



Introduction
India is home to almost one-fifth of the world's population. People living in each of its twenty-nine states and seven union territories differ in ethnic origins, cultures, religions and socio-economic means, which are exposed to a wide variety of often difficult climatic and ecological conditions as well as to numerous other factors affecting their health(Reference Bush, Luber and Kotha1,Reference Narain2) . A recent survey(Reference Simon Iain Hay, Ashkan and Sanjay3) shows that the overall disease burden per person varies considerably between states, the burden rate due to the major diseases ranging five to ten times amongst states. However, contrarily to the all too often repeated view presenting India as the ‘diabetes capital of the world’(Reference Yajnik4,Reference Wells, Pomeroy and Walimbe5) , it is maternal and child malnutrition and anaemia which are the leading risk factors for the burden of health problems in India(Reference Simon Iain Hay, Ashkan and Sanjay3). The primary consequences of these are insulin resistance and infantile stunting and wasting; diabetes and obesity appearing as low prevalence secondary consequences and certainly not as primary causes for the disease burden(Reference Simon Iain Hay, Ashkan and Sanjay3). Dietary behaviours, including foods preferentially consumed, meal frequency and timing, are heavily influenced by the ecology, demography, regions, religions, traditions, seasons, cultural specificities, economic burden and psychosocial beliefs(Reference Green, Milner and Joy6). Such beliefs around food choices are extremely deep-rooted and are mostly practised by women especially during pregnancy and lactation(Reference Sharma, Akhtar and Singh R7). These practices determine what they eat, how much, why and when. Consequences of such beliefs are seen in health of women of childbearing age, in newborn babies and in infants and adolescents(Reference Zaveri, Paul and Saha8).
Across India, dietary intakes of children and adults in rural and urban areas show gross inadequacy of all nutrients and poor quality of protein(Reference Hemalatha, Toteja and Bhargava9). Maternal and child malnutrition is characterised by low energy intake (eating less often and small portions) and low dietary diversification. The usual diets are low in proteins, vitamins and micronutrients but rich in carbohydrates and saturated fats. Concurrently, hygiene conditions can vary from very poor to excellent not only between rural areas but within urban centres as well(Reference Kumar, Kar and Jain10). A suboptimal prenatal environment, in particular global nutrient restriction during the periods of placental and embryonic development, is increasingly being recognised as programming physiology, enhancing predisposition for metabolic diseases in adult life(Reference Ojha, Robinson and Symonds11,Reference Symonds, Budge and Stephenson12) .
Low birth weight (LBW) offspring of women suffering from malnutrition, clinical anaemia and chronic micronutrient shortages, including vitamins, are characterised by elevated subcutaneous adiposity but very low visceral adiposity (thin-fat phenotype)(Reference Yajnik, Fall and Coyaji13,Reference Kurpad, Varadharajan and Aeberli14) . They also exhibit insulin resistance and infantile stunting and wasting, together with increased risk of developing cardiometabolic disorders in adulthood, hence promoting a self-perpetuating, highly multifactorial disease burden which cannot be remedied through uniform dietary recommendations(Reference Joy, Green and Agrawal15). To propose coherent modes of interventions likely to alleviate this multifactorial disease burden, it appears necessary to first understand the physiological roots of the pathological phenotypes encountered within affected populations and communities. To this effect, we implemented a previously described(Reference Iris, Beopoulos and Gea16,Reference Iris, Gea and Lampe17) systems-analytical approach (Computer-Assisted Deductive Integration [CADI]) which had already proven its efficacy in multiple biological contexts(Reference Gadal, Starzec and Bozic18–Reference Troisi, Autio and Beopoulos21). We utilised the results of previously published field studies(Reference Katre, Bhat and Lubree22–Reference Katre, Joshi and Bhat27), undertaken in rural as well as urban populations, addressing qualitative phenotypic biomarkers together with the corresponding maternal and infantile metabolic and nutritional parameters (glucose tolerance, circulating levels of individual amino acids, haematocrit, haemoglobin levels, morphological indices and inflammatory parameters) to precisely define the range of pathological phenotypes encountered and their individual biological characteristics. These characteristics were then integrated, via extensive literature searches, into metabolic and physiological mechanisms to identify the dysregulations most likely to underpin the ‘thin-fat’ phenotype and its associated self-perpetuating high disease burden.
Our studies reveal hitherto poorly understood maternal nutrition-dependent mechanisms most likely to promote and sustain this self-perpetuating situation, suggesting clear avenues for interventions likely to significantly alleviate the leading risk factors for health deterioration in India.
Maternal characteristics in the populations studied
The data used in this analysis was obtained from Indian population, and all women who participated in the previously published field studies(Reference Katre, Bhat and Lubree22–Reference Katre, Joshi and Bhat27), whether pregnant or not, were characterised by low body weight, elevated subcutaneous adiposity (thin-fat phenotype), significant anaemia, malnutrition, insulin resistance, low micronutrients and vitamin B12 levels together with low circulating glutathione (GSH), high homocysteine (Hcy), triacylglycerols (TGs) and 5-methyltetrahydrofolate (5-mTHF) levels.
However, in this context, since B12-dependent physiological processes such as odd carbon chain-length fatty acid (FAs) metabolism, serine-glycine interconversion (see later) and nucleic acids synthesis are clearly functional, low vitamin B12 circulating levels, while certainly indicative of vitamin intake deficiency(Reference Green28), may be representative of high cellular uptake for metabolic purposes rather than low availability(Reference Hannibal, Lysne and Bjorke-Monsen29). Furthermore, it seems highly unlikely that deficiency in vitamin intake could address B12 only. It appears more likely that all essential vitamins would be similarly affected, in particular vitamins A, B1, B2 B6, B8, B9 and C(Reference Nunn, Kehoe and Chopra30). Circulating levels of non-essential and essential amino acids (NEAA and EAA, respectively) were generally low, with the notable exception, most particularly in pregnant women, of aspartic acid which was extremely elevated, followed by elevated serine, threonine and histidine (in descending order, respectively, Table 1).
Table 1. Trends in circulating levels of non-essential and essential amino acids during pregnancy

CIT, citrulline; TAU, taurine; ABA, α-aminobutyric acid; ORN, ornithine.
The data presented were collected during previously published field studies(Reference Katre, Bhat and Lubree22–Reference Katre, Joshi and Bhat27). V1 = enrolled at 1st trimester (n 75); V2 = 2nd trimester + 30 newly enrolled during their 2nd trimester of pregnancy (n 72 + 30); 34w = 34th week of pregnancy (n 30 remaining out of 92).
a Essential amino acids.
Integration
Micronutrient deficits and their effects
Micronutrients, and in particular zinc and selenium, act as key regulators of metabolic and immune functions(Reference Elmadfa and Meyer31,Reference Kieliszek32) . Zinc deficiency in human subjects is now known to be an important malnutrition problem worldwide. It is more prevalent in areas of high cereal and low animal food consumption not because the diet could be low in zinc and selenium but because phytic acid is the main known inhibitor of zinc absorption(Reference Oberleas and Harland33), while selenocysteine, the organic form of selenium most easily absorbed by human subjects, dominates in products of animal origin(Reference Kieliszek32). Compared to adults, infants, children, adolescents, pregnant and lactating women have increased requirements for zinc and selenium and are at increased risk of deficiencies. Zinc deficiency results in growth failure, while epidermal, gastrointestinal, central nervous, immune, skeletal and reproductive systems are the organs clinically most affected by zinc and selenium deficiencies(Reference Xu, Hao and Li34,Reference Roohani, Hurrell and Kelishadi35) .
