


Hepatocellular carcinoma (HCC) is the third most frequent cause of death due to malignancies in men, and its incidence is increasing worldwide(Reference El-Serag and Mason1–Reference Taylor-Robinson, Foster, Arora, Hargreaves and Thomas3). High incidences, easy metastasis and frequent recurrence even after ablation mean that patients with advanced HCC have little chance of survival. Infection with hepatitis B and C viruses and ingestion of aflatoxin B1-contaminated food remain the major risk factors for HCC in Asia(Reference Chen, Yu and Liaw4). Age, sex and alcohol-related cirrhosis are associated with HCC development(Reference Chen, Yu and Liaw4, Reference Koike, Shiratori and Sato5). Relatively little is known about the nutritional status of HCC patients and its relationship with HCC development. A recent prospective cohort study showed an association of low blood folate levels with risks for liver damage and HCC(Reference Welzel, Katki, Sakoda, Evans, London, Chen, O'Broin, Shen, Lin and McGlynn6), suggesting a possible role of folate in the carcinogenesis of human HCC.
Folate plays a central role in one-carbon metabolism in the liver. An adequate folate status supplies the liver with available one-carbon carriers for de novo thymidylate and purine synthesis, amino acid interconversion and methylation of macromolecules(Reference Shane and Bayley7). Chronic liver diseases such as viral hepatitis(Reference Kao8, Reference Tkaczewski, Niedzielska, Malafiej, Dworniak and Dramiski9), alcoholic liver disorder(Reference Wu, Chanarin, Slavin and Levy10) and liver cirrhosis(Reference Bosy-Westphal, Petersen, Hinrichsen, Czech and Muller11–Reference García-Tevijano, Berasain, Rodríguez, Corrales, Arias, Martin-Duce, Caballeria, Mato and Avila13) may compromise the folate status, as low blood folate levels are commonly found in patients with those diseases. In animal studies, folate deficiency leads to genomic instability, including increased DNA strand breaks(Reference Pogribny, Basnakian, Miller, Lopatina, Poirier and James14), hypomethylation within the p53 tumour suppressor gene(Reference Pogribny, Basnakian, Miller, Lopatina, Poirier and James14–Reference Pogribny, Miller and James16) and altered expression of genes involved in cell-cycle regulation(Reference Crott, Choi, Ordovas, Ditelberg and Mason17), all of which have been proposed as plausible mechanisms that link folate deficiency to liver tumour progression(Reference James, Pogribny, Pogribna, Miller, Hernigan and Melnyk18). Disturbance of one-carbon metabolism by a methyl-deficient diet (deficient in folate, choline and methionine) has been shown to promote tumour progression in rat liver(Reference Ghoshal and Farber19, Reference Mikol, Hoover, Creasia and Poiner20). The folate status of HCC patients in relation to tumour progression, however, has not been assessed.
Accordingly, the aims of the present study were to investigate the folate status of HCC patients and its relationship to tumour progression. Ninety HCC patients recruited through the Department of Internal Medicine, Chi-Mei Hospital, participated in this cross-sectional study. We studied serum folate levels, dietary folate intake, general nutritional status and clinical data, including liver damage and tumour progress.
Materials and methods
Subjects
Between April 2005 and October 2006, 120 potentially eligible patients with HCC were recruited through the Department of Internal Medicine of Chi-Mei Hospital (Tainan, Taiwan). Chi-Mei Hospital provides medical services to a defined population base in southern Taiwan. Patients were diagnosed with HCC by imaging examinations, including B-type ultrasonography, computed tomography, MRI and angiography. The diagnosis of HCC progression (size, number and metastasis) was made by two physicians specialised in hepatology and oncology. For patients with a tumour size of 1–2 cm, the presence of HCC was histologically confirmed. All studied patients had primary HCC. Diagnosis of liver cirrhosis was also histologically proven. HCC patients with cardiac or renal diseases, overt diabetes, or active intravenous drug abuse were excluded. Patients with severe illness or who were unwilling to donate extra blood samples withdrew from the study. In total, ninety HCC patients participated in the entire study. Informed consent was secured from all study participants. The protocol was approved by the Committee on Medical Research of Chi-Mei Hospital and the Ethical Review Board of Fu-Jen University.
