


Isothiocyanates are promising chemopreventive agents that are present in substantial concentrations in brassica cruciferous vegetables; in epidemiological studies, intake of these vegetables was found to afford protection against a number of cancersReference van Poppel, Verhoeven, Verhagen and Goldbohm1–Reference Zhang3. Isothiocyanates occur as glucosinolates but are released following exposure to the enzyme myrosinase (β-thioglucoside glucohydrolase), which comes into contact with these compounds during the harvesting, chopping and mastication of these vegetables. Moreover, microbial myrosinase activity in the human intestine contributes to the release of isothiocyanates from their glucosinolate precursorsReference Getahun and Chung4.
Epidemiological studies revealed the potential of isothiocyanates to protect against tumorigenesis at a number of sitesReference Lin, Probst-Hensch, Louie, Kau, Witte, Ingles, Frankl, Lee and Haile5, Reference Spitz, Duphorne, Detry, Pillow, Amos, Lei, de Andrade, Gu, Hong and Wu6. Furthermore, studies conducted in animal models established that isothiocyanates can antagonise the carcinogenicity of chemicals, including dietary carcinogens such as polycyclic aromatic hydrocarbonsReference Hecht7. A number of mechanisms appear to contribute to their anticarcinogenic activity including impairment of the bioactivation of carcinogens and increased detoxication of their reactive intermediates, suppressed cellular proliferation and increased apoptosisReference Zhang3, Reference Singh, Srivastava and Choi8–Reference Talalay and Fahey10. As most of these mechanistic studies have been conducted in in vitro systems, utilising a range of concentrations, in order to extrapolate such data to the in vivo situation it is essential that the pharmacokinetic fate of isothiocyanates, following dietary levels of intake, including their bioavailability and plasma concentrations, are ascertained.
One such isothiocyanate is sulforaphane [1-isothiocyanato-4-(methylsulphinyl) butane], found particularly in broccoli, where it is present as the glucosinolate, glucoraphanin. In animal studies it efficiently antagonised the carcinogenicity of polycyclic aromatic hydrocarbonsReference Zhang and Talalay11 and reduced the number of azoxymethane-induced aberrant crypt foci in rat colonReference Chung, Conaway, Rao and Reddy12. Although its mechanism of action appears to be multifactorial, its chemopreventive effect also involves impairment of the initiation stage of carcinogenesis. Sulforaphane inhibited the DNA binding of benzo[a]pyrene and 1,6-dinitropyrene in human mammary epithelial cellsReference Singletary and MacDonald13 and the formation of DNA adducts with the heterocyclic amine 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in human hepatocytesReference Bacon, Williamson, Garner, Lappin, Langouët and Bao14. Even when administered at dietary doses, sulforaphane stimulates detoxication enzymes such as quinone reductaseReference Yoxall, Kentish, Coldham, Kuhnert, Sauer and Ioannides15 and at higher doses it also enhances the activity of other deactivating enzyme systems such as glutathione S-transferaseReference Zhang and Talalay11, Reference Zhang and Talalay16. However, sulforaphane also acts at the post-initiation stages, for example, suppressing the conversion of lung benign tumours to carcinomas in miceReference Conaway, Wang, Pittman, Yang, Schwarz, Tian, McIntee, Hecht and Chung17. An interesting observation is that mercapturates, the principal metabolites of isothiocyanates, may retain the anticarcinogenic activity of the parent compoundsReference Conaway, Wang, Pittman, Yang, Schwarz, Tian, McIntee, Hecht and Chung17, Reference Tang, Li, Song and Zhang18.
In the present study, a liquid chromatography-MS/MS method has been developed to determine sulforaphane in rat plasma and was used to determine the absolute bioavailability of dietary doses of sulforaphane following oral and intravenous administration.
Experimental methods
Materials
R,S-Sulforaphane was purchased from LKT Laboratories (St Paul, MN, USA). All solvents were of HPLC grade (Fisher Scientific, UK).
Treatment of animals
Male Wistar albino rats (200 g) were obtained from B&K Universal Ltd (Hull, East Yorkshire, UK). The animals were housed at 22 ± 2°C, 30–40 % relative humidity in an alternating 12-h light–dark cycle with light onset at 07.00 hours. The animals were allowed to acclimatise for at least 4 d before commencement of the study; they were then treated, four per group, with either a single intravenous dose of sulforaphane (0·5 mg/kg) in a volume of 100 μl water or single oral doses of 0·5, 1·0 and 5·0 mg/kg, corresponding to 2·8, 5·6 and 28 μmol/kg, dissolved in 1 ml water. Blood samples (100 μl) were withdrawn from the rat tail at regular time intervals for 8 h and placed into lithium-heparinised centrifuge tubes. A sample was also obtained 24 h after administration as well as from untreated rats.
Sample preparation
Aliquots of the plasma (40 μl) were made to 1 ml with 0·01 m-PBS (pH 7·3) and were subsequently extracted with chloroform (5 ml) twice for 30 min; the layers were separated by centrifugation at 510 g for 10 min. The combined chloroform extracts were evaporated under N using an N-EVAP® model 111 (Organomation Assoc. Inc, USA) and redissolved in 40 μl HPLC mobile phase (10 % acetonitrile in water containing 0·1 % formic acid) and 20 μl was injected for analysis. Quality control sulforaphane solutions of known concentrations (0·05, 0·5 and 1·8 μg/ml) were carried through the same procedure.
Determination of sulforaphane in rat plasma
Plasma concentrations of sulforaphane in the rat plasma were determined by liquid chromatography-MS/MS, using methodology developed in our laboratories. Separation of sulforaphane was achieved using an Ultimate 3000 (Dionex, Camberley, UK) HPLC employing a Synergi 4u Fusion-RP analytical column (80 Å particle size, 150 × 1 mm) fitted with a KrudKatcher in-line filter, both supplied by Phenomenex (Macclesfield, UK). Mobile phase consisted of solvent A (10 % acetonitrile in water containing 0·1 % formic acid) and solvent B (90 % acetonitrile in water containing 0·1 % formic acid). The analyte was eluted with a linear gradient (0 to 20 %) of organic solvent B over 22 min. The column was then washed with 80 % solvent B over 3 min and re-equilibrated for a further 10 min; the eluent flow rate was 0·05 ml/min. Under these conditions, the retention time of sulforaphane was about 18 min in a total run time of 35 min.
Sulforaphane was detected on-line using an API 2000 triple quadrupole mass spectrometer (Applied Biosystems, Warrington, UK) equipped with a turbo-ion electrospray probe, operating in the positive ionisation mode. The analyte was detected by monitoring the m/z 178 → 114 and 178 → 72 mass transition (Fig. 1) in the multiple reaction monitoring (MRM) scan mode, under the following conditions: turbo ion-spray interface temperature, 350°C; collision-activated dissociation gas pressure and curtain gas pressure, 3 and 40 psi respectively; turbo ion-spray voltage, 5000 V; declustering, entrance and focusing potential, 11, 10 and 300 V respectively; collision energy 18 and 41 V for m/z 114 and 72 transitions respectively; collision cell exit potential, 15 V. Acquisition of data was achieved using the Analyst software (version 1.4; Applied Biosystems).
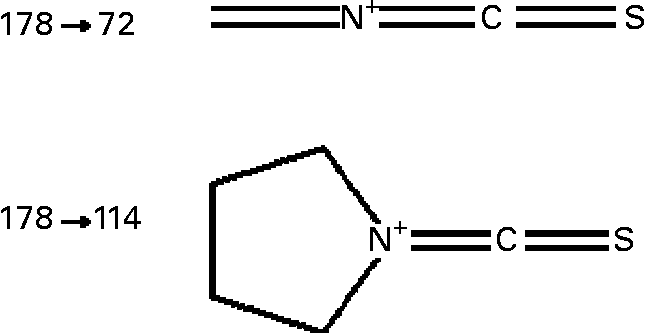
Fig. 1 Structure of sulforaphane transitions monitored in the MRM mode. For details of procedures, see Experimental methods.
Pharmacokinetic analysis
Pharmacokinetic analysis was carried out using PK solutionsTM software package (version 2.0, Summit Research Services, Ohio, USA). Because of the very long terminal phase (t ½ 65·6 h after intravenous administration), pharmacokinetic parameters were calculated from the better defined area under the curve (AUC)0–24, rather than from AUC0-∝, to avoid introducing an inaccuracy in extrapolating the terminal phase to time infinity. AUC0-24 was calculated using the trapezoidal rule utilising the time data determined experimentally. Absolute bioavailability (F) was determined from the ratio of the oral to intravenous dose-normalised AUC0-24 values. Apparent volume of distribution (V d) was calculated from the equation V d = FD/AUC0-24k el, where FD is fraction of dote absorted k el is the elimination rate constant. Plasma clearance (Cl) was determined using the equation Cl = FD/AUC0-24. Finally, C max and T max were determined graphically from the plasma concentration v. time plots.
Animal data were analysed individually and are presented as means and standard deviations; statistical evaluation was performed using the Student's t test, for four animals per group.
Results
A liquid chromatography-MS/MS method for the measurement of sulforaphane in rat plasma has been developed and validated. No sulforaphane was detected in rat plasma prior to administration of the isothiocyanate. Limit of detection, defined as the lowest concentration of analyte that generates a minimum signal:noise ratio of 3, and limit of quantification, defined as the lowest concentration of analyte that gives rise to an instrument response with a minimum signal:noise ratio of 5, were 5 and 15 ng/ml respectively, following a 20 μl injection (Fig. 2(A),(B)). The calibration curve, at a plasma concentration range of 0·01 to 2·0 μg/ml, showed excellent linear relationship, with an R value of 0·99. Recovery of sulforaphane at three plasma concentrations, 0·05, 0·5 and 1·8 μg/ml, was 91, 91 and 88 % respectively (n 6). At the same concentrations inter-assay variation was 1·0, 2·2 and 3·2 % respectively, whereas intra-assay variation, established at the same concentrations, was 1·5, 6·5 and 5 % respectively (n 4). Finally, accuracy was established by comparing calculated and theoretical values at two plasma concentrations (0·5 and 1·8 μg/ml); calculated and theoretical values did not differ more than 11 % (n 4).

Fig. 2 Determination of sulforaphane in rat plasma. (A) Representative chromatogram of sulforaphane in spiked plasma extracts at the limit of detection; (B) representative chromatogram of sulforaphane in spiked plasma extracts at the limit of quantification. The outer peak is the more abundant m/z 178 to 114 transition whereas the inner peak represents that of m/z 114 to 72. CPS, counts per s. For details of procedures, see Experimental methods.
Fig. 3 shows the time-course changes in the plasma concentrations of sulforaphane, plotted using a semi logarithmic plot, following intravenous administration to rats; the plasma profile is best described by a two-compartment pharmacokinetic model. Within 2 h, plasma concentrations decline to about 10 % of the levels present 0·5 h after administration; the concentrations then remain fairly constant, indicating a long terminal phase. Similