


- IGF
-
insulin-like growth factor
- MetSyn
-
metabolic syndrome
- PI3K
-
phosphoinositide 3-kinase
- RR
-
relative risk
The prevalence of obesity has increased rapidly in recent years( 1 ). Being overweight or obese is now the most prevalent body composition in some countries, accounting for 71% of males and 62% of females in the United States( Reference Ogden, Carroll and Curtin 2 ). Similar trends exist in Western Europe with greater than 66% of men and 49–57% of women being overweight or obese in the UK and Ireland( 3 , Reference McCarthy, Gibney and Flynn 4 ). This pattern shows no signs of abating as obesity rates are increasing among children( Reference Jackson-Leach and Lobstein 5 ) and overweight children tend to become overweight adults( Reference Serdula, Ivery and Coates 6 ). In countries where sedentary lifestyles and high-energy foods are abundant it is easy to see how energy intake exceeds that expended. It has been estimated that ingestion of 5% more energy than expended may result in an accumulation of 5 kg adipose tissue in a single year( Reference Klein, Warden and Superman 7 ).
Epidemiological studies have demonstrated a robust link between obesity and cancer development at numerous sites, in particular the oesophagus, pancreas, colorectum, breast (postmenopausal), endometrium and kidney( Reference René an, Tyson and Egger 8 , 9 ). This association carries relative risk (RR) estimates of 1·1–1·6 per 5 kg/m2 incremental increase in BMI( Reference René an, Tyson and Egger 8 ). Obesity also increases cancer-related mortality with studies reporting that obesity could account for 14% of all deaths from cancer in men and 20% in women( Reference Calle, Rodriguez and Walker-Thurmond 10 ). The causative link between obesity and cancer is further strengthened by research in animal models which demonstrates that energy restriction decreases spontaneous tumour occurrence( Reference Dirx, Zeegers and Dagnelie 11 , Reference Mai, Colbert and Berrigan 12 ). Furthermore, emerging clinical research suggests weight loss following bariatric surgery leads to a reduction in cancer incidence( Reference Sjostrom, Gummesson and Sjostrom 13 ). Thus, the potential mechanisms by which obesity increases both the incidence of certain malignancies, and cancer deaths, have become the focus of considerable research interest in recent years.
Adipose tissue and the metabolic syndrome
WHO defines obesity as an abnormal or excessive fat accumulation in adipose tissue, to the extent that health is impaired( 1 ). Fat is principally deposited in two compartments; subcutaneously and viscerally. Attention was first drawn to the different patterns of fat distribution over 60 years ago by Jean Vague. He described two distinct types of fat deposition; upper-body, male-type fat (visceral) and lower-body, female-type fat (subcutaneous), and the association of visceral fat with type 2 diabetes, atherosclerosis and gout( Reference Vague 14 , Reference Vague 15 ). Visceral adipose tissue largely comprises omental adipose tissue and also includes other intra-abdominal fat sources such as mesenteric fat and is more metabolically active than subcutaneous adipose tissue( Reference Fox, Massaro and Hoffmann 16 ). Visceral adipose tissue has multiple endocrine, metabolic and immunological functions and has been shown to be central to the pathogenesis of the metabolic syndrome (MetSyn), a pro-inflammatory, pro-coagulant state associated with insulin resistance( Reference Galic, Oakhill and Steinberg 17 ). The multiple risk factors that commonly appear together as the MetSyn include abdominal obesity, atherogenic dyslipidaemia (raised TAG and reduced HDL cholesterol), elevated fasting plasma glucose and hypertension( Reference Alberti, Zimmet and Shaw 18 ). The importance of adipose tissue location in terms of dysmetabolism risk is evident as an increased ratio of visceral fat area to subcutaneous fat area has been shown to be strongly related to disorders of glucose and lipid metabolism in obese subjects( Reference Fujioka, Matsuzawa and Tokunaga 19 ). Furthermore, visceral obesity is more strongly associated with increased risk of insulin resistance, the MetSyn and CVD than BMI alone( Reference Nedungadi and Clegg 20 ).