Risk factors and dietary assessment
Within 1 week following the diagnosis of HCC and before treatment in scheduled consultations, patients were asked to donate fasting blood samples and to complete questionnaires concerning their medical history, personal habits and use of medications. Demographic data, smoking status and alcohol consumption were recorded. Experienced dietitians helped patients complete a semi-quantitative FFQ covering the previous year to assess their habitual dietary folate intake. Their current folate intake was assessed by a 24 h recall at the time of the HCC diagnosis. The questionnaire was developed in our laboratory for assessing the folate intake of Taiwanese(Reference Lee, Lee, Wong, Tzeng and Huang21). It has been validated by multiple 24 h recalls (r 0·86; P < 0·001)(Reference Lee, Lee, Wong, Tzeng and Huang21) and by plasma folate levels (r 0·57; P < 0·001)(Reference Kao8).
Blood biochemical determinations
Peripheral blood samples were taken after a 12 h fasting period, chilled, and transported to the laboratory, where serum samples were immediately separated upon arrival. The time between blood sampling and separation of serum for total homocysteine (tHcy) determinations was restricted to 2 h. tHcy levels were measured by fluorescence polarisation immunoassay (Becton Dickinson, Orangeburg, NY, USA). Serum folate was determined with a commercial RIA kit (Becton Dickinson). Serum glutamic-oxaloacetic transaminase (GOT), alanine aminotransferase (ALT) and albumin concentrations were measured by standard techniques (ITC Diagnostics, Taiwan). Hb was measured in a Coulter STKS counter (Beckman Coulter, Miami, FL, USA).
Classification of tumour progression
HCC progression was classified according to recognised criteria by the tumour, regional lymph node, and metastases (TNM) system(Reference Greene, Balch, Page, Haller, Fleming, Morrow, Fritz and Greene22). The classification considers the presence or absence of vascular invasion, the number of tumour nodules (single v. multiple) and the size of the largest tumour ( < 5 v. ≥ 5 cm). In brief, primary tumour progression was categorised as T1 (solitary tumour without vascular invasion), T2 (solitary tumour with vascular invasion or multiple tumours none more than 5 cm), T3 (multiple tumours more than 5 cm or a tumour involving a major branch of the portal or hepatic veins) or T4 (with metastasis). Tumour stage grouping was defined as stages I–III for T1, T2 and T3, respectively, with no regional lymph node metastasis or distant metastasis. Stage IV was defined as any primary tumour with metastasis in any regional lymph node or distant metastasis.
Statistical analysis
Data are presented as mean values and standard deviations. According to the clinical criteria, serum folate status of individuals was classified as normal (serum folate>14 nmol/l or 6 ng/ml), marginally deficient (7–14 nmol/l or 3–6 ng/ml) or deficient ( < 7 nmol/l or 3 ng/ml)(Reference Waters, Mollin, Pope and Towler23, Reference Herbert24). As serum folate levels of patients were stratified into various folate statuses, the absolute frequencies of categorical variables such as sex, viral infection and the presence of clinical complications were compared using the χ2 test. Demographic and laboratory data of continuous variables were compared using one-way ANOVA followed by Duncan's multiple range test. Dependence between the folate status and tumour progression markers was evaluated using Pearson's correlation coefficient. Logistic regression models were used to estimate the OR and 95 % CI for large tumours ( ≥ 5 cm), tumour multiplicity ( ≥ 2), and metastasis with respect to normal folate status (serum folate ≥ 14 nmol/l) v. deficient folate status (serum folate < 14 nmol/l). Non-normally distributed dependent variables were first log-transformed. Statistical analyses were performed using the Statistical Analysis System (SAS/STAT version 6.12; SAS Institute, Cary, NC, USA). Differences were considered to be statistically significant at P < 0·05.
