction peaks of β-, γ-MgH2, and MgO are observed for sample S1 (Figure 1) at RT. There is no observed diffraction from V2O5, which suggests that a redox reaction has occurred during mechano-chemical treatment [see reaction scheme (3)]. Decomposition of MgH2 initiates at 270 °C by decreasing excess hydrogen pressure below equilibrium at this temperature. All γ-MgH2 disappears within <1 min of heating at 270 °C and most of the present β-MgH2 (90%) is decomposed after 10 min (during measurement of 40 PXD patterns). The PXD data show the presence of Mg, MgO, and possibly V at 270 °C. The latter two phases are difficult to distinguish owing to overlapping Bragg reflections.
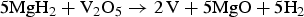 $$5{\rm Mg}{{\rm H}_ 2}+{{\rm V}_ 2}{{\rm O}_ 5} \to 2\, {\rm V}+5{\rm MgO}+5{{\rm H}_ 2}$$
$$5{\rm Mg}{{\rm H}_ 2}+{{\rm V}_ 2}{{\rm O}_ 5} \to 2\, {\rm V}+5{\rm MgO}+5{{\rm H}_ 2}$$

Figure 1. In-situ SR-PXD data measured for sample MgH2–V2O5 (S1) heated under p(H2) = 100 bar from RT to 270 °C (15 °C min−1) and subsequently dehydrogenated and hydrogenated at this temperature applying alternately p(H2) = 10−2 and 100–150 bar (λ = 0.946 08 Å).

Figure 2. In-situ SR-PXD data measured for sample MgH2–ZrH2 (S2) heated under p(H2) = 100 bar from RT to 270 °C (15 °C min−1) and subsequently dehydrogenated at this temperature applying p(H2) = 10−2 (λ = 0.946 08 Å).

Figure 3. In-situ SR-PXD data measured for sample MgH2–TiH2 (S3) heated under p(H2) = 100 bar from RT to 270 °C (15 °C min−1) and subsequently cycled at this temperature applying alternately p(H2) = 10−2 and 100–150 bar (λ = 0.946 08 Å).

Figure 4. In-situ SR-PXD data measured for sample MgH2–ScCl3 (S4) heated under p(H2) = 100 bar from RT to 270 °C (15 °C min−1) and subsequently cycled at this temperature applying alternately p(H2) = 10−2 and 100–150 bar (λ = 0.946 08 Å).

