


INTRODUCTION
Salmonellosis is a leading cause of gastroenteritis that is frequently caused by Salmonella enterica subspecies enterica serovar Typhimurium (S. Typhimurium) [Reference Rabsch, Tschape and Baumler1]. Most human cases of salmonellosis are thought to occur following ingestion of contaminated food or water [Reference Rabsch, Tschape and Baumler1], with some large foodborne outbreaks reported [Reference Kivi2–Reference Luzzi4]. The impact of a food- or waterborne disease outbreak on a population can be reduced if the outbreak is detected early and the contaminated source identified and removed from distribution. Many surveillance systems used to both detect and investigate outbreaks of food- and waterborne disease rely, in part, on laboratory characterization or typing data [Reference Fisher and Threlfall5–Reference Koopmans7].
Bacterial typing and subtyping facilitate identification of common-source outbreaks and enhance epidemiological investigations, by differentiating outbreak-related cases from sporadic cases and confirming the implicated reservoir. The gold standard methods for typing S. Typhimurium are phage typing [Reference Anderson8, Reference Rabsch, Schatten and Eisenstark9] and macrorestriction using pulsed-field gel electrophoresis (PFGE) [Reference Fisher and Threlfall5, Reference Gerner-Smidt6]. These methods have recognized limitations, with the most important being inadequate discriminatory power to distinguish common-source S. Typhimurium outbreaks from sporadic cases [Reference Ribot10–Reference Ross and Heuzenroeder13]. More discriminatory methods need to be used if laboratory data are to contribute more useful information to outbreak investigations. It is also advantageous to use methods that are easy to perform, easy to interpret, easily standardized, and generate rapid results that are easily comparable between laboratories.
Multiple-locus variable-number tandem-repeat analysis (MLVA) is an alternative, PCR-based subtyping method that utilizes the variation in multiple tandem-repeat sequences to distinguish non-clonal isolates [Reference Vergnaud and Pourcel14]. Methods are available for a number of pathogens [Reference Vergnaud and Pourcel14], including S. Typhimurium [Reference Lindstedt15, Reference Lindstedt16]. Reports suggest that MLVA produces rapid results, has high throughput potential, and is highly discriminatory. For S. Typhimurium the discriminatory power of MLVA is dependent on the phage type being assessed [Reference Lindstedt16, Reference Torpdahl17]. In New Zealand a large proportion of the reported S. Typhimurium infections are caused by a small number of definitive phage types (DT) [18]. As different S. Typhimurium phage types cause infections in New Zealand compared to other countries, it was important to determine if MLVA is suitable for analysis of these isolates. This will ensure that the power of MLVA is understood for these phage types and that results can be interpreted correctly during outbreak investigations, both in New Zealand and internationally.
Currently routine laboratory surveillance of salmonellae from both human infections and animal sources in New Zealand is undertaken by New Zealand's Enteric Reference Laboratory (ERL) at the Institute of Environmental Science and Research Ltd (ESR). Serotyping and phage-typing are the primary laboratory-based methods used to detect clusters of cases with further characterization using single-enzyme PFGE undertaken if an outbreak is suspected. This is in contrast to some other reference laboratories that use PFGE for primary subtyping and MLVA for finer discrimination [Reference Hyytia-Trees19]. The purpose of this study was to determine if MLVA would be suitable for routine subtyping of S. Typhimurium isolates in New Zealand [20]. Four phage types commonly isolated in New Zealand were used to assess this [S. Typhimurium DT101, DT104, DT160, and an RDNC phage type (reacts but does not conform to a recognized Typhimurium phage pattern) first observed in May 2006 (RDNC-May 2006)]. To further evaluate the suitability of MLVA for outbreak investigations we used this method to analyse isolates possibly associated with two distinct outbreaks.
METHODS
Bacterial strains
All S. Typhimurium isolates used in this study were referred to New Zealand's ERL for serotyping [Reference Grimont and Weill21] and phage-typing [Reference Anderson8, Reference Rabsch, Schatten and Eisenstark9] using a standard set of typing phages (Central Public Health Laboratory, Health Protection Agency, London, UK). Phage types are reported as a number except if the phage pattern does not correspond to a recognized phage type, when it is reported as phage type RDNC followed by the month it was first observed in our laboratory. Isolates were stored long-term on Dorset egg slopes at room temperature.