Visceral fat has been identified as an independent risk factor for breast cancer( Reference Schapira, Clark and Wolff 21 ), oesophageal adenocarcinoma( Reference Beddy, Howard and McMahon 22 ), colorectal adenocarcinoma( Reference Schoen, Tangen and Kuller 23 ) and colorectal adenomas( Reference Yamaji, Iwasaki and Sasazuki 24 , Reference Nam, Kim and Han 25 ). The other elements of the MetSyn, i.e. dyslipidemia, hypertension and insulin resistance have also been independently linked with increased cancer risk( Reference Cowey and Hardy 26 – Reference Bowers, Albanes and Limburg 29 ). The synergistic impact of these factors on cancer risk is the focus of the Me-Can Study, a prospective international population-based study of 580 000 people( Reference Stocks, Borena and Strohmaier 30 ). Initial findings suggest that a combination of components of the MetSyn is associated with increased RR of colorectal cancer development (men: RR 1·25, 95% CI 1·18, 1·32; women: RR 1·14, 95% CI 1·02, 1·18)( Reference Stocks, Lukanova and Bjørge 31 ), endometrial cancer (RR 1·37, 95% CI 1·28, 1·46)( Reference Bjørge, Stocks and Lukanova 32 ), bladder cancer in men (RR: 1·1, 95% CI 1·01, 1·18)( Reference Häggström, Stocks and Rapp 33 ) and pancreatic cancer in women (RR 1·58, 95% CI 1·34, 1·87)( Reference Johansen, Stocks and Jonsson 34 ). Similarly, in studies of pancreatic and colorectal cancer a high prevalence of MetSyn, central obesity and systemic inflammation has been identified( Reference Russo, Autelitano and Bisanti 35 – Reference Siegel, Ulrich and Poole 37 ) and has been shown to be associated with advanced tumour stage and reduced survival( Reference Healy, Howard and Ryan 38 – Reference Moon, Ju and Jeong 41 ).
Oesophageal adenocarcinoma: a model of visceral obesity, metabolic syndrome and cancer
The link between visceral adiposity, the MetSyn and cancer is probably best illustrated in the oesophageal adenocarcinoma model. Oesophageal adenocarcinoma develops along a predefined sequence of events. It begins with the development of Barrett's oesophagus in response to chronic reflux of acid and bile, where the normal squamous lining of the oesophagus is replaced with specialised intestinal metaplasia, which then progresses through to dysplasia and ultimately adenocarcinoma( Reference Jankowski, Wright and Meltzer 42 ). The incidence of oesophageal adenocarcinoma in Ireland has increased by nearly 50% in recent years( 43 ), a trend mirrored across developed countries( Reference Bollschweiler, Wolfgarten and Gutschow 44 ) and coincides with the sharp increase in the prevalence of obesity( Reference McCarthy, Gibney and Flynn 4 ). Epidemiological studies highlight a marked association between oesophageal adenocarcinoma and obesity, and a recent meta-analysis reported that a BMI≥30 kg/m2 is associated with a RR of 3·0 for developing oesophageal adenocarcinoma, a higher association than for any other cancer( Reference Calle and Kaaks 45 ).
Visceral obesity itself may promote gastro-oesophageal reflux disease and predispose to Barrett's oesophagus, and this may be one contributory factor to the association with oesophageal adenocarcinoma( Reference Friedenberg, Xanthopoulos and Foster 46 ). However, the observation that obesity is a risk factor independent of reflux( Reference Lagergren, Bergstrom and Nyren 47 , Reference Ryan, Rowley and Fitzgerald 48 ) suggests other mechanisms are at play. A study at this centre in 2008 reported that the prevalence of the MetSyn in Barrett's oesophagus patients far exceeded population norms with the MetSyn present in 46% of patients and central obesity in 78%. The syndrome was also significantly associated with the length of specialised intestinal metaplasia; 60% of patients with long-segment Barrett's oesophagus had MetSyn and 92% were centrally obese compared with 23·8% and 62% (P=0·007 and P=0·005) respectively in short-segment Barrett's oesophagus. Long-segment Barrett's oesophagus was also associated with systemic inflammation, with significantly higher serum levels of IL-6 observed in patients (P=0·03)( Reference Ryan, Healy and Power 49 ). The obesity, inflammation, malignancy association is further strengthened by studies which have identified increased visceral adiposity and altered secretion of adipose-derived hormones in oesophageal adenocarcinoma( Reference Beddy, Howard and McMahon 22 , Reference Howard, Beddy and Ennis 50 ).