Figure 5. In-situ SR-PXD data measured for sample MgH2-VCl3 (S5) heated under p(H2) = 100 bar from RT to 270 °C (15 °C min−1) and subsequently cycled at this temperature applying alternately p(H2) = 10−2 and 100–150 bar (λ = 0.946 08 Å).
PXD patterns of the samples S2 and S3 measured at RT reveal diffracted intensities from the reactants only, i.e., no reaction has occurred between ZrH2 or TiH2 and MgH2. Notice that there is no observed diffraction from MgO in sample S2 and S3. This indicates that the presence of MgO in S1 is because of a reaction between V2O5 and MgH2. Magnesium metal formation in both samples, S2 and S3, initiates at 270 °C when the pressure is reduced.
The as-milled chloride doped sample S4 reveals diffraction from MgH2 and scandium(II) hydride ScH2 and no diffraction from the additive, ScCl3. This indicates that a redox reaction has occurred during ball-milling [see reaction scheme (4)]. Formation of magnesium initiates when the pressure is reduced at 270 °C. Formation of MgCl2 is observed during the rehydrogenation at p(H2) = 100 bar.
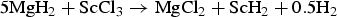 $$5{\rm Mg}{{\rm H}_ 2}+{\rm ScC}{{\rm l}_ 3} \to {\rm MgC}{{\rm l}_ 2}+{\rm Sc}{{\rm H}_ 2}+0.5{{\rm H}_2}$$
$$5{\rm Mg}{{\rm H}_ 2}+{\rm ScC}{{\rm l}_ 3} \to {\rm MgC}{{\rm l}_ 2}+{\rm Sc}{{\rm H}_ 2}+0.5{{\rm H}_2}$$
Sample S5 (MgH2 − VCl3) behaves in a similar way, i.e., no observation of VCl3 after milling, crystallization of MgCl2 during rehydrogenation (see Figure 5). Furthermore, unidentified reflection reveals a continuous change in composition at 2θ = 24.9–25.4° under vacuum.
B. Evaluation of additives kinetic effect
In situ SR-PXD data are used to investigate the effect of selected early d-block hydrides, oxides, and chlorides additives on the mechanism of hydrogen release and uptake in magnesium hydride system at 270 °C. The diffracted intensities from MgH2 are integrated, normalized, and converted to phase fraction and used to visualize decreasing amounts of MgH2 during dehydrogenation reactions (Figure 6). At 270 °C the weakest kinetic effect for hydrogen desorption is provided by ScCl3 (S4) and TiH2 (S3) and the strongest influence is provided by VCl3 (S5) and V2O5 (S1). In the early stages of MgH2 decomposition V2O5 demonstrates the fastest kinetics, which is somewhat slowed down in the late stage of the decomposition. In contrast, the effect from VCl3 additive is also significant in the late stage of the decomposition. Zirconium hydride (S2) has an intermediate kinetic effect. A similar analysis of additives effect was also performed at higher temperature of 320 °C and the results are presented in Figure 7. The time scale for these experiments is clearly shorter as compared with those performed at 270 °C. In addition, the additives ability to accelerate the reaction kinetics follows the sequence: ScCl3 < TiH2 < ZrH2 < VCl3 < V2O5. These experiments reveal that the effect of increasing temperatures on the acceleration of reaction rate is more pronounced than the different additives effect. It should be emphasized that commercial MgH2 ball-milled without additives in the same conditions does not desorb hydrogen at 270 nor at 320 °C. Slight yield of hydrogen begins to be observed only at temperatures above 350 °C.
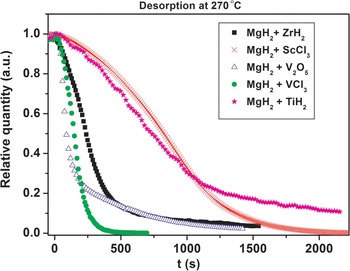
Figure 6. (color online) Integrated normalized diffracted intensities for samples S1–S5 showing changes in the relative amount of MgH2 at 270 °C as a function of time.

Figure 7. (color online) Integrated normalized diffracted intensities for samples S1–S5 showing changes in the relative amount of MgH2 at 320 °C as a function of time.
C. Activation of magnesium hydride via a chemical reaction
Analysis of the additives effect (Section B) revealed that the best results were obtained using the addition of V2O5. Here we further analyse the mechanism for this effect. Bragg diffraction peaks of β-, γ-MgH2 and MgO are observed for MgH2–V2O5 (S1) (see Figure 1), at RT. There is no observed diffraction from V2O5, which suggest that a redox reaction has occurred during mechano-chemical treatment [see reaction scheme (3)]. To crystallize possible reaction products the same sample was heated to 450 °C and 2 cycles of hydrogen release and uptake were conducted (see Figure 8). Diffraction peaks from V2H are clearly observed at 2θ = 24.5° and 35.9° in the hydrogenated state when the temperature reaches 450 °C and for V in the dehydrogenated state at 2θ = 25.4° and 36.4°. This leads to the splitting of the peak previously assigned to MgO and V, at 2θ ≈ 26° in Figure 1. Apparently, the heating procedure fully reduced the possibly remaining V2O5 and facilitated crystallization of V2H/V. The experiment was continued using the same sample (see Figure 9), the temperature was decreased to 270 °C and one dehydrogenation was conducted similar to what is shown in Figure 1. Presence of vanadium and vanadium hydride is more clearly observed in Figure 9 as compared with Figure 8 and during dehydrogenation diffraction peaks from V2H (at 2θ = 24.4°) continuously change position to 2θ = 25.5° because of the release of hydrogen and formation of V.

Figure 8. In-situ SR-PXD data of the activation process for sample MgH2–V2O5 (S1) heated under p(H2) = 100 bar from 320 to 450 °C (15 °C min−1) and subsequently cycled at this temperature applying alternately p(H2) = 10−2 and 100–150 bar (λ = 0.946 08 Å).