The isolates used for assessing the suitability of MLVA were selected according to their phage type. All S. Typhimurium DT104 (1997–2007 inclusive) and RDNC-May 2006 isolates in the ERL culture collection were characterized (37 and 71 isolates, respectively). DT104 isolates from cases that occurred prior to 1997 were not available for analysis. Due to the large number of DT160 and DT101 isolates in the culture collection ten human isolates and ten isolates from non-human sources were characterized from each year for 5 years (2003–2007 inclusive). The isolates analysed were from cases in diverse areas of the country and occurred in different months of the year.
Isolates from two separate outbreaks of S. Typhimurium were also characterized by MLVA. All 149 DT42 isolates referred to ERL during 2008 and January 2009 were characterized, as were 49 potentially outbreak-associated DT1 isolates.
Preparation of DNA template
S. Typhimurium isolates were cultured on tryptic soy agar plates (Fort Richard Laboratories, New Zealand) at 37°C for 18 h. Four to five colonies were suspended in 500 μl sterile distilled water that was heated to 95°C for 10 min prior to the debris being pelleted by centrifugation (5 min at 13 768 g). The neat supernatant was used as a source of DNA template for PCR amplification.
PCR amplification
The multiplex MLVA PCR reactions were performed as described previously [Reference Lindstedt16] with reaction 1 amplifying STTR3 and STTR6 amplicons and reaction 2 amplifying STTR5, STTR9, and STTR10pl amplicons. All forward primers were fluorescently labelled (STTR3F-Pet, STTR5F-Pet, STTR6F-6FAM, STTR9F-6FAM, and STTR10plF-NED; Applied Biosystems, USA). All non-labelled primers were synthesized by Invitrogen (USA). Amplifications were performed in 10 μl reactions prepared using Qiagen Master Mix (Qiagen, Germany) and a primer concentration of 0·4 μm except for the STTR3 primers that were used at 0·2 μm. Reactions were cycled in a GeneAmp® 9700 thermocycler (Applied Biosystems). The amplification conditions for both reactions 1 and 2 were: an initial denaturation step at 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 63°C for 30 s, and 72°C 60 s, and a final extension at 72°C for 5 min. If amplification products needed to be visualized standard gel electrophoresis techniques were used.
Fragment analysis
PCR products were diluted 1/50 with DNase/RNase free water and analysed on an ABI 3130xl Genetic Analyzer (Applied Biosystems) using the GeneScan™ 600 LIZ® standard (Applied Biosystems) for comparison. Raw data were analysed using GeneMapper® software v. 4.0 (Applied Biosystems) to determine the size and dye label associated with each amplicon. This information along with the size of the flanking region, which is the size of the amplicon for a given locus excluding any repeat sequences, was used to determine the number of repeat sequences present and assign an allele number. The flanking regions used were: STTR9, 144 bp; STTR5, 181 bp; STTR6, 264 bp; STTR10pl, 311 bp; and STTR3, 106 bp, which were similar to those used in other studies [Reference Gilbert22, Reference Larsson23]. Sequencing a range of alleles at every locus confirmed the correct alleles had been assigned. All MLVA results were reported as a string of five numbers (STTR9-STTR5-STTR6-STTR10pl-STTR3) using nomenclature described previously [Reference Larsson23]. Not all S. Typhimurium isolates contain all five alleles, with STTR10pl and STTR6 missing in 48% and 16%, respectively, of isolates tested by Lindstedt et al. [Reference Lindstedt16]. If no amplicon was generated the allele ‘NA’ was assigned [Reference Larsson23]. If necessary the absence of an amplicon at a given locus was confirmed using single-plex PCR.
Sequence analysis
Sequence analysis was performed according to manufacturer's instructions using the BigDye® Terminator v. 3.1 Cycle Sequencing kits (Applied Biosystems) and an ABI 3130xl Genetic Analyzer (Applied Biosystems). Sequencing primers were the (non-labelled) MLVA amplification primers [Reference Lindstedt16].