Mechanisms underlying obesity and tumorigenesis
The mechanisms by which visceral adiposity and the MetSyn are thought to promote tumorigenesis are manifold. Adipose tissue may promote tumour development in a paracrine manner (Fig. 1). This is supported by the observation that epithelial tumour cell growth is enhanced by injection into fat pads rather than subcutaneously( Reference Elliott, Tam and Dexter 51 ) suggesting that chemokine production within the adipose tissue provides conditions which enhance tumour cell growth. Furthermore, a proteomic study of mammary fat revealed the production of a wide variety of proteins involved in diverse processes such as cell communication, growth, immune response, apoptosis and numerous signalling molecules including hormones, cytokines and growth factors( Reference Celis, Moreira and Cabezon 52 ). The paracrine influence of adipose tissue on tumorigenesis is less fully investigated, whereas much research has focused on the systemic effects of visceral adiposity that are putatively involved in cancer biology. These include alterations in adipokine production, insulin and insulin-like growth factor (IGF) pathways, cancer cell signalling and inflammatory pathways (Fig. 1) and are discussed in further detail below.
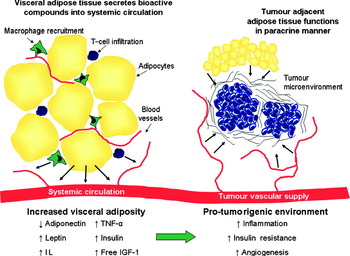
Fig. 1. (Colour online) Increased visceral adiposity creates a pro-tumorigenic environment. As visceral adipose tissue expands it becomes infiltrated with macrophages and T-cells. These immune cells and the adipocytes produce adipokines including leptin, IL and TNF-α. In addition, increasing visceral adiposity leads to hyperinsulinaemia and increased levels of free insulin-like growth factor-1 (IGF-1). Consequently, there is a pro-tumorigenic state of inflammation, angiogenesis and insulin resistance. The tumour microenvironment is influenced by adipose-derived factors secreted into systemic circulation and also via paracrine effects of tumour adjacent adipose tissue.
Altered adipokine production
The existence of adipose tissue-derived factors that may influence metabolism has been suggested since the 1980s( Reference Cook, Min and Johnson 53 , Reference Flier, Cook and Usher 54 ); however, it was the discovery of the adipokines leptin( Reference Zhang, Proenca and Maffei 55 ) and adiponectin( Reference Scherer, Williams and Fogliano 56 ) which led to the concept of adipose tissue as an endocrine organ. The altered secretion of these and other adipokines in obesity has been implicated in the pathogenesis of obesity-associated pathologies, including cancer( Reference Ouchi, Parker and Lugus 57 ). Excess adiposity, in particular visceral obesity, results in a state of chronic systemic low-grade inflammation, attributed to production of these inflammatory cytokines by both adipocytes and infiltrating immune cells creating a pro-tumorigenic environment( Reference Harvey, Lashinger and Hursting 58 ).
Adiponectin is the most abundant adipokine and is secreted mainly from adipocytes in visceral fat; however, circulating levels are inversely correlated with obesity( Reference Kadowaki and Yamauchi 59 ). It has been shown to be both anti-angiogenic and anti-inflammatory, can inhibit tumour growth in animal models and circulating levels are lower in cancer patients( Reference Rose, Komninou and Stephenson 60 ). Leptin may be considered the antithesis of adiponectin. It is an adipokine that acts centrally to regulate appetite and bodyweight( Reference Cummings and Foster 61 ) and circulating levels are positively associated with adiposity( Reference Pär, Annekatrin and Carine 62 ). In vitro studies confirm promotion of cell proliferation, angiogenesis and matrix metalloproteinase expression in oesophageal and colonic cancer cell lines( Reference Somasundar, McFadden and Hileman 63 , Reference Howard, Pidgeon and Reynolds 64 ). Furthermore, serum levels of leptin are positively correlated with cancer risk( Reference Garofalo and Surmacz 65 ). Increased production of leptin and decreased production of adiponectin, correlating with amounts of visceral adiposity and the MetSyn, have been associated with cancer promoting pathways including cellular proliferation, angiogenesis and metalloproteinase expression across a wide variety of cancer subtypes( Reference Howard, Beddy and Ennis 50 , Reference Kadowaki and Yamauchi 59 , Reference Somasundar, McFadden and Hileman 63 ).