Figure 9. In-situ SR-PXD data of post-activation desorption measured for sample MgH2–V2O5 (S1) cooled from 450 to 270 °C under p(H2) = 100 bar (15 °C min−1) where the pressure was decreased to p(H2) = 10−2 bar (λ = 0.946 08 Å).
The activation procedure, i.e., cycling at 450 °C clearly improves the desorption kinetics. This is illustrated in Figure 10 where extracted decomposition curves for MgH2 are shown using data from Figures 1 and 9. After the activation process (Figure 8) the release of hydrogen from MgH2 is both faster and more complete. This may be related to the formation of more V2H/V in the sample and the fact that the hydrogen permeability of vanadium is large and exceeds that of palladium (Dolan, Reference Dolan2010). It is known that Nb and V based alloys are promising membranes materials, which currently are developed instead of Pd based alloys (Yukawa et al., Reference Yukawa, Nambu and Matsumoto2012). Therefore, the V2H/V system may be the active catalytic component for the MgH2–V2O5 system.
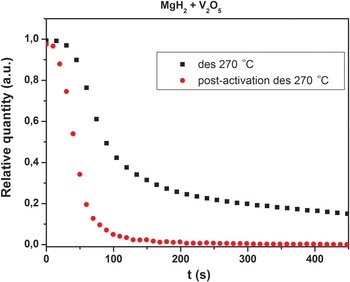
Figure 10. (color online) Comparison of the kinetic curves for MgH2 decomposition at 270 °C before and after the activation of MgH2–V2O5 (S1).
D. Apparent activation energy for the MgH2 decomposition
The decomposition curves for MgH2–V2O5 (S1) can be fitted well using the contracting surface model (two-dimensional growth) (Bamford and Tipper, Reference Bamford, Tipper, Brown, Dollimore and Galwey1980), e.g., kinetic shrinking core model, which suggests that hydrogen desorption is interface controlled (Liang et al., Reference Liang, Huot, Boily and Schulz2000; Varin et al., Reference Varin, Czujko and Wronski2009).
An Arrhenius plot of the natural logarithm to the rate constant as a function of inverse temperature is provided in Figure 11. The calculated apparent activation energy E A = 43 kJ mol−1 is obtained for the MgH2–V2O5 sample S1 after milling. This value compares well to the activation energy for long range hydrogen diffusion in bulk Mg determined from experimental data to be E A = 40.2 kJ mol−1 (Renner and Grabke, Reference Renner and Grabke1978). For the fully activated sample S1 the compounds V/V2H are identified to be the active catalytic compounds, which facilitate hydrogen release. The in situ SR-PXD data also allow to extract an apparent activation energy for the activated sample, E A = 34 kJ mol−1 (see Figure 11). These measured values are low, in particular when compared with that for bulk (unmilled) and air-exposed MgH2, E A = 300 kJ mol−1 (Jensen et al., Reference Jensen, Andreasen, Vegge, Andreasen, Stahl, Pedersen, Nielsen, Molenbroek and Besenbacher2006). Apparent activation energies of E A = 52 and 120 kJ mol−1 are measured for mechano-chemically treated MgH2 with and without vanadium (5 mol%) as additive (Liang et al., Reference Liang, Huot, Boily, Van Neste and Schulz1999b, Reference Liang, Huot, Boily and Schulz2000).