Simpson's diversity index (DI)
The variation in the number of repeats at each locus was assessed using Simpson's DI [Reference Hunter and Gaston24] via the online tool available at the Health Protection Agency website (http://www.hpa-bioinformatics.org.uk/cgi-bin/DICI/DICI.pl). Simpson's DI ranges from zero (no diversity) to one (extreme diversity).
RESULTS
Analysis of selected S. Typhimurium isolates from New Zealand
A total of 304 isolates were characterized using MLVA (Table 1). The isolates were selected according to phage type and included all 71 RDNC-May 2006 and all 37 DT104 isolates (1997–2007 inclusive) in the culture collection as well as 100 DT101 and 96 DT160 isolates. None of the 304 isolates selected for analysis were identified as being part of an outbreak using epidemiological data.
Table 1. MLVA data for S. Typhimurium DT101, DT104, DT160 and RDNC May-2006 case isolates from New Zealand
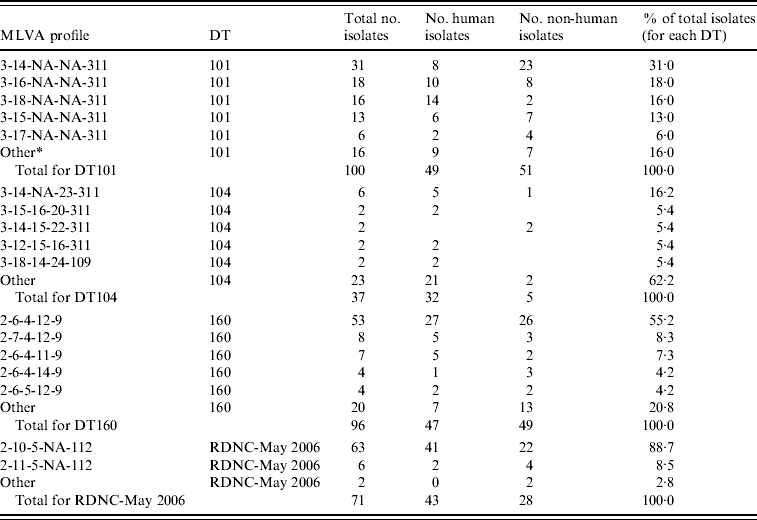
MLVA profiles are expressed according to nomenclature described by Larsson et al. [Reference Larsson23].
* A MLVA profile different from the other profiles listed for a given phage type. They were found in three or less isolates for DT101 and DT160, one or two isolates for DT104 and RDNC-May 2006.
Fifteen different profiles were found within the 100 DT101 isolates assessed (Table 1) although 90% of isolates assessed had identical profiles at all loci except STTR5 (3-x-NA-NA-311, where x is variable). This suggests that the allele at STTR5 is very important for assessing variability in DT101 isolates using the MLVA methodology described by Lindstedt et al. [Reference Lindstedt16]. Seventy-eight percent of DT101 isolates had one of four MLVA profiles: 3-14-NA-NA-311 (31%), 3-16-NA-NA-311 (18%), 3-18-NA-NA-311 (16%), and 3-15-NA-NA-311 (13%).
As reported previously [Reference Lindstedt16, Reference Torpdahl17], we found that MLVA discriminated between DT104 isolates with 28 different profiles within the 37 isolates assessed (Table 1). The most common profile (3-14-NA-23-311) was found in six isolates and a total of 23 profiles were identified in one isolate each. Very little variation was found in STTR3 and STTR9 with most isolates having 14 (three 27-bp and eleven 33-bp repeats) and three repeats, respectively, at these loci. Most variation was found in STTR10pl. Of the 37 isolates assessed six were from animal (five isolates) or environmental (one isolate) sources. Of the remaining 31 isolates, ten isolates were from cases that had a history of recent overseas travel. Travel information was not available for cases associated with 21 isolates.
The most common MLVA profile in DT160 isolates was 2-6-4-12-9, which was found in 53 of the 96 (55%) DT160 isolates assessed (Table 1). Most of the other isolates (41/43, 95%) assessed had one of 15 profiles identified that differed from 2-6-4-12-9 at one or two loci.
There was limited variability in MLVA profiles from the RDNC-May 2006 isolates characterized (Table 1). Of the 71 isolates assessed 63 (89%) had profile 2-10-5-10-112. There were three other profiles identified that differed from the most common profile at either one or two loci.