Hyperinsulinaemia, insulin resistance and the insulin-like growth factor axis
A core component of the MetSyn is insulin resistance. Nutritionally induced insulin resistance develops as a metabolic adaptation to increased circulating levels of NEFA, which are constantly released from adipose tissue. Increased NEFA levels force liver, muscle and other tissues to shift towards increased storage and oxidation of fats for their energy production. Consequently, insulin secretion rises to compensate for the decreased capacity to handle glucose( Reference Calle, Rodriguez and Walker-Thurmond 10 , Reference Bergman and Ader 66 , Reference Ebeling and Koivisto 67 ). Furthermore, there is a decreased expression of insulin-receptor levels and reduced intracellular insulin signalling in response to insulin receptor binding( Reference Moller and Flier 68 ) resulting in hyperinsulinaemia and insulin resistance.
The ratio of visceral to subcutaneous fat is a proxy measure of insulin resistance. The rate of lipolysis is higher in visceral adipose tissue than subcutaneous adipose tissue, leading to increased circulation of NEFA( Reference Wajchenberg 69 ). Therefore, the risk of developing insulin resistance and hyperinsulinaemia increases with increasing visceral adipose tissue accumulation. Adipokines are believed to be central to the pathogenesis of insulin resistance( Reference Fasshauer and Paschke 70 ), with high concentrations of TNF-α, IL-6, IL-1β, and low concentrations of adiponectin, having deleterious effects on glucose homoeostasis leading to chronic hyperinsulinaemia and insulin resistance( Reference Greenberg and McDaniel 71 ). A number of epidemiological studies have reported that insulin resistance status, characterised by hyperinsulinaemia, is associated with an increased risk of malignancy, including carcinomas of the breast, prostate and colon( Reference Giovannucci 27 , Reference Rose, Komninou and Stephenson 60 , Reference Stocks, Rapp and Bjørge 72 , Reference Hsing, Gao and Chua 73 ). Data from the Me-Can study reported that every 1 mmol/l increment in glucose increased risk of incident cancer in men (RR 1·05, 95% CI 1·01, 1·1) and women (RR 1·11, 95% CI 1·05, 1·16) and further increased RR for fatal cancer (men: RR 1·15, 95% CI 1·07, 1·22; women: RR 1·21, 95% CI 1·11, 1·33)( Reference Stocks, Rapp and Bjørge 72 ). Interestingly, studies of type 2 diabetics have found that the risk of colorectal cancer is higher in those treated with insulin( Reference Yang, Hennessy and Lewis 74 ).
The tumorigenic effects of insulin could be directly mediated by insulin receptors in the pre-neoplastic target cells, or may relate to changes in endogenous hormone metabolism, secondary to hyperinsulinaemia( Reference Schoen, Tangen and Kuller 23 ). In vitro studies demonstrate that insulin increases the neoplastic proliferation of cell lines at both physiological and pharmacological doses, and the insulin receptor is commonly expressed in human neoplasms( Reference Osborne, Bolan and Monaco 75 ). Whether there are differential downstream signalling effects in normal or transformed epithelial cells compared to insulin-responsive tissues (such as fat, liver and muscle) is the subject of current research, addressing in particular whether receptor activation results in cell survival and proliferation rather than altered energy metabolism( Reference Pollak 76 ).
The IGF system may also be relevant to the effects of hyperinsulinaemia in pro-tumour pathways. In the IGF-axis, binding of IGF-1 or IGF-2 to the IGF-1 receptor leads to cell proliferation, differentiation and protection from apoptosis and a dysregulation in this pathway can give rise to malignancy( Reference Frasca, Pandini and Sciacca 77 , Reference Samani, Yakar and LeRoith 78 ). Levels of IGF are influenced by circulating insulin levels, with increasing insulin leading to decreased levels of IGF-binding proteins 1 and 2, thus increasing the bioavailability of IGF( Reference Jones and Clemmons 79 ). The IGF-1 axis has been implicated in the progression of breast, pancreatic, colorectal and oesophageal cancer( Reference Coussens and Werb 80 – Reference Renehan, Frystyk and Flyvbjerg 83 ). Obesity increases levels of bioactive-free IGF-1( Reference Frystyk, Vestbo and Skjaerbaek 84 , Reference Nam, Lee and Kim 85 ), high levels of which are associated with increased risk of malignancy.