Hence, in a context characterised by significant maternal malnutrition, micronutrient deficiency during the periconceptional period, and in particular zinc and selenium deficiencies, is likely to result in widespread, low level but persistent maternal as well as foetal metabolic dysregulations with deleterious consequences upon placentation and embryogenesis, negatively affecting foetal development as a whole(Reference Cetin, Berti and Calabrese36). Indeed, an adequate supply of these trace elements is essential for healthy foetoplacental development. For instance, zinc plays major functional roles in zinc-dependent enzymes, zinc-binding factors and zinc transporters required in a variety of complex mechanisms during cell replication, maturation and adhesion, such as DNA and RNA metabolism, signal recognition and transduction, gene expression and hormone regulation(Reference Donangelo and King37). Among the proteins encoded in the human genome which require zinc for their physiological function are 397 hydrolases, followed by 302 ligases, 167 transferases, 43 oxidoreductases and 24 lyases/isomerases. Proteinases include carboxypeptidases, aminopeptidases, matrix metalloproteinases and peptide hormone processing enzymes/convertases, indicating a wide role of zinc in proteostasis. Hydrolases also include zinc-dependent phosphodiesterases, phospholipases, alkaline and acid phosphatases and pyrophosphatases with roles in regulating second messenger metabolism and signal transduction pathways. Zinc is used in DNA and protein (histone) modification in demethylases and deacetylases, in DNA and RNA metabolism and in DNA repair enzymes. Another significant group of zinc enzymes is involved in regulation: transferases such as geranyl and farnesyl transferase, palmitoyl transferases, ligases such as E3 ubiquitin-protein ligases, SUMO conjugating enzymes and the corresponding hydrolases(Reference Maret38,Reference Andreini, Banci and Bertini39) .
In parallel, selenium is vital for efficient antioxidant defence in both mother and foetus. Weak placental antioxidant defence due to low maternal plasma selenium concentration increases the risk of small for gestational age infants(Reference Iqbal, Ali and Rust40). Many of the beneficial effects of selenium are attributable to its presence as selenocysteine in the selenoproteins, a small but vital group of proteins(Reference Pappas, Zoidis and Surai41,Reference Roman, Jitaru and Barbante42) . Selenoproteins W and N (SELENOW and SELENON) are both required for muscle growth, differentiation and regeneration, as well as satellite cell maintenance in skeletal muscle(Reference Castets, Bertrand and Beuvin43–Reference Brown and Arthur46). Selenoprotein S (SELENOS) is involved in the degradation process of misfolded endoplasmic reticulum (ER) luminal proteins(Reference Williams and Dhoot47), while selenoprotein T (SELENOT) is involved in the control of glucose tolerance by contributing to prolonged adenylate cyclase-activating polypeptide 1 (ADCYAP1/PACAP)-induced insulin secretion(Reference Prevost, Arabo and Jian48) while also contributing to increased quantitative insulin sensitivity(Reference Tabrizi, Akbari and Moosazadeh49). Growth retardation, poor appetite and mental lethargy are some of the manifestations of chronically zinc-deficient human subjects(Reference Prasad50). Furthermore, low serum zinc has been reported as a major predictor of anaemia mediating the effects of low selenium upon oxidative stress-dependent haemoglobin denaturation and erythrocytes osmotic fragility(Reference Houghton, Parnell and Thomson51), whereas vitamin B12 and folate deficiencies were found not to be associated with anaemia(Reference Wirth, Woodruff and Engle-Stone52). Table 2 lists the selected key enzymes that are either zinc-/selenium-dependent or the activities of which are controlled by zinc/selenium and are of prime relevance in the context of maternal malnutrition.
Table 2. List of the key enzymes that are either zinc-/selenium-dependent or the activities of which are controlled by zinc/selenium and are of prime relevance in the context of maternal malnutrition

In selenium deficiency, there is a strict hierarchy of selenium supply to specific tissues and also to different selenoenzymes within a tissue. Concentrations of selenium and selenoenzymes are greatly decreased in liver, kidney and muscle, whereas those in the brain and endocrine organs such as the thyroid gland are less affected. Within different organs, specific selenoproteins are retained at the expense of others, presumably to preserve the most important aspects of metabolism in selenium deficiency. For example, in the selenium-deficient rat, DIO1 is better retained than cytoplasmic glutathione peroxidase in thyroid, liver and kidney, presumably in order to preserve thyroid function and iodothyronine de-iodination and to limit changes in plasma T4, T3 and TSH(Reference Arthur and Beckett76). Many of the beneficial effects of selenium are attributable to its presence as selenocysteine in the selenoproteins, a small but vital group of proteins(Reference Pappas, Zoidis and Surai41,Reference Roman, Jitaru and Barbante42) . Selenoproteins W and N (SELENOW and SELENON) are both required for muscle growth, differentiation and regeneration, as well as satellite cell maintenance in skeletal muscle(Reference Castets, Bertrand and Beuvin43–Reference Brown and Arthur46). Selenoprotein S (SELENOS) is involved in the degradation process of misfolded endoplasmic reticulum luminal proteins(Reference Williams and Dhoot47), while selenoprotein T (SELENOT) is involved in the control of glucose tolerance by contributing to prolonged adenylate cyclase-activating polypeptide 1 (ADCYAP1/PACAP)-induced insulin secretion(Reference Prevost, Arabo and Jian48) while also contributing to increased quantitative insulin sensitivity(Reference Tabrizi, Akbari and Moosazadeh49).
Growth retardation, poor appetite and mental lethargy are some of the manifestations of chronically zinc-deficient human subjects(Reference Prasad50). Furthermore, low serum zinc has been reported as a major predictor of anaemia mediating the effects of low selenium upon oxidative stress-dependent haemoglobin denaturation and erythrocytes osmotic fragility(Reference Houghton, Parnell and Thomson51), whereas vitamin B12 and folate deficiencies were found not to be associated with anaemia(Reference Wirth, Woodruff and Engle-Stone52). The foods with the highest zinc contents include meat, shellfish, eggs, nuts and seeds such as hemp, flax, pumpkin or squash. Legumes, such as chickpeas, lentils and beans, all contain substantial amounts of zinc, while whole grains like wheat, quinoa, rice and oats contain some zinc (USDA Food Composition Databases). The foods with the highest selenium content include Brazil nuts, fish, meat, dairy products, eggs and bananas (USDA Food Composition Databases). However, low-quality and less varied plant-based diets in low-income communities have a high content of phytic acid [myo-inositol hexaphosphate (InsP6)] and associated magnesium, potassium and calcium salts, termed ‘phytate’, which together constitute potent inhibitors of iron and zinc absorption and bioavailability(Reference Gupta, Gangoliya and Singh79–Reference Gibson, Raboy and King81).
Hence, the origin of high to severe anaemia in Indian women and children stands to be a direct consequence of malnutrition compounded by low micronutrient intake, and in particular low availability of zinc and selenium resulting in suboptimal haemoglobin synthesis associated with oxidative stress-dependent haemoglobin denaturation and osmotic fragility of erythrocytes (see earlier). Furthermore, maternal micronutrient deficiency in association with chronic malnutrition will most probably provoke significant maternal metabolic skewing.