Approximately equal numbers of DT160 and DT101 isolates from human and non-human sources were characterized (Table 1). There was no evidence to suggest that there were differences between the S. Typhimurium isolates from human and non-human sources.
Investigation of isolates with double peaks at a single locus
One isolate had an unusual MLVA profile as two peaks were identified at the STTR10pl locus. The peaks differed in size by two repeat units (STTR10pl-12 and STTR10pl-14). The presence of these double peaks may indicate that each bacterium in the culture has two copies of the locus of interest. Alternatively each bacterium may have one copy of the STTR10pl locus but different alleles may be present in the population. To investigate the reason for the presence of the double peaks, single colonies from a plate culture were assessed to determine what STTR10pl allele(s) they contained. All 18 colonies assessed contained only one STTR10pl allele, with 12 encoding STTR10pl-12, five encoding STTR10pl-14, and one encoding STTR10pl-15. To investigate the stability of the STTR10pl locus, plate cultures of the single colonies were made and single colonies from these cultures tested to determine what STTR10pl allele(s) they contained. All single colonies taken from a parent colony determined to encode the STTR10pl-12 allele also contained the STTR10pl-12 allele. Similarly all single colonies taken from a parent culture determined to encode the STTR10pl-14 allele contained the STTR10pl-14 allele.
Locus characteristics
When MLVA results from all 304 S. Typhimurium isolates were considered together the greatest number of different alleles (22 in total) was found in STTR10pl, followed by STTR5 and STTR6 that both had 13 different alleles (Table 2). When all isolates assessed were considered together STTR5 had the greatest diversity, determined using Simpson's DI, followed by STTR6 (Table 2). The DI for a given VNTR locus was dependent on the phage type being assessed (Table 2). This was partially due to some loci not being present in some phage types although there was large variation in the diversity indices calculated for STTR3, STTR5, and STTR9, which were present in all isolates.
Table 2. Diversity of the S. Typhimurium MLVA data determined using Simpson's diversity index (DI)
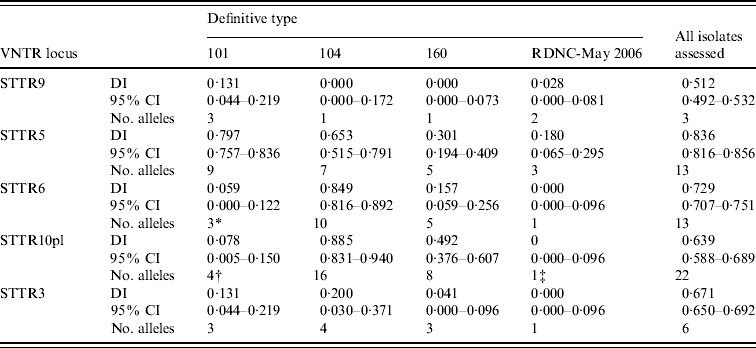
CI, Confidence interval.
* Absent in 97 of the 100 isolates assessed.
† Absent in 96 of the 100 isolates assessed.
‡ Absent in all isolates assessed.
Use of MLVA to investigate salmonellosis outbreaks
To assess the usefulness of MLVA for outbreak investigation purposes it was used to characterize isolates associated with recent S. Typhimurium outbreaks in New Zealand.
S. Typhimurium DT42 outbreak
In November 2008 an increased number of S. Typhimurium DT42 isolates were received by the ERL, predominantly from cases in the South Island of New Zealand. Initially MLVA analysis was performed on eight case isolates thought to be outbreak-associated. These isolates all had MLVA profile 3-15-NA-NA-311, whereas two unrelated isolates had distinct profiles that differed by the number of repeats at the STTR5 locus. To further investigate the significance of differences at the STTR5 locus, all 61 S. Typhimurium DT42 isolates referred to the ERL in 2008 between January and October, inclusive, were characterized using MLVA. Of these, 36 were case isolates from human infections and 25 were from animal or environmental sources. Most isolates (59/61) had MLVA profile 3-x-NA-NA-311, where x is variable. Twenty-two isolates had the same MLVA profile as the outbreak-associated isolates (3-15-NA-NA-311). These isolates were from human cases (11), poultry feed (8), poultry (2) or offal (1). Of the 11 isolates from human cases five were linked by time and place and one further isolate was thought to be associated with the current outbreak being investigated. PFGE analysis was performed on 12 isolates from the beginning of the outbreak that were thought to be outbreak-associated. Single-enzyme PFGE analysis was not used during the outbreak investigation as it failed to identify a difference between these 12 isolates, which all had MLVA profile 3-15-NA-NA-311, and four sporadic DT42 isolates. The ability of MLVA to provide some discrimination between DT42 isolates meant that this method was used to characterize all DT42 isolates suspected of being associated with the outbreak.