In experimental murine models, diet-induced obesity increased both local tumour growth and metastases in wild-type mice compared with lean controls, an effect not seen in liver-specific IGF-1-deficient (LID) mice. In addition, chronic IGF-1 deficiency negates the enhanced expression of inflammatory cytokines and cell adhesion molecules normally up-regulated in obese compared with lean mice( Reference Wu, Brodt and Sun 86 – Reference Olivo-Marston, Hursting and Lavigne 88 ). Hence, it is possible that in obesity IGF-1 can affect tumour development both directly, by stimulating tumour growth and indirectly, by creating a microenvironment that is permissive for tumour growth.
Cancer cell signalling
Eukaryotic cells coordinate cell growth in line with the availability of nutrients in their environment( Reference Shaw and Cantley 89 ). Obesity as a condition of both systemic insulin resistance and nutrient excess may lead to the activation of intracellular pathways that promote tumour growth and progression. The systemic alterations associated with obesity include a change in inflammatory, sex hormone, insulin and adipokine secretion which may directly influence the tumour microenvironment. Cancers that arise in this environment may go on to develop progressive mutations and epigenetic alterations which are influenced by this obese milieu. The concept of oncogene addiction describes the apparent dependency of some tumours on one or a few genes to maintain the malignant phenotype( Reference Weinstein 90 ). Clinical evidence of this is cited as the ability of therapies targeting specific genes or pathways to inhibit cancer cell growth or improve survival rates( Reference Weinstein and Joe 91 ). Whole genome arrays have demonstrated that breast cancer may be subdivided into a number of types based on the genes over-expressed in certain subtypes and that targeting these predominant pathways provides avenues for chemotherapy treatment for each subtype( Reference Perou, Sorlie and Eisen 92 – Reference Sawyers 94 ). Molecular characterisation of tumours has been exploited to develop assays to predict subgroups of patients with poorer prognosis who may benefit from adjuvant therapy( Reference Paik, Shak and Tang 95 ).
Cancers that develop within an obese environment may become selectively altered to signal via specific pathways. For examples, in patients with non-small cell lung cancer, only a subset (approximately 10–20%) respond to the epidermal growth factor receptor targeted therapy gefitinib, and these patients often have an activating mutation of epidermal growth factor receptor( Reference Lynch, Bell and Sordella 96 ). Patients with activating mutations are more likely to have adenocarcinomas, and to be female, non-smokers and Japanese( Reference Taron, Ichinose and Rosell 97 ). Similarly, obesity-related cancers may have a specific set of targets (malfunctioning molecules or pathways), which may be exploitable in clinical practice. Certainly, a number of the putatively dysregulated adipokines and growth factors in obesity may signal via the same intracellular signalling pathways. Candidate pathways include the phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase and signal transducer and activator of transcription 3 pathways. Activation of these pathways leads to multiple downstream effects that underpin cancer progression and metastasis( Reference Huang, Han and Hui 98 – Reference Aggarwal, Kunnumakkara and Harikumar 100 ). Importantly, inhibitors of these pathways are under development at present in order to provide new therapeutic avenues( Reference Sebolt-Leopold and Herrera 101 – Reference Jing and Tweardy 103 ).