Maternal metabolic skewing and its consequences
Transmethylation cycle (one-carbon metabolism) alterations
Maternal dietary methyl donor intake (methionine, folate and choline) and cofactor (zinc and vitamins B2, B6 and B12) play crucial roles in one-carbon metabolism and DNA methylation in the foetus and placenta, impacting foetal growth and lifelong health outcomes(Reference McGee, Bainbridge and Fontaine-Bisson82). However, in a context characterised by significant maternal malnutrition, not only such dietary intakes will be highly restricted, but the deficiencies in cofactors intake apparently promote down-regulation of the THF-transmethylation cycle. The resulting elevation in Hcy and 5mTHF concurrently with low GSH circulating levels observed in the populations studied probably does not arise from low vitamin B12 availability but from attenuation of zinc-dependent methionine synthase and betaine–homocysteine S-methyltransferase enzymatic activity. Here, high Hyc circulating levels lead to GSH depletion through uncoupling of the activities of NOX (NADPH-dependent) from those of XO [S-adenosylhomocysteine (SAH)-dependent] and NOS (BH4-dependent), both of which are negatively affected by dysregulation of the transmethylation pathway(Reference Topal, Brunet and Millanvoye83). This dysregulation, resulting in S-adenosyl methionine (SAM) deficiency, besides reducing methylation potential, also has an effect upon choline biosynthesis from phosphatidylethanolamine (PE), each step of which requires methyl groups donated by SAM(Reference Obeid, Awwad and Knell84) (Fig. 1). In pregnancy, increased maternal Hcy levels are associated with increased risk of adverse pregnancy outcomes such as intrauterine growth restriction leading to small size for gestational age at birth and LBW(Reference Azzini, Ruggeri and Polito85). Dietary protein restriction in animals and marginal protein intake in human subjects cause characteristic changes in one-carbon metabolism that are further exacerbated by micronutrient deficiency, negatively impacting the health of the mother, impairing growth and reprogramming metabolism of the foetus, and causing long-term morbidity in the offspring(Reference McGee, Bainbridge and Fontaine-Bisson82,Reference Kalhan86) .
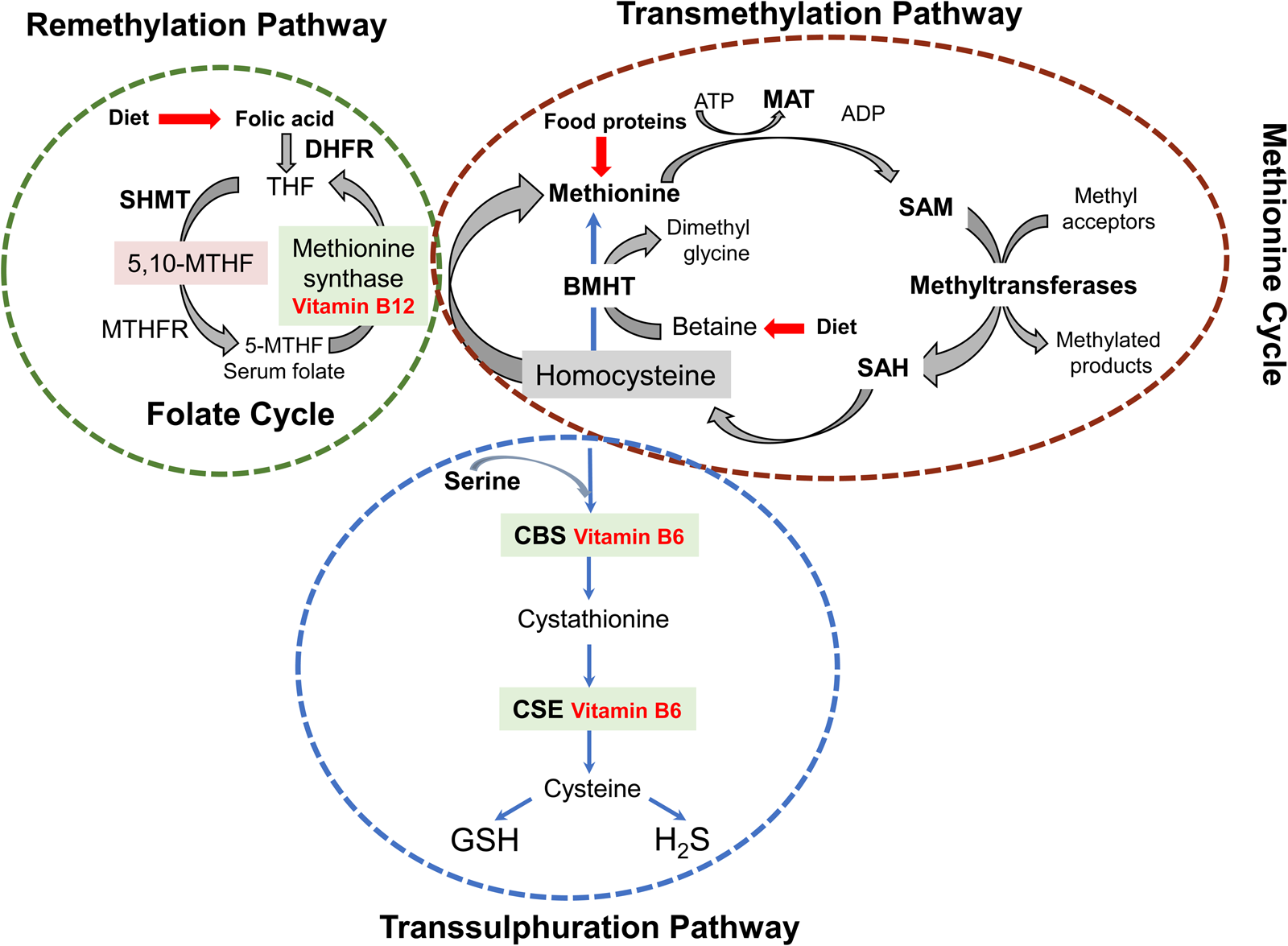
Fig. 1. Schematic representation of the interconnected transmethylation cycle/transsulphuration pathway, a critical branching point in metabolism connecting sulphur metabolism, cofactor chemistries, biological methylations and redox homoeostasis. In these interconnected pathways, methionine synthase (MS) and betaine–homocysteine S-methyltransferase (BHMT) are zinc-dependent enzymes. In parallel, methionine adenosyltransferase (MAT), N-glycine methyltransferase (GNMT), SAH hydrolase, cystathionine β-synthetase (CBS), γ-glutamylcysteine synthetase (glutamate-cysteine ligase, GCL), methylenetetrahydrofolate reductase (MTHFR) and serine hydroxymethyltransferase (SHMT) are all regulated by zinc-dependent transcription factors. Additional zinc enzymes are histone and DNA methyltransferases and adenosine deaminase(Reference Maret38,Reference Andreini, Banci and Bertini39) . DHFR, dihydrofolate reductase; THF, tetrahydrofolate; MTHF, methylenetetrahydrofolate; ATP, adenosine triphosphate; ADP, adenosine diphosphate; SAM, S-adenosyl methionine; SAH, S-adenosylHcy; CSE, cystathionase; GSH, glutathione; H2S, hydrogen sulphide. Figure adapted from Azzini et al. (Reference Azzini, Ruggeri and Polito85).