Results
Baseline and clinical characteristics of the studied hepatocellular carcinoma patients
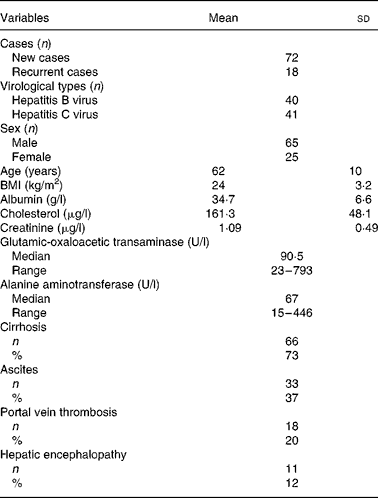
Table 1 presents the baseline and clinical data of the HCC patients. Of the ninety patients, 80 % had a first-time diagnosis of HCC and 20 % had recurrent HCC within an interval of 6–24 months following their first surgical resection or transarterial chemoembolisation. A total of 44 % of patients were seropositive for hepatitis B surface antigen, 46 % were positive for serum anti-hepatitis C virus antibody, and the remaining 10 % were non-B, non-C. The mean age of patients was 62 (sd 10) years. No obvious disorders with cholesterol metabolism or kidney dysfunction were observed. Seventy-three percent had liver cirrhosis. Clinical complications included ascites (37 %), portal vein thrombosis (20 %) and hepatic encephalopathy (12 %).
Table 1 Baseline and clinical data of ninety patients with hepatocellular carcinoma
(Mean values and standard deviations)
Demographic, lifestyle and nutritional variables by serum folate status
According to the clinical criteria, 44 % of HCC patients showed marginal folate deficiency (serum folate 7–14 nmol/l) and 16 % were folate deficient ( < 7 nmol/l) (Table 2). Patients with deficient serum folate had a significantly lower habitual folate intake (207 (sd 113) v. 336 (sd 190) μg/d; P = 0·032), higher serum tHcy concentrations (14·1 (sd 6·2) v. 9·6 (sd 4·8) μmol/l; P = 0·001) and higher frequencies of alcohol intake (36 v. 14 %; P = 0·019) than those with normal serum folate levels. Serum folate levels were not significantly different by age, BMI, smoking habit, albumin level or Hb level.
Table 2 Demographic, lifestyle and nutritional variables by serum folate status of patients with hepatocellular carcinoma (HCC)†
(Mean values and standard deviations)

DRI, dietary reference intake.
* Value was significantly different from that of the normal-folate group (P < 0·05).
† Data of continuous variables were compared using one-way ANOVA followed by Duncan's multiple range test. The χ2 test was used for categorical variables.
‡ Serum folate status of individuals was classified into normal ( ≥ 14 nmol/l), marginal deficiency (7–14 nmol/l) or deficiency ( < 7 nmol/l) according to the clinical criteria(Reference Waters, Mollin, Pope and Towler23, Reference Herbert24).
§ Regular alcohol intake was defined as one or more drinks per week.
∥ Smoking habit was defined as never smoking, or not smoking in the 6 months before the diagnosis of HCC.
¶ Current dietary intake was assessed by 24 h recall. DRI of elderly healthy individuals for folate is 400 μg/d.
†† Habitual folate intake was the dietary intake in the last year, as assessed by a semi-quantitative frequency questionnaire.
Hepatic damage, clinical complications and tumour markers in hepatocellular carcinoma patients with various folate statuses
As shown in Table 3, neither liver injury (GOT and ALT levels) nor clinical complications (cirrhosis and ascites) were correlated with serum folate levels. Patients with folate deficiency had significantly higher α-fetal protein levels (1137 (sd 3838) v. 8 (sd 28) μg/l; P = 0·037), larger tumours (10 (sd 14·3) v. 3·0 (sd 2·7) cm; P = 0·0003), more tumours (3 (sd 0·6) v. 2·2 (sd 1·0); P = 0·042) and higher metastasis rate (64 v. 11 %; P = 0·0002) than those with normal folate levels ( ≥ 14 nmol/l). Values of the clinical parameters in patients with marginal folate deficiency lay between those of the folate-deficient and normal-folate groups. Pearson's correlation coefficient revealed inverse correlations between serum folate and tumour size (r − 0·29; P = 0·005), tumour multiplicity (r − 0·24; P = 0·018) and metastasis rate (r − 0·391; P = 0·0001).