The first S. Typhimurium DT42 isolate with MLVA profile 3-15-NA-NA-311 thought to be associated with the outbreak was from a case that became symptomatic on 13 October 2008. From this date until the end of January 2009 a total of 72 S. Typhimurium DT42 case isolates were received by ERL. Of these, 65 had the MLVA outbreak profile 3-15-NA-NA-311 and seven had a distinct profile and were not considered a case. All seven distinct profiles differed from the outbreak profile at the STTR5 locus only, i.e. they had MLVA profile 3-x-NA-NA-311, where x is variable but does not equal 15 (three STTR5-16, one STTR5-17, and three STTR5-18). MLVA results correlated well with epidemiological investigations as most isolates with a MLVA profile that differed from 3-15-NA-NA-311 were isolated from cases that did not appear to be part of the outbreak, for example they were from the North Island of New Zealand, not the South Island where the majority of cases were occurring. Epidemiological analysis and microbiological investigation confirmed exposure to contaminated flour as the source of the outbreak. In total 43 isolates were tested from animal and environmental sources. Twenty-five isolates described above were received by ERL prior to the start of the outbreak. The 18 isolates received during the outbreak were from flour (15), poultry (2) and an environmental sample (1). More information on the descriptive epidemiology of this outbreak will be described in a forthcoming paper.
S. Typhimurium DT1 outbreak
In early 2009 an increased number of S. Typhimurium DT1 isolates were received by the ERL. A total of 49 cases were reported over a 2-month period. The majority of cases were found in Auckland (19 cases) and Gisborne (17 cases), which are cities in New Zealand's North Island separated by over 450 km. MLVA analysis concluded that the cases in Auckland were likely to be exposed to a different source than the cases in Gisborne as the MLVA outbreak profile for all case isolates tested from Auckland was 5-19-NA-NA-200 and the profile for all case isolates tested from Gisborne was 3-16-NA-NA-311. PFGE analysis also detected a difference between the isolates from cases in Auckland and Gisborne.
DISCUSSION
Characterization of S. Typhimurium isolates using MLVA is slowly gaining international acceptance and is being used more widely for both routine surveillance and outbreak investigations. Although the method is being more widely used we do not have a complete understanding of the discriminatory power of the technique for all phage types and we do not fully understand the implications of finding MLVA profiles that differ at one or two loci from an outbreak profile. For these reasons more studies are required to determine the power and limitations of S. Typhimurium MLVA analysis. There is limited published information about the discriminatory power of MLVA for a number of phage types that are problematic in New Zealand. As the discriminatory power of MLVA is related to the phage type of the isolate being assessed [Reference Lindstedt16, Reference Torpdahl17], it was important to determine the discriminatory power of MLVA for these phage types so that this information was available to assist outbreak investigations both in New Zealand and internationally.
Initially four S. Typhimurium phage types (DT101, DT104, DT160, RDNC-May 2006) were characterized in this study. DT101 isolates are problematic in New Zealand [20] but cause limited disease in other areas of the world, DT104 isolates are found world-wide and frequently exhibit antibiotic resistance [Reference Cooke25], DT160 is the most common S. Typhimurium phage type identified in New Zealand [20], and isolates with phage type RDNC-May 2006 have become more prevalent in New Zealand since they was first identified in 2006 (C. Nicol, personal communication). To assist interpretation of MLVA profiles from a potential outbreak all DT42 isolates from 2008 were also tested. It has previously been reported that MLVA can discriminate within DT104 isolates [Reference Lindstedt16, Reference Torpdahl17]. Information from a limited number of isolates suggests that MLVA may discriminate between DT101 isolates, as the five DT101 isolates tested had five distinct MLVA profiles [Reference Wang26]. By contrast, in the same study, all four DT160 isolates tested had the same MLVA profile [Reference Wang26].