Amino acid metabolism alterations
Four amino acids (aspartic acid, serine, threonine and histidine) show elevated serum levels in malnourished pregnant women, and in particular aspartic acid is extremely elevated (see Table 1). Under protein deficiency, the NEAA aspartate becomes a significant metabolic hub, a major product of the oxidative glutaminolysis pathway and a required substrate for other anabolic pathways, including the synthesis of purines and pyrimidines(Reference Vettore, Westbrook and Tennant87). Besides playing a key role in the urea cycle as well as in pyrimidine synthesis, aspartate carries reducing equivalents in the malate–aspartate shuttle, which utilises the ready interconversion of aspartate and oxaloacetate in order to maintain mitochondrial oxidative phosphorylation (Fig. 2). Hence, extremely elevated serum aspartate levels could be a consequence of high biosynthesis and interconversion rates. Aspartate can be synthesised by the transamination of oxaloacetate using either alanine or glutamine, yielding aspartate and an α-keto acid(Reference Levin88–Reference Nielsen, Stottrup and Lofgren90). Interestingly, in malnourished pregnant women alanine, and even more so glutamine, show low circulating levels, possibly comforting hypothetically increased aspartate de novo synthesis and interconversion levels.
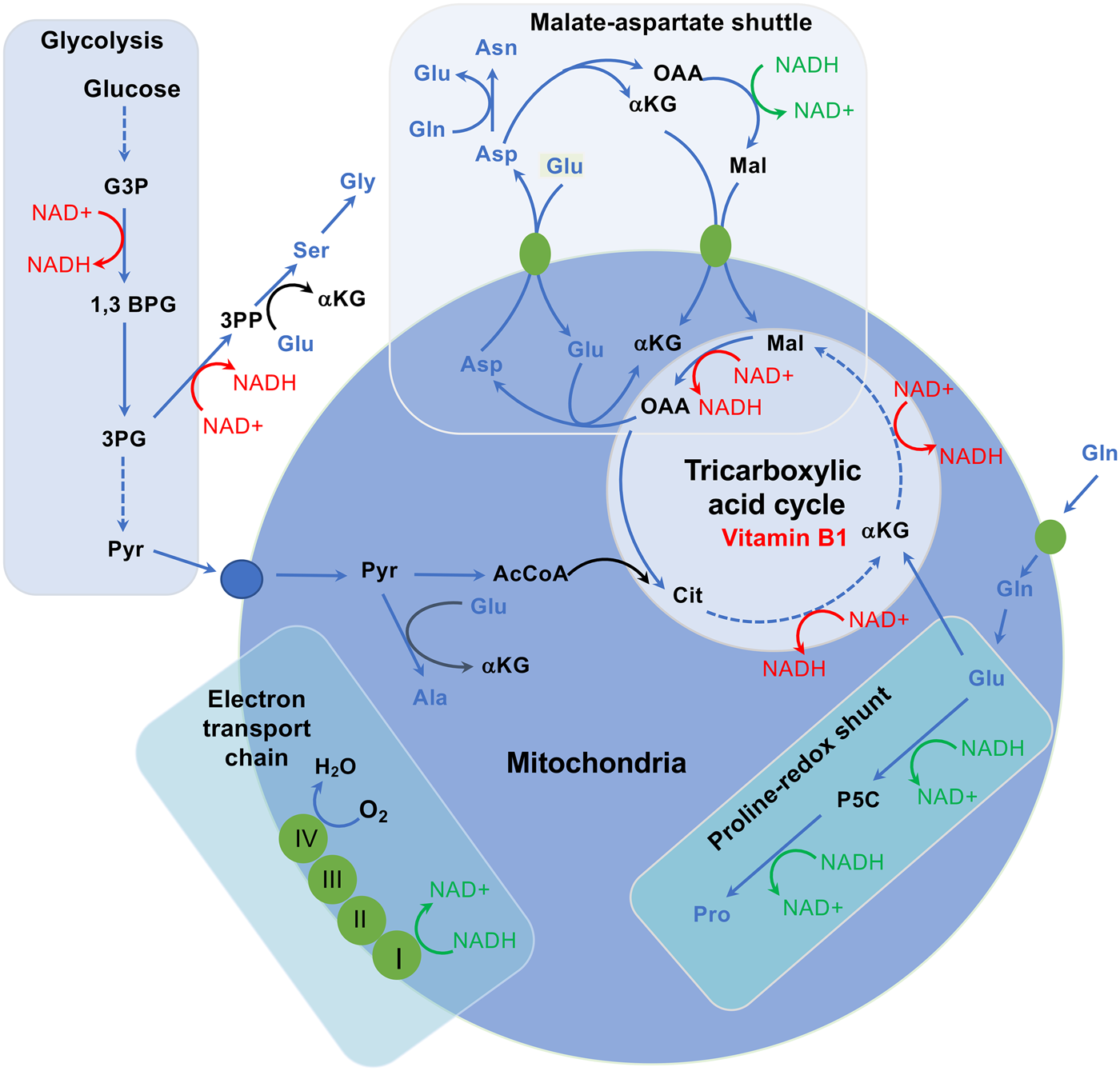
Fig. 2. Interplay between amino acid metabolism and redox homoeostasis. The synthesis and catabolism of amino acids is interwoven into redox homoeostasis. The malate–aspartate shuttle, besides transporting NADH between the cytosol and the mitochondrial matrix, also moves the amino acids glutamate and aspartate between the two compartments and is functionally connected to the TCA cycle. When aspartate is removed from this cycle to synthesise asparagine, arginine or nucleosides, this would disrupt the cycle, requiring additional carbon input. Coupling oxaloacetate (OAA) production in oxidative TCA cycle activity with glutamate production by mitochondrial glutaminase maintains flux of α-ketoglutarate into the TCA cycle while removing OAA to permit continued activity, producing reducing potential to generate ATP. In this way, two metabolites central to homoeostasis, ATP and aspartate, are synthesised in parallel. These links go further, given that aspartate, glutamate, α-ketoglutarate and malate are functionally coupled through the malate–aspartate shuttle, which is important for moving reducing potential between the matrix and the cytosol. NADH oxidation reactions and NAD+ reduction reactions, which affect the connectivity of this network, are shown in green and red, respectively. Amino acids are represented in blue, while proteins (transporters and electron carriers) are in orange. αKG, α-ketoglutarate; 1,3 BPG, 1,3-bisphosphoglycerate; 3PG, 3-phosphoglycerate; 3PP, 3-phosphopyruvate; AcCoA, acetyl CoA; Ala, alanine; Asn, asparagine; Asp, aspartate; Cit, citrate; G3P, glyceraldehyde 3-phosphate; Gln, glutamine; Glu, glutamate; Gly, glycine; Lac, lactate; Mal, malate; NAD+, nicotinamide adenine dinucleotide; NADH, reduced NAD+; OAA, oxaloacetate; P5C, pyrroline 5-carboxylate; Pro, proline; Pyr, pyruvate; Ser, serine. Figure adapted from Vettore et al. (Reference Vettore, Westbrook and Tennant87).
However, such mechanisms would be at the expense of amino acid supply and cannot be sustained indefinitely. Furthermore, glutamate dehydrogenase, which, in this scheme, catalyses the oxidative deamination of glutamate to α-ketoglutarate and ammonia, is zinc-dependent and its activity would be down-regulated by zinc deficiency. This could be alleviated by increased FA oxidative catabolism, provided that sufficient L-carnitine is available. Since diets low in meat and dairy products lead to low L-carnitine uptake, this could be compensated by de novo L-carnitine biosynthesis, which takes place mainly in skeletal muscle, kidney and liver, using L-lysine, an EAA, as primary substrate(Reference Bjorndal, Brattelid and Strand91) (Fig. 3). In malnourished pregnant women, serum lysine and methionine levels are lower than those of most other EAAs, indicative of active de novo L-carnitine biosynthesis. However, this methylation-dependent mechanism implicates SAM as methyl group donor(Reference Paik and Kim92), hence placing further demands on the transmethylation cycle.