Table 3 Hepatic injuries, clinical complications and tumour progression by serum folate status of patients with hepatocellular carcinoma‡
(Mean values and standard deviations)
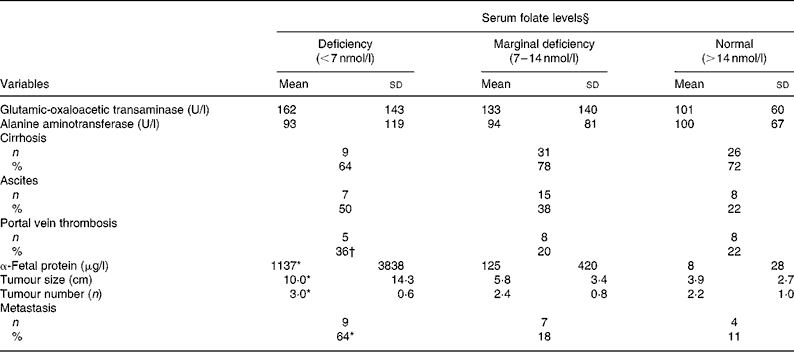
* Value was significantly different from that of the normal-folate group (P < 0·05).
† Value was significantly different from that of the marginal folate-deficient group (P < 0·05).
‡ Data of continuous variables were compared using one-way ANOVA followed by Duncan's multiple range test. The χ2 test was used for categorical variables.
§ Serum folate status of individuals was classified into normal ( ≥ 14 nmol/l), marginal deficiency (7–14 nmol/l) or deficiency ( < 7 nmol/l).
Folate status and clinical factors in relation to hepatocellular carcinoma stages
When HCC progression was categorised into stages I to IV(Reference Greene, Balch, Page, Haller, Fleming, Morrow, Fritz and Greene22), tumour progression was associated with lower serum folate levels (stage I 24·5 (sd 11·5) v. stage IV 10·3 (sd 3·3) nmol/l; P = 0·032), independent of folate intake (Table 4). Advanced tumour staging was associated with elevated α-fetal protein levels (P = 0·001) and lower BMI (P = 0·032). No significant differences in general nutritional status (albumin and Hb levels) and liver injury (GOT and ALT levels) between tumour stages were observed.
Table 4 Folate status and clinical factors in relation to hepatocellular carcinoma (HCC) stages†
(Mean values and standard deviations)

* Mean value was significantly different from that of the stage I group (P < 0·05).
† Statistical difference was determined by one-way ANOVA followed by Duncan's multiple range test.
‡ HCC staging was classified according to recognised criteria of the tumour, regional lymph node, and metastases (TNM) system. Detailed descriptions are given in Materials and methods.
§ Current dietary intake was assessed by 24 h recall.
∥ Habitual folate intake was the dietary intake in the last year, as assessed by a semi-quantitative frequency questionnaire.
¶ Data of α-fetal protein levels were log-transformed.
Odds ratios of hepatocellular carcinoma progression by blood folate status
We used a blood folate level of less than 14 nmol/l (the cut-off point for marginal and deficient folate levels) to represent low blood folate status for further analysis (Table 5). The OR of advanced tumour progression associated with low blood folate levels relative to normal serum folate status ( ≥ 14 nmol/l) was 7·1 for large tumours (95 % CI 2·27, 21·9; P = 0·007), 3·2 for multiple tumours (95 % CI 1·07, 3·51; P = 0·04) and 4·5 (95 % CI 1·11, 18·4; P = 0·03) for metastasis after controlling for age, sex, lifestyle and dietary factors (model A; Table 5). Further controlling for liver injury, tumour proliferation and tumour stages, however, negated the effect of folate on advanced tumour progression (models B and C).
Table 5 Risk of hepatocellular carcinoma (HCC) progression by blood folate level
(Odds ratios and 95 % confidence intervals)
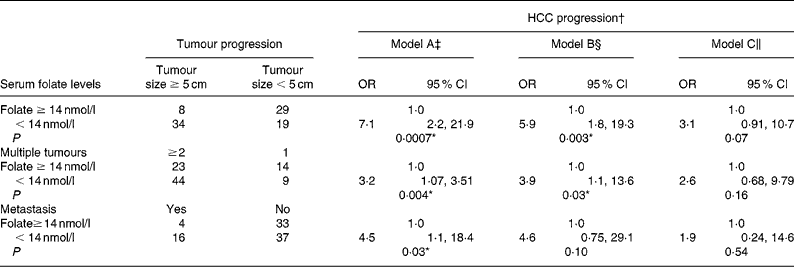
* Statistically significant at P < 0·05.