Of the five phage types investigated (DT101, DT104, DT160, DT42, RDNC-May 2006) only MLVA data for DT104 provided a reasonable level of discrimination with 28 different MLVA profiles found within the 37 isolates assessed. The variability observed in the MLVA profiles of DT104 isolates may be related to the fact that a large portion of DT104 infections are acquired overseas. By contrast, there was limited variation in the MLVA profiles for the other four S. Typhimurium phage types investigated, which we assumed were largely acquired in New Zealand. The lack of diversity found in the New Zealand S. Typhimurium isolates is in contrast to that found in other areas of the world [Reference Torpdahl17, Reference Wang26]. The limited variability may be due to factors such as New Zealand's geographic isolation and efficient border control that have led to the introduction of S. Typhimurium into New Zealand on a limited number of occasions [Reference Crump, Murdoch and Baker27]. This may also mean that there are a limited number of reservoirs of S. Typhimurium, which would be beneficial for implementation of public health control measures.
In order to interpret MLVA data appropriately we require more information on what S. Typhimurium MLVA variants should be considered part of an outbreak. Outbreak investigations overseas have highlighted that S. Typhimurium MLVA loci appear stable over short periods of time although single-locus variants have been identified [Reference Ethelberg28–Reference Nygard31]. Further information on the mutation rate or the stability of VNTR loci would assist epidemiological investigations by providing information about the likelihood of a mutation occurring and enabling predictions about the significance of any variants that are identified [Reference Hopkins29, Reference Noller32–Reference Vogler34]. For example the significance of the variability observed in the STTR5 locus in our S. Typhimurium DT42 outbreak investigation was initially unclear although subsequent investigations determined that this variability was significant. The variability observed in New Zealand DT101, DT160, DT42 and RDNC-May 2006 isolates is similar to what would be expected in an outbreak situation, with most isolates sharing a common profile except for single- or double-locus variants. The limited variability in the New Zealand S. Typhimurium isolates means that it will often be challenging to differentiate sporadic cases from outbreak-related cases using the MLVA methodology described by Lindstedt et al. [Reference Lindstedt16]. Improved differentiation may be achieved by assessing additional loci, such as those used in the PulseNet MLVA scheme for S. Typhimurium [35], or using multiple typing methods. The use of additional loci would be particularly useful for phage types without amplifiable products for STTR6 and/or STTR10pl.
Currently the gold standard method used to characterize S. Typhimurium is PFGE. In New Zealand S. Typhimurium isolates referred to the ERL are characterized using serological typing and phage typing. PFGE is only used to characterize isolates during outbreak investigations and it is anticipated that if MLVA was used it would also be used solely for outbreak investigation purposes. During outbreak investigations it is important that laboratory-characterization data is capable of discriminating between outbreak-related and sporadic cases. This ensures that epidemiological investigations, which try to identify exposures that the cases have in common, are more accurate as the data is not contaminated with irrelevant information from sporadic cases. However, it is challenging to find a typing method that facilitates discrimination of all outbreak-related and sporadic cases for all S. Typhimurium phage types [Reference Foley, Zhao and Walker36]. PFGE has proven useful for a large number of investigations although there are some phage types where the discrimination of PFGE is not sufficient for outbreak investigations. For example, S. Typhimurium DT104 isolates show a high level of genetic homogeneity, with epidemiologically related and unrelated isolates having indistinguishable PFGE profiles [Reference Ribot37]. Similarly PFGE profiles from DT160 isolates in New Zealand show a high degree of similarity and are often indistinguishable [Reference Alley38], as are PFGE profiles for RDNC-May 2006, DT42 and DT101 isolates (ERL, ESR, New Zealand, unpublished data). The inability of PFGE to provide the level of discrimination needed for outbreak investigations within particular phage types led to our investigation of MLVA as a potential outbreak investigation method.