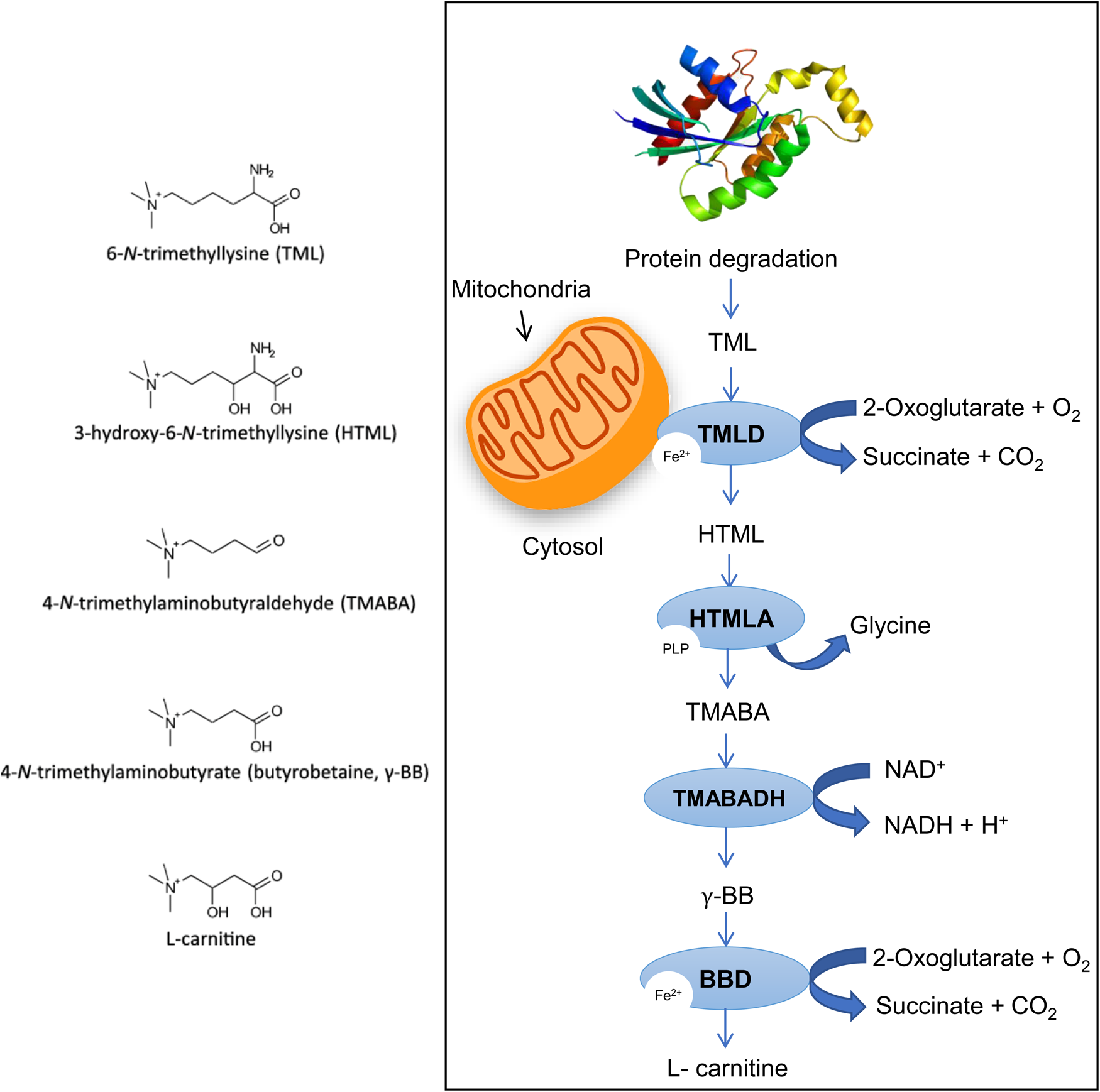
Fig. 3. De novo L-carnitine biosynthesis. (A) Carnitine is synthesised from the amino acids’ lysine and methionine. Lysine provides the carbon backbone of carnitine, and the 4-N-methyl groups originate from methionine via the transmethylation pathway (see earlier). (B) In mammals, certain proteins such as calmodulin, myosin, actin, cytochrome c and histones contain N'-trimethyl-lysine (TML) residues. N-methylation of these lysine residues occurs as a post-translational event. This reaction is catalysed by specific methyltransferases, which use S-adenosyl methionine as a methyl donor. Lysosomal hydrolysis of these proteins results in the release of TML, which is the first metabolite of carnitine biosynthesis. TML is first hydroxylated on the three-position by TML dioxygenase (TMLD) to yield 3-hydroxy TML (HTML). Aldolytic cleavage of HTML yields 4-trimethylaminobutyraldehyde (TMABA) and glycine, a reaction catalysed by HTML aldolase (HTMLA). Dehydrogenation of TMABA by TMABA dehydrogenase (TMABA-DH) results in the formation of 4-Ntrimethylaminobutyrate (butyrobetaine). In the last step, butyrobetaine is hydroxylated on the three-position by γ-butyrobetaine dioxygenase (BBD) to yield carnitine. Figure adapted from Vaz and Wanders(Reference Vaz and Wanders93).
In malnourished pregnant women, serum serine levels also are elevated, while glycine levels are not, suggesting down-modulated activity in serine hydroxymethyltransferase-mediated interconversion to glycine (synthesis of 5,10-methylene tetrahydrofolate from tetrahydrofolate) for the cytoplasmic synthesis of thymidylate, purines and methionine regeneration(Reference Zheng, Gupta and Patterson-Fortin94–Reference Giardina, Brunotti and Fiascarelli96). Furthermore, high circulating levels of serine might also be indicative of down-regulated PE biosynthesis(Reference Pacana, Cazanave and Verdianelli97) which, together with SAM deficiency, could lead to low phosphatidylcholine synthesis. This may take particular importance in a context where the availability of choline-rich food items such as fish, crustaceans, meat and eggs are highly limited, since, in pregnancy, choline deficiency could worsen placental dysfunctions while promoting foetal slow growth as gestation progresses.
In human subjects, choline and phosphatidylcholine are synthesised de novo via the PE N-methyltransferase (PEMT) pathway(Reference Zeisel98) but biosynthesis is not enough to meet physiological requirements(Reference Zeisel and da Costa99). In the hepatic PEMT pathway, 3-phosphoglycerate (3PG) receives two acyl groups from acyl-CoA forming a phosphatidic acid. It reacts with cytidine triphosphate to form cytidine diphosphate-diacylglycerol. Its hydroxyl group reacts with serine to form phosphatidylserine which decarboxylates to ethanolamine and PE forms. A PEMT enzyme moves three methyl groups from three SAM donors to the ethanolamine group of the PE to form choline in the form of a phosphatidylcholine. Three SAHs are formed as a by-product(Reference Zeisel and da Costa99). Most of the physiological requirements for phosphatidylcholine are met by channelling dietary choline through the CDP pathway which utilises adenosine triphosphate (ATP), cytidine triphosphate (CTP) and diacylglycerol to generate phosphatidylcholine (Fig. 4).