† Advanced tumour progression is defined as tumour size ≥ 5 cm, tumour number ≥ 2 and metastasis development.
‡ Model A: adjusted for age, sex, alcohol intake and habitual folate intake.
§ Model B: adjusted for all parameters in model A with the addition of albumin, alanine aminotransferase levels and portal vein thrombosis.
∥ Model C: adjusted for all parameters in model B with the addition of α-fetal protein levels and tumour stages.
Discussion
According to the clinical criteria(Reference Waters, Mollin, Pope and Towler23, Reference Herbert24), 44 % of HCC patients showed marginal folate deficiency, and 16 % were folate deficient. Elevated tHcy concentrations in these patients confirmed a functional folate insufficiency(Reference Selhub, Jacques, Wilson, Rush and Rosenberg25). The causes of low blood folate status in HCC patients are unknown. Several factors may contribute. Malnourishment is commonly found in some cancer patients(Reference Shimizu, Nagaya, Isobe, Imazu, Okumura, Hosoda, Kojima, Kangawa and Kohno26), and may compromise the serum folate status. Impaired liver function is reported to accompany a low folate status and high tHcy levels, especially in patients with viral hepatitis(Reference Kao8, Reference Tkaczewski, Niedzielska, Malafiej, Dworniak and Dramiski9), alcoholic liver disorder(Reference Wu, Chanarin, Slavin and Levy10) and liver cirrhosis(Reference Bosy-Westphal, Petersen, Hinrichsen, Czech and Muller11–Reference García-Tevijano, Berasain, Rodríguez, Corrales, Arias, Martin-Duce, Caballeria, Mato and Avila13). In the present study, the low serum folate levels were not associated with changes in BMI, levels of albumin, Hb, GOT or ALT, or liver cirrhosis (Tables 2 and 3). It is likely that low folate status was associated with general ill-health of HCC patients, yet was not detected due to low numbers of the study or a low predictive value of the nutritional marker. A recent study reported that folate and B vitamin status of HCC patients were reduced as HCC progressed(Reference Lin and Yin27). Our data confirmed that serum folate decreased as HCC stage progressed (Table 4). The findings raise the possibility that low folate status could be an intrinsic consequence of tumour growth. There may be an increased demand for folate as tumours grow. Alternatively, we found that the low serum folate in HCC patients was associated with a low habitual folate intake, suggesting the possible contribution of chronic insufficient dietary folate intake to their low folate status. Whether variations in eating habits and dietary patterns or germline methylenetetrahydrofolate reductase C677T genotype (a marker of long-term folate status) affect the blood folate status of HCC patients is under our investigation.