Is there any evidence that cancers which develop within the obese milieu are ‘addicted to’ or preferentially signal via these pathways? Whole genome analysis of breast cancer tumours divided according to BMI demonstrate that an obesity-associated gene signature pattern is associated with a shorter time to metastasis and is associated with an IGF signalling signature in multiple publically available breast cancer genome arrays( Reference Creighton, Sada and Zhang 104 ). Most data come from murine models. PI3K activity (measured by phosphorylated protein kinase B and mammalian target of rapamycin protein levels) is increased in diet-induced obesity in mice and is associated with an increased level of circulating IGF-1 compared with controls( Reference Moore, Beltran and Carbajal 105 ). Mice fed a high-energy diet have twice the volume of tumours 17 d after colon cancer cell injection versus controls. PI3K pathway activity was demonstrated by increased phosphorylated protein kinase B protein levels. The tumour growth effect was abrogated by metformin treatment, which led to decreased phosphorylated protein kinase B levels( Reference Algire, Amrein and Zakikhani 106 ). In a mouse model of obesity-related skin cancer, obese mice had higher PI3K activity after UV exposure than lean mice( Reference Sharma and Katiyar 107 ). Activity of mitogen-activated protein kinase phosphorylation and NF-κB signalling were also higher following UVB irradiation in the obese mice( Reference Katiyar and Meeran 108 ). In breast and colorectal cancer, over-expression of the leptin receptor has been identified. As leptin has mitogenic and anti-apoptotic effects on cancer cells lines it is postulated that it acts through mitogen-activated protein kinase and PI3K pathways. This hypothesis is strengthened by the observation that inhibition of these pathways attenuates the growth promoting effects of leptin( Reference van Kruijsdijk, van der Wall and Visseren 109 ). In an obesity-associated hepatoma model, IL-6 and TNF-α induce the development of cancer via activation of signal transducer and activator of transcription 3 pathway( Reference Park, Lee and Yu 110 ). Excess energy balance associated with the obese state may also influence tumour growth. Mouse tumour xenografts have decreased incidence and slower growth in mice that are fed an energy-restricted diet. Tumours that are resistant to dietary restriction have constitutive activation of the PI3K pathway( Reference Kalaany and Sabatini 111 ). The proposed cell signalling pathways that are altered in the obese state and associated with tumorigenesis are illustrated in Fig. 2.
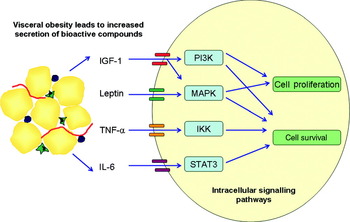
Fig. 2. (Colour online) Visceral adiposity effects cancer cell signalling pathways. Increased visceral adipose tissue leads to increased circulation of a number of bioactive compounds such as IL-6, TNF-α, leptin and insulin-like growth factor-1 (IGF-1). Binding of these compounds to their respective receptors on tumour cells leads to the activation of cell signalling pathways including the phosphatidylinositol 3-kinase (P13K), mitogen-activated-protein-kinase (MAPK), signal transducer and activator of transcription 3 (STAT3) and IkB kinase (IKK) pathways. The cascade of downstream signalling leads to increased cell survival and proliferation thus promoting tumour progression.
Visceral obesity, inflammation and cancer
As early as the nineteenth century it was perceived that cancer is linked to inflammation and an inflammatory component is present in the microenvironment of most neoplastic tissues. Key features of cancer-related inflammation include the infiltration of immune cells, the presence of cytokines and chemokines, including TNF-α, IL-1β, IL-6 and chemokines such as monocyte chemotactic protein-1 and CXCL8( Reference Colotta, Allavena and Sica 112 ). Thus, cancer-related inflammation is a key component of the tumorigenic process and is increasingly referred to as the seventh hallmark of cancer.
NF-κB, first described in 1986 by Ranjan Sen and David Baltimore, is a central coordinator for multiple inflammatory and anti-inflammatory signalling pathways and has become one of the most investigated transcription factors( Reference Sen and Baltimore 113 ). Activation of NF-κB has been shown to control a range of cellular processes in cancer including inflammation, proliferation, angiogenesis, metastasis, chemoresistance and radioresistance( Reference Chaturvedi, Sung and Yadav 114 , Reference Ben-Neriah and Karin 115 ). Obesity has been shown to increase the systemic activation of NF-κB ( Reference Cai, Yuan and Frantz 116 ). It is constitutively active in a wide range of human tumour cells, and its suppression has been shown to inhibit their growth, leading to the ‘NF-κB addiction’ theory in tumour cells( Reference Chaturvedi, Sung and Yadav 114 ).