Fig. 4. Intersection between pathways of choline and methionine metabolism in the transmethylation cycle and key enzymes that control methyl group transfer: methionine adenosyltransferase 1A (MAT1A), betaine–homocysteine S-methyltransferase (BHMT), 5-methyltetrahydrofolate-homocysteine methyltransferase (MTR), methylenetetrahydrofolate reductase (MTHFR), microsomal TG transfer protein (MTTP), S-adenosylhomocysteine hydrolase (SAHH), glycine-N-methyltransferase (GNMT), guanidinoacetate N-methyltransferase (GAMT), phosphatidylethanolamine N-methyltransferase (PEMT), vitamin B12 (B12), dimethylglycine (DMG), glutathione (GSH), homocysteine (HCY), S-adenosyl methionine (SAM), S-adenosylhomocysteine (SAH), tetrahydrofolate (THF), phosphatidylethanolamine (PE), phosphatidylcholine (PC), very low-density lipoprotein (VLDL). Figure adapted from Chandler and White(Reference Chandler and White100).
In a context dominated by maternal malnutrition, low protein diet will lead to inhibition of mammalian target of rapamycin complex 1 (mTORC1), thereby promoting autophagy as a mechanism maintaining EAA availability for protein synthesis and protective mechanisms (see later) while supplying ketogenic and glucogenic precursors, such as glutamine and alanine, for ATP generating pathways(Reference Panchaud, Peli-Gulli and De Virgilio101–Reference Henagan, Laeger and Navard103). These effects would be amplified during pregnancy, triggering the placental mammalian amino acid response pathway and thereby programming the growth capacity of offspring not only in utero but also long after gestational protein restriction(Reference Wang, Wilson and Zhou104,Reference Strakovsky, Zhou and Pan105) . Elevated plasma threonine, a member of the EAA group, could reflect zinc and/or vitamin B6 deficiencies, since the initial step in threonine catabolism to Kreb's cycle precursors requires vitamin B6, the activation of which, via pyridoxal kinase, is zinc-dependent(Reference Navarro, Ramirez-Sarmiento and Guixe106,Reference Chiba, Terada and Kameya107) . However, this would also suggest sufficient dietary supply in EAAs which, in the context addressed here, is highly improbable. Hence, it appears more likely that elevated plasma threonine, arising from maternal autophagy, could supply the developing foetus with an immunostimulant which promotes thymus growth while concurrently promoting maternal innate immune defence functions(Reference Okabe, Sano and Nagata108). Elevated plasma histidine, another member of the EAA group, might also play significant protective roles benefiting both the mother and the foetus, particularly in a context dominated by chronic anaemia. Histidine is essential in globin synthesis and erythropoiesis and has also been implicated in the enhancement of iron absorption from human diets. Histidine-deficient diets predispose healthy subjects to anaemia and accentuate anaemia in chronic uraemic patients(Reference Vera-Aviles, Vantana and Kardinasari109). Furthermore, histidine plays key roles in the detoxification of cytotoxic oxidative stress metabolites such as reactive carbonyls(Reference Song, Joo and Aldini110).
FA metabolism alterations
Under conditions characterised by deficit in methyl donors and increased homocysteine levels, β-oxidation becomes deficient and hypertriglyceridaemia ensues(Reference Pooya, Blaise and Moreno Garcia111) as reflected by high circulating TGs in anaemic Indian women (see earlier). Low amino acid availability, and in particular lysine, may further contribute to high circulating TG levels. Low lysine levels would lead to low L-carnitine de novo synthesis, while diets low in meat and dairy products would lead to low L-carnitine uptake(Reference Longo112). This, in turn, would further impede mitochondrial β-oxidation of FAs without affecting cytoplasmic FA synthesis from excess carbohydrates intake(Reference Nalecz and Nalecz113). Under these conditions, mitochondrial β-oxidation of medium-chain FA, including odd medium-chain FA, the catabolism of which requires the activity of B12-dependent methylmalonyl-CoA mutase, an enzyme indispensable in human metabolism(Reference Banerjee114), would remain functional. However, both microsomal α-oxidation, which requires Fe2+, vitamin C/GSH and thiamine (vitamin B1) as cofactors(Reference Van Veldhoven, Mannaerts and Casteels115,Reference Casteels, Foulon and Mannaerts116) , as well as ω-oxidation, which requires haem iron protein such as microsomal or mitochondrial cytochrome P-450(Reference Miura117) together with zinc-dependent alcohol dehydrogenase, are also likely to be impeded. Hence, peroxisome-mediated β-oxidation of dietary long- and very long-chain FA and α-oxidation of dietary branched-chain FA stand to be favoured. However, this process would also increase oxidative stress since the first step in peroxisome-mediated β-oxidation results in the generation of H2O2 and subsequent increase in Fe2+-dependent catalase activity(Reference Bonekamp, Volkl and Fahimi118). Here, the activity of the main ROS-controlling enzymes (SODs, GLO1, GPXs and TXNRDs) will be attenuated through zinc and selenium deficiencies, while GSH will be subjected to Hcy-mediated depletion. These phenomena stand to exert negative impacts upon placental functions.
Functional placental alterations
During pregnancy, the characteristics of maternal blood biochemistry will necessarily constitute the nutritional supply provided to the developing foetus. Maternal hypertriglyceridaemia during pregnancy is correlated with foetoplacental endothelial dysfunction(Reference Leiva, Salsoso and Saez119). This can be expected to result in constitutive mild placental (and consequently foetal) hypoxia and subsequent ER stress, which would then affect metabolic control via ATF4 and ATF6β(Reference Mizuuchi, Cindrova-Davies and Olovsson120). This would stand to further worsen the direct effects of maternal anaemia. Additionally, most of the serological maternal characteristics will also be transferred to the foetus via the foetoplacental endothelial system. Hence, in a context dominated by maternal malnutrition, the developing foetus will be constitutively supplied with low vitamins, micronutrients, unbalanced amino acid supply, high TGs and Hcy. Furthermore, due to maternal selenium deficiency, the foetus will also experience a drastically reduced supply of thyroid hormones and in particular low bioactive T3 supply. Furthermore, maternal deficiencies in protein intake will trigger the placental mammalian amino acid response pathway, thereby programming the growth capacity of offspring not only in utero but also long after gestational protein restriction(Reference Wang, Wilson and Zhou104,Reference Strakovsky, Zhou and Pan105) .
Foetal developmental dysregulations leading to the LBW ‘thin-fat’ phenotype
The cord blood of LBW infants is characterised by low adiponectin levels which correlate with hyperinsulinaemia and differential distribution of fat depots giving rise to the newborn's thin-fat phenotype characterised by insulin resistance and increased subcutaneous fat but decreased intra-abdominal fat. Given the marked differences in metabolic and pathophysiological characteristics which differentiate subcutaneous and visceral fat depots(Reference Hamdy, Porramatikul and Al-Ozairi121,Reference Tchkonia, Thomou and Zhu122) , this situation is radically different from that observed in normal-weight obese individuals of Asian and Indian descent(Reference Kapoor, Furler and Paul123,Reference Kapoor, Lotfaliany and Sathish124) , characterised by high visceral adiposity and disproportionately lower subcutaneous adiposity(Reference Kapoor, Lotfaliany and Sathish124,Reference Kapoor, Furler and Paul125) . The fat overflow hypothesis invoked to explain this phenotype(Reference Anand, Tarnopolsky and Rashid126) does not correlate with the anatomical and pathological consequences observed in association with the ‘thin-fat’ phenotype of these LBW infants studied here.