One of our major findings is that low blood folate status could be a risk factor for tumour progression. After adjustment for age, sex, lifestyle and dietary factors, we found increased risks for tumour progression in large tumours (OR 7·1; 95 % CI 2·27, 21·9), tumour multiplicity (OR 3·2; 95 % CI 1·07, 3·51) and metastasis (OR 4·5; 95 % CI 1·11, 18·4) in HCC patients with deficient blood folate levels. The finding is consistent with results from the folate/methyl-deficient rat model of hepatocarcinogenesis showing that a methyl-deficient diet promoted tumour progression in rat liver(Reference James, Pogribny, Pogribna, Miller, Hernigan and Melnyk18–Reference Mikol, Hoover, Creasia and Poiner20). Chronic dietary folate/methyl deficiency is known to result in genomic DNA strand breakage(Reference Pogribny, Basnakian, Miller, Lopatina, Poirier and James14), uracil misincorporation, hypomethylation within the p53 tumour suppressor gene(Reference Pogribny, Basnakian, Miller, Lopatina, Poirier and James14–Reference Pogribny, Miller and James16) and altered expression of genes involved in cell-cycle regulation(Reference Crott, Choi, Ordovas, Ditelberg and Mason17), all of which contribute to in vivo carcinogenesis(Reference James, Pogribny, Pogribna, Miller, Hernigan and Melnyk18) and possibly to multiple tumour development. Genome-wide hypomethylation and decreased expression of tumour suppressor genes (p53 and p16 INK4A) were observed in early pre-neoplastic liver tumours in rats fed a folate/methyl-deficient diet(Reference Pogribny and James28, Reference Pogribny, James, Jernigan and Pogribna29) and in patients with HCC(Reference Zhang, Ahsan, Chen, Lunn, Wang, Chen, Lee, Chen and Santella30, Reference Zhang, Chen, Ahsan, Lunn, Lee, Chen and Santella31), and may deregulate apoptotic and proliferation processes(Reference James, Miller, Basnakian, Pogribny, Pogribna and Muskhelishvili32) to favour the development of large tumours. Our data together with results of folate-deficient animal studies support the hypothesis that HCC progression may be associated with low blood folate status. However, the possibility of reverse causality cannot be excluded in the present study given the fact that serum folate decreased as HCC stage progressed, independent of dietary folate intake (Table 4). Whether the mechanistic relationships exist between deficient folate levels, large tumours and tumour multiplicity in human HCC, as proposed in rodent models of hepatocarcinogenesis, will depend on future research results.
It is notable that adjustment for liver injury (ALT levels), tumour proliferation (α-fetal protein levels) and tumour staging, however, negated the effect of folate on the increased risk of advanced tumour progression. The data suggest that clinical lesions present in HCC patients are important confounders that modulate the effect of low folate status in advanced HCC progression. Although the mechanisms that promote human HCC malignancy remain unclear, cancer metastasis is thought to involve complex multi-steps including growth, angiogenesis, dissemination, invasion and survival of cancer cells(Reference Hanahan and Weinberg33, Reference Budhu, Zipser, Forgues, Ye, Sun and Wang34). In response to the in vivo microenvironment, differential changes of the primary tumours including a mixture of tumour cells, stromal cells and endothelial cells may play a role in tumour progression to metastasis(Reference Steeg35). It has been proposed that the degree of viral hepatitis-mediated liver damage may affect intrahepatic metastasis(Reference Budhu, Zipser, Forgues, Ye, Sun and Wang36). Thus, changes of the in vivo clinical microenvironment in HCC appear to work together and override the effects of folate deficiency on advanced HCC progression. Future studies with larger sample sizes are warranted to confirm this result and to further elucidate the combined effects of low blood folate level, liver injury and tumour behaviour in the cascade of events driving HCC malignancy.
Our findings should be interpreted in the context of a few limitations. The most important one is the relatively small sample size, which reduces the statistical power for subgroup analysis. Insufficient statistical power in multivariate analysis may provide only pilot results. The second is the possibility of error associated with dietary assessments using a 24 h recall or FFQ. Participants may misreport their daily intake or underestimate portion sizes, particularly when ill. Finally, the inherent limitations associated with retrospective study designs cannot conclude folate insufficiency as a cause of advanced HCC progression.
Despite these limitations, our data provide several implications for HCC prognosis. For clinical treatment of advanced HCC, hepatic resection followed by intrahepatic arterial chemotherapy using the antifolate drug 5-fluorouracil may place more metabolic pressure on the unidentified folate deficiency of HCC patients(Reference Damdinsuren, Nagano and Monden37, Reference Robien38). Persistent folate deficiency during the clinical course of HCC could aggravate nutrition problems if left untreated. Further longitudinally designed and interventional studies with a large sample size may help determine the optimal strategies to improve low folate status of HCC patients and to explore the possible interactions with combined clinical conditions for better prognosis.
Acknowledgements
The authors gratefully acknowledge the assistance of Ms Wan Shen Lin in sample collection and lymphocyte extraction. The study was supported by grants from the Department of Health, Taiwan (DOH95-TD-F-113-008 to R.-F. S. H.). There are no financial or other contractual agreements that might cause conflicts of interest. C.-S. K. and C.-Y. L. contributed equally to the study.