Even though initial results indicated that MLVA may not facilitate differentiation of all common-source outbreaks, the methodology proved to be very useful in two outbreaks. MLVA played an important role in the successful identification of the source of S. Typhimurium DT42 and its removal from the food supply. MLVA enabled sporadic cases to be differentiated from outbreak-related cases, which made the epidemiological investigation more accurate as the data was not tainted with irrelevant information from unrelated cases. The use of MLVA meant secondary laboratory characterization data was available within 24 hours of serotyping and phage-typing data confirming the isolate was potentially outbreak-related. This enabled the laboratory data to play a more significant role in the investigation, for example the 3-15-NA-NA-311 MLVA profile was one component of the case definition. As PFGE was not able to differentiate sporadic and outbreak-associated S. Typhimurium DT42 case isolates but MLVA profiles showed some variability, MLVA provided more useful information in this particular outbreak. MLVA also provided useful information for investigation of an outbreak of S. Typhimurium DT1 isolates although similar information was provided by PFGE.
Even if there is only a small improvement in discriminatory ability it may be preferable to use MLVA over PFGE as results are available rapidly, MLVA has high throughput potential, and is less labour intensive. In our laboratory we do not routinely characterize S. Typhimurium isolates using PFGE. The ability to produce characterization data rapidly with significantly less staff time means that data for outbreak investigations can be provided in a more timely manner and it may be possible to introduce this level of characterization into routine typing of case isolates. The usefulness of MLVA in outbreak investigations was highlighted by application of the methods to two recent outbreaks.
We identified one culture that appeared to have multiple copies of one VNTR region. This could be due to a duplication of the VNTR region, although the data from single colonies do not support this. The more likely explanation for the presence of two different VNTR alleles is that one copy of the VNTR region is present in each bacterium although two different alleles are found within the population. It is likely that the case was infected with bacteria containing one allele and a mutation has occurred either in vivo or during laboratory culture. The identification of a third allele from this culture (STTR10pl-15) during the single colony analysis is most likely the result of a mutation that has occurred in the laboratory.
One of the advantages of MLVA is the possibility of representing the data as a string of five numbers, which is easily comparable between laboratories. However, if MLVA is to be truly portable between laboratories then the method used to derive the string of five numbers needs to be standardized, such that all laboratories generate the same result for any given isolate. It is unlikely that all the variables affecting the reported size of an amplicon following capillary electrophoresis can be controlled between laboratories so it is important to understand these variables and allow for them when assigning alleles. In this study we used sequence data to confirm that the correct alleles had been assigned although reference isolates with known MLVA profiles could also be used. The meaning of a given allele assignment also needs to be standardized, as different nomenclatures have been proposed [Reference Lindstedt16, Reference Gilbert22, Reference Larsson23]. For example, Gilbert et al. [Reference Gilbert22] proposed that an isolate with no amplicon, an isolate with an amplicon containing no repeat sequences and an isolate with an amplicon containing one repeat sequence should be given allele numbers of 0, 1, and 2, respectively. By contrast most published papers use allele 1 when there is one repeat sequence and allele 2 when two repeat sequences are present. Similarly most published papers use an allele number for STTR3 representing the total number of repeat sequences, which does not differentiate between the 27-bp and 33-bp repeats. The nomenclature described by Larsson et al. [Reference Larsson23] outlines both the total number of repeat sequences as well as providing information about the number of 27-bp repeats and the number of 33-bp repeats. Until a standardized nomenclature is agreed upon the method used to derive the string of numbers needs to be clearly stated.
The purpose of this study was to determine if MLVA would be suitable for routine subtyping of S. Typhimurium isolates, with particular focus on the common phage types isolated in New Zealand. This investigation shows that MLVA has significant potential as a discriminatory typing method for S. Typhimurium outbreak investigations, although the limitations of the methodology need to be more completely understood. It would be worthwhile conducting additional investigations to compare the discriminatory power of PFGE and MLVA. Future use of this methodology in New Zealand will be partially dependent on the acceptance of the methodology internationally.
ACKNOWLEDGEMENTS
The authors thank the ERL staff for the phage-typing data and the diagnostic laboratories around New Zealand for referring isolates used in this study for confirmatory testing. This project was funded by Capability Development funding (Ministry of Research, Science and Technology) and the Ministry of Health. E. Turbitt is currently affiliated with Murdoch Children's Research Institute, Melbourne, Australia.
DECLARATION OF INTEREST
None.