The omentum is one of the largest components of the visceral adipose depot in human subjects and has been the subject of research for more than 3600 years( Reference Walker 117 ). The word ‘omentum’ has its origins in the practice of Egyptian priests examining the abdominal viscera during embalming the body. They would tell fortunes by it and give good or bad ‘omens’ for the deceased( Reference Walker 117 ). Adipose tissue, including the omentum, is thought to be a primitive immune organ, but unlike other adipose tissue depots the omentum has many unique functions( Reference Caspar-Bauguil, Cousin and Galinier 118 ). It migrates to and surrounds areas of inflammation and infection within the peritoneal cavity and is a rich source of pro-angiogenic and inflammatory mediators( Reference Casten and Alday 119 ). Dense lymphoreticular structures within the human omentum, termed milky spots contain a range of innate and adaptive immune cells, namely macrophages, mast cells, T- and B-cells( Reference Platell, Cooper and Papadimitriou 120 ). More recent studies have also demonstrated the presence of natural killer and invariant natural killer T-cell populations within the human omentum, which are altered by obesity and cancer( Reference Lynch, O'Shea and Winter 121 ). These innate lymphocytes are capable of killing virally infected or tumour cells in a non-antigen-specific manner. In addition to an important role in immunosurveillance of the peritoneal cavity, another key function of the human omentum is to support B-cell function. The omentum collects antigens and cells from the peritoneal cavity and supports T-dependent B-cell responses, including isotype switching, somatic hypermutation and limited affinity maturation, despite the lack of identifiable follicular dendritic cells( Reference Rangel-Moreno, Moyron-Quiroz and Carragher 122 ).
Macrophages are the most abundant leukocyte population in adipose tissue( Reference Morris, Singer and Lumeng 123 ). Infiltration of inflammatory macrophages into adipose tissues correlates with increasing obesity levels and triggers insulin resistance and fuels inflammation. A significant number of macrophages are also present in lean, metabolically healthy human subjects suggesting that not all adipose tissue macrophages are programmed to be pro-inflammatory. Current evidence indicates that macrophages undergo a functional conversion to promote inflammation during obesity( Reference Lumeng, Bodzin and Saltiel 124 ). In visceral fat, obesity alters the state of adipose tissue macrophages from an anti-inflammatory M2 phenotype to a pro-inflammatory M1 state( Reference Morris, Singer and Lumeng 123 ), further potentiating metabolic dysfunction, insulin resistance and inflammation in obese individuals.
The majority of studies so far evaluating the immune cell properties of visceral adipose tissue have predominantly focused on macrophages. T-cells, however, may be key regulators of adipose tissue inflammation, and a recent study in a murine model revealed a significant increase in CD8+ T-cell infiltration into expanding visceral adipose tissue, which preceded macrophage infiltration( Reference Nishimura, Manabe and Nagasaki 125 ). These T-cells were found to be responsible for both the establishment and maintenance of adipose tissue inflammation in obesity( Reference Nishimura, Manabe and Nagasaki 125 ). We have recently shown that the human omentum is a particularly rich source of CD8+ T-cells in patients with obesity-associated oesophageal adenocarcinoma. A high percentage of CD4+ and CD8+ omental T-cells were activated with an inflammatory T-helper 1 phenotype (CD69+CD45RO+IFN-γ+)( Reference Lysaght, Allott and Donohoe 126 ). Interestingly, we also demonstrated that significantly more interferon-γ+ T-cells were present in the omentum of cancer patients compared with non-cancer controls( Reference Lysaght, Allott and Donohoe 126 ). The role of T-cells in adipose tissue inflammation and cancer has not received a lot of attention so far but offers potential immunotherapeutic targets for obesity-associated morbidities.
Conclusion
Adipose tissue is not simply an energy store but acts in a co-ordinated fashion to exert systemic hormonal and metabolic effects. There is sound epidemiological evidence relating excess adiposity and associated dysmetabolism to a broad range of cancer subtypes and emerging clinical research continues to bolster this observation. The recognition of visceral obesity as a major contributing factor to the development of cancer mandates further research into pathophysiological mechanisms. Much work remains to elucidate the independent and synergistic effects of inflammation, insulin resistance, the IGF-axis and adipokines on carcinogenesis in obesity. Clearly, the association between obesity and cancer is an important area to target in public health policy and one that will assume increasing importance over the coming years.
Acknowledgements
This work was funded by an Irish Council for Science, Engineering and Technology, EMBARK scholarship, an Irish Cancer Society scholarship, a Health Research Board Post-Doctoral Fellowship (PD/2009/35) and CROSS charity (Reg. Charity 389874). The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported. J.V.R. presented the work and was responsible for the concept, direction and supervision of the manuscript. C.L.D. and J.L. researched and prepared sections for the manuscript. S.L.D. researched and prepared sections for the manuscript and was responsible for manuscript assembly and construction of figures.