Figure 11. (color online) Arrhenius plot of the kinetic data for dehydrogenation of MgH2–V2O5 (S1).
Thus, this kinetic analysis reveals that in situ synthesis of small particles of V/V2H in the sample during cycling appears to have a stronger catalytic effect on hydrogen release and uptake in magnesium/magnesium hydride system. This may be because of a more uniform distribution of catalytic active particles compared with mechano-chemically prepared samples of MgH2–V. There are inherent difficulties with obtaining uniform distribution of ductile metals in MgH2 in contrast to brittle oxides that tend to form more homogenous distributed samples. The drawback is that considerable amounts of inert MgO are formed during reduction of V2O5 in the MgH2–V2O5 system.
E. On the catalytic properties of vanadium pentoxide and vanadium metal
There are several studies that report high catalytic activity of V2O5 for release and uptake in magnesium hydride (Oelerich et al., Reference Oelerich, Klassen and Bormann2001a, Reference Oelerich, Klassen and Bormann2001b; Barkhordarian et al., Reference Barkhordarian, Klassen and Bormann2006; Jung et al., Reference Jung, Lee and Lee2006). Vanadium(V) oxide, similar to niobium(V) oxide, is readily reduced in contact with hydrogen or mechano-chemically treated with MgH2 (Jung et al., Reference Jung, Lee and Lee2006; Nielsen and Jensen, Reference Nielsen and Jensen2012). Several lower oxides may act as intermediates during the formation of MgO. The final product is shown here to be vanadium, which also participate in hydrogen release and uptake via V/V2H system.
As mentioned above vanadium does not form alloy or intermetallic compounds with Mg (Schimmel et al., Reference Schimmel, Huot, Chapon, Tichelaar and Mulder2005) thus it can act as a pure catalyst. MgH2-5 mol%V composite desorbs hydrogen at 473 K (under vacuum) and re-absorbs hydrogen rapidly even at RT (Liang et al., Reference Liang, Huot, Boily, Van Neste and Schulz1999b). The desorption rate was the highest for this mixture and decreased in the following order of additives: Ti, Mn, Fe, and Ni (Liang et al., Reference Liang, Huot, Boily, Van Neste and Schulz1999a).
Moreover, MgH2-5 mol%V system has the fastest kinetics for hydrogen release and uptake in magnesium hydride in comparison with Ti and is similar to Nb additive (Charbonnier et al., Reference Charbonnier, de Rango, Fruchart, Miraglia, Pontonnier, Rivoirard, Skryabina and Vulliet2004). Mg-10 wt.%V system exhibits the highest rate of hydrogen desorption and shows the lowest activation energy among Y and Zr additives (Czujko et al., Reference Czujko, Varin, Chiu and Wronski2006). In addition, vanadium itself reversibly absorbs and desorbs 2.3 wt% of hydrogen at RT, which is also larger than that of the known alloys, LaNi5, FeTi (Varin et al., Reference Varin, Czujko and Wronski2009; Nakamura et al., Reference Nakamura, Kato, Takamatsu, Fuura, Oosawa and Tsunokake2012).
IV. CONCLUSION
Using the same conditions for the synthesis (mechano-chemical treatment) and evaluation of properties (hydrogen-vacuum cycling) a comparative analysis was carried out and the best from the studied catalysts for desorption/absorption of hydrogen for commercial powder MgH2 was determined. It was found that the fastest kinetics at 320 °C shows the sample with additive of 5 mol% V2O5. Additional activation of the system (2 cycles at 450 °C) leads to the significant improvement and acceleration of kinetics even at lower 270 °C temperature. Observed effect is achieved through the full reduction of vanadium oxides, the formation of new efficient catalyst in the form of ultrafine particles of vanadium and purification of interface MgH2/V in the process of heating to 450 °C in dynamic vacuum. This reduces the apparent activation energy of hydrogen desorption below the values of the activation energy of hydrogen diffusion in Mg, owing to the joint effect of nanostructuring and high hydrogen permeability of vanadium catalyst, which strongly bound to the surface of the MgH2 crystallites. Thus the real catalyst for the hydrogen release/uptake in the MgH2 is nanoparticles of pure vanadium obtained by the full reduction of V2O5 that was used originally.
ACKNOWLEDGEMENTS
The authors acknowledge funding for this research from the Danish Research Council for Natural Sciences (Danscatt). Moreover, the work was supported by the Danish National Research Foundation (Centre for Materials Crystallography), the Danish Strategic Research Council (Centre for Energy Materials), and the Carlsberg Foundation and the Swedish Research Council. The access to beam time at the MAX II synchrotron, Lund, Sweden in the research laboratory MAX-lab is gratefully acknowledged. Support from EU COST action MP1103 is also acknowledged.