Indeed, the lipid overflow/ectopic fat model states that excess visceral fat accumulation, while causally related to the features of insulin resistance, might also be a marker of a dysfunctional adipose tissue being unable to appropriately store the excess calories. According to this model, the body's ability to cope with the surplus of energy (resulting from excess caloric consumption, a sedentary lifestyle or a combination of both factors) might ultimately lead to metabolic syndrome presentation. There is evidence suggesting that if the extra energy is channelled into insulin-sensitive subcutaneous adipose tissue, the individual, although in positive energy balance, will be protected against the development of metabolic syndrome. However, in cases in which adipose tissue is absent, deficient or insulin-resistant with a limited ability to store the energy excess, the triacylglycerol surplus will be deposited at undesirable sites such as the liver, the heart, the skeletal muscle and in visceral adipose tissue, a phenomenon described as ectopic fat deposition. The resulting metabolic consequences include visceral obesity, insulin resistance, atherogenic dyslipidaemia and a pro-thrombotic, inflammatory profile(Reference Despres and Lemieux127). This clearly cannot be invoked to explain the thin-fat phenotype addressed here, characterised by elevated subcutaneous adiposity but very low visceral adiposity (thin-fat phenotype), exhibiting infantile insulin resistance, stunting and wasting, together with increased risk of developing cardiometabolic disorders in adulthood.
Roles of adipokines in the inception of the LBW ‘thin-fat’ phenotype
Adiponectin is an adipocyte-derived plasma protein with insulin-sensitizing and anti-atherosclerotic properties. There is no correlation between cord adiponectin levels and maternal body mass index, cord leptin or insulin levels, and there is no correlation between cord and maternal adiponectin levels. However, high cord blood adiponectin levels, compared with serum levels in children and adults, positively correlate with foetal birth weights. Taking fat mass-related parameters such as the birth weight/birth length ratio into consideration, plasma adiponectin concentrations exhibit a significant inverse correlation with insulin concentrations. The high adiponectin levels in newborns may be due to lack of negative feedback on adiponectin production resulting from lack of adipocyte hypertrophy, low percentage of body fat or a different distribution of fat depots in the newborns as compared to children and adults(Reference Sivan, Mazaki-Tovi and Pariente128–Reference Tsai, Yu and Hsu131). This indicates that adiponectin in cord blood is derived from foetal and not from placental or maternal tissues.
During pregnancy, leptin and adiponectin seem to act in an autocrine/paracrine fashion on the placenta and adipose tissue, playing a role in the maternal–foetal interface and contributing to glucose metabolism and foetal development(Reference Lecke, Morsch and Spritzer132). Hence, the low cord blood adiponectin levels observed in LBW births and its correlation with hyperinsulinaemia and differential distribution of fat depots giving rise to the newborn's thin-fat phenotype clearly suggest that the metabolic skewing resulting from maternal malnutrition and anaemia induce significant changes in foetal metabolism.
Cumulative effects of maternal malnutrition and placental alterations in the development of the LBW ‘thin-fat’ phenotype
Maternal nutrition, particularly micronutrients, vitamins and omega-3 FAs play a role in modulating the activity of peroxisome proliferator-activated receptors (PPARs) during placentation and angiogenesis, which affects placental and foetal growth(Reference Meher, Sundrani and Joshi133). In placental angiogenesis, PPARγ signalling causes increased vascular endothelial growth factor receptor 2 (VEGFR2) expression. VEGF binding to VEGFR2 then mediates angiogenic signalling involving increases in NOS activity(Reference McCarthy, Drewlo and English134,Reference Viita, Markkanen and Eriksson135) . Hcy suppresses PPARγ signalling and expression(Reference Wang, Dou and Yao136) while impeding NO production. Hence, during pregnancy, the multiple methylation network dysregulations resulting from micronutrient and vitamin deficiencies are likely to primarily result in placental vascularisation defects, while endothelial ER-stress resulting from chronically elevated Hcy levels(Reference Wang, Sun and Yu137) is in turn likely to result in elevated placental leptin production. These factors, together with the low circulating levels of vitamins, micronutrients and amino acids together with high TGs, are now likely to have serious consequences upon foetal development, predisposing the newborn to insulin resistance, dyslipidaemia(Reference Houde, Guay and Desgagne138) and preferential subcutaneous adipocyte patterning, whereas imbalance in the availability of amino acids and low T3 hormone production, resulting from selenium deficiency, will promote growth retardation(Reference van Gucht, Meima and Moran73).
Differential adipocyte patterning and dyslipidaemia mechanisms in the development of the LBW ‘thin-fat’ phenotype
In mammals, individual white and brown adipocyte tissues (WAT and BAT, respectively) depots appear at different times in development and have unique functional characteristics. The distinction between subcutaneous and visceral fat may be oversimplified because evidence suggests that metabolic properties vary between some visceral fat depots, while heterogeneity exists even within a single fat depot(Reference Lee, Wu and Fried139). Furthermore, metabolic and environmental challenges highlight the extraordinary plasticity of the mammalian adipose organ. Two distinct subtypes of preadipocytes have been characterised in human fat (Myf5+ and Myf5−), the proportions of which vary among depot locations(Reference Cinti140,Reference Giralt and Villarroya141) . Despite the heterogeneity in the adipocyte precursor cell compartment, it appears that the Myf5+ lineage may selectively differentiate in the BAT, subcutaneous and retroperitoneal WAT (sWAT and rWAT, respectively), while Myf5− lineages selectively give rise to most adipocytes in the inguinal and visceral WAT (ingWAT and vWAT, respectively)(Reference Sanchez-Gurmaches, Hung and Sparks142). Up-regulation of the PI3K-Akt-mTORC1 pathway (PTEN silencing) dramatically redistributes body fat such that interscapular WAT (iWAT), sWAT and rWAT (the Myf5+ lineage depots) expand, while the ingWAT and vWAT (the Myf5− lineage depots) disappear(Reference Sanchez-Gurmaches, Hung and Sparks142). In other words, the adipocytes of Myf5+ lineage expand (causing lipohypertrophy of BAT, sWATs and rWAT) at the expense of Myf5− lineage (i.e. inguinal and visceral WAT).
Hence, the thin-fat phenotype of LBW infants, characterised by increased subcutaneous fat but decreased intra-abdominal fat, is clearly indicative of metabolic skewing towards AKT-mediated mTORC1 up-regulation, probably as a result of elevated Hcy and insulin supply, concurrently with foetal hypoxia and oxidative stress, as a result of placental dysfunction cumulatively with significant maternal anaemia, during in utero development(Reference Yates, Zafar and Hubbard143). As a direct consequence, adipogenesis (lineage commitment, clonal expansion and terminal differentiation of preadipocytes) and lipogenesis in adipose tissue will be promoted, while β-oxidation and ketogenesis will be attenuated, leading to dyslipidaemia and insulin resistance(Reference Ricoult and Manning144), a situation which shall remain dominant after birth. Should the affected individual be then exposed to chronic malnutrition, these developmental phenomena will then have predisposing effects towards stunting/wasting and the development of cardiovascular diseases later in life.
Post-birth nutrition and stunting/wasting in ‘thin-fat’ phenotype individuals
Following weaning, LBW infants are subjected to the same restrictive dietary conditions experienced by their parents, namely malnutrition characterised by diets low in proteins, vitamins and micronutrients but rich in carbohydrates and saturated fats, leading to metabolic dysfunctions, including significant anaemia, which will be considerably worsened by increased consumption of low-quality processed products rich in saturated fats, salt and sugars and low in vitamins and micronutrients (ill-nutrition, or so-called junk food). In this context, the deficiencies in micronutrients and metabolic cofactors, and in particular selenium and vitamins, appear to play a key role. Recurrent nightly leg muscle cramps, muscle weakness and fatigue, are indicative of significant L-carnitine deficiency(Reference Nakanishi, Kurosaki and Tsuchiya145) and subsequent dysregulation of FA metabolism (see earlier), non-ketotic hypoglycaemia and muscle wasting(Reference Engel and Angelini146). The latter effect stands to be further reinforced by selenium deficiency. Selenium bioavailability, which, in muscles, plays a key role in oxidative stress defence and calcium transport control, and the development of nutritional muscular dystrophies affecting cardiac and skeletal muscles have long been established both in human subjects and livestock. Skeletal muscle degeneration leads to muscle weakness or stiffness, postural instability or walking disability, while cardiac muscle degeneration is associated with respiratory distress, cardiogenic shock, enlarged heart, cardiac arrhythmias, congestive heart failure and ultimately sudden death(Reference Oropeza-Moe, Wisloff and Bernhoft147). Concurrently, selenium deficiency will also result in deficient thyroid hormone supply(Reference van Gucht, Meima and Moran73) which, conjunctly with malnutrition, will promote stunting.
It is important to note that the results of malnutrition, such as deficiencies in amino acids, vitamins and micronutrients together with an oversupply of saturated fats, do not lead to the full inhibition or full activation of metabolic reactions or pathways. These deficiencies and/or oversupplies are not absolute but only relative and act as ‘rheostats’, slowing down or facilitating particular metabolic reactions or pathways. These will in turn have discrete metabolic skewing effects, the cumulative result of which will swing development in a particular direction while opening the door to predisposing effects towards context-dependent pathologies over longer period.
Conclusion
According to the earlier analysis, significant maternal malnutrition leads to deficiencies in amino acids, vitamins and micronutrients together with an oversupply of saturated fats. These deficiencies result in alterations affecting key maternal metabolic processes, and in particular the amino acid interconversion, transmethylation, FA oxidation and redox control pathways. These alterations, together with the deficiencies in micronutrients, result in high maternal anaemia, exacerbated during pregnancy and placental dysfunctions. The ensuing alterations in oxygen and nutrients supply to the embryo give rise to in utero foetal metabolic alterations. This results in LBW children characterised by high subcutaneous but low abdominal adipocyte deposits and already existing insulin resistance (Figure 5). Being raised in the same environment as their parents promote significant anaemia, accompanied by stunting and wasting in late childhood, repeating the same cycle as in their parents. Should these individuals shift, during mid to late childhood, to a food environment which primarily consists of processed foods low in proteins, vitamins and micronutrients but rich in saturated fats, salt and sugar (so-called junk food), their condition stands to be worsened by the appearance of significantly increased insulin resistance and the pathogenesis of cardiovascular and metabolic disorders. These individuals, after reaching sexual maturity, are now likely to perpetuate this deleterious cycle via their own children.

Fig. 5. The mechanisms whereby maternal chronic malnutrition compounded by ill-nutrition leads to the ‘thin-fat’ phenotype in newborn. Maternal diet chronically low in proteins, micronutrients and carbohydrates but high in fat, compounded by deficient hygiene (left panel) leads to multiple maternal metabolic alterations resulting in high maternal anaemia, exacerbated during pregnancy, together with placental dysfunction (middle panel). Subsequently, these maternal metabolic and placental dysfunctions induce in utero foetal development alterations resulting in low birth weight children, characterised by high subcutaneous but low abdominal adipocyte deposits and already existing insulin resistance (‘thin-fat’ phenotype, right panel). Being raised in the same environment as their parents promote significant anaemia, accompanied by stunting and wasting in late childhood, repeating the same cycle as in their parents. Should these children then be exposed, during mid-to-late childhood, to a food environment primarily consisting in highly processed products low in vitamins and micronutrients but rich in salt, sugar and fat (ill-nutrition, so-called junk food), their condition stands to be worsened by the appearance of significantly increased insulin resistance and heightened susceptibility to the pathogenesis of cardiovascular (CVD) and metabolic disorders. These individuals, after reaching sexual maturity, are likely to perpetuate this deleterious cycle via their own children.
However, attempts to remedy this situation must take into consideration the history of Indian diets. This history demonstrates gradual transitions over the centuries from a low energy diet of large quantities of indigestible fibre carbohydrate, small amounts of digestible carbohydrate, moderate fat and moderate protein, to an increasing intake of low fibre and refined carbohydrates associated with increased fat and decreasing intake of animal proteins interspersed with variable periods of starvation. There were fourteen recorded famines in India between the 11th and 17th centuries, while those that took place over the course of the 18th, 19th and early 20th centuries resulted in more than 60 million deaths(Reference Bhattacharya148). Currently, food intake patterns show that most Indians are vegetarians consuming poor and monotonous cereals-based diets and that food items rich in micronutrients (pulses, other vegetables, fruits, nuts, oilseeds and animal foods) are generally consumed less frequently(Reference Vecchio, Paramesh and Paramesh149). However, from 1947 onwards there has been an increase in the frequency of intake and quantities of low fibre and refined carbohydrates, with protein intake improving only marginally while intakes of industrially processed foods containing high salt, high saturated fats and high sugar but low micronutrients kept increasing(Reference Bhattacharya148,Reference Gulati, Misra and Sharma150,Reference Misra, Singhal and Sivakumar151) . Hence, Indian populations are most probably genetically as well as epigenetically adapted to forms of ‘chronic malnutrition’ characterised by low energy, low refined carbohydrates, low protein and moderate fat intake with seasonal variation in micronutrient intake (thrifty metabolism). Hence, it most probably is the metabolic consequences of the recent and rapid dietary shifts over an adaptation to ‘thrifty metabolism’ which must be addressed.
The task is made more challenging by the complexity of political, economic, climatic, social and cultural factors twined together. The resolution of malnutrition-associated problems will require a product well suited to the cultures addressed and developed in light of the actual needs as depicted by the biological evidence. To alleviate the consequences of malnutrition, improvement in affordability, accessibility, delivery to end-users, knowledge and awareness of social and cultural constrains and, most importantly, in the convergence between demand and supply must also be addressed. The solution therefore cannot merely consist in providing a ‘one size fits all’ supplement but in the use of a well-designed supplement which suits the nutritional requirements along with a social and behaviour change approach, training the beneficiaries in the basics of nutrition based on what is locally available and accessible.
Acknowledgments
The authors wish to thank Fatema Rajgara and Ayush Madhok for help with formatting and references.
The present study was partly funded by the internal research programmes of BM-Systems Pvt Ltd and Arbuza Regenerate Pvt Ltd. Work in SG laboratory was partly supported by research grant from the Department of Biotechnology (DBT), Government of India (No. CEIB-BT/PR12629/MED/97/364/2016). SG is recipient of the JC Bose National Fellowship (No. JCB/2019/000013) from the Science and Engineering Research Board, Government of India.
P.P., F.I. and S.G. conceived and coordinated the study. P.P. collected and formatted the data, F.I. performed the systems analyses, and S.G., P.P. and F.I. wrote the MS.
Ethical standards disclosure is not applicable; analysis performed using data from previously published studies.
The authors are founding directors of Arbuza Regenerate Pvt Ltd. F.I. is the Chief Scientific Officer at BM-Systems Pvt Ltd.








 Open access
Open access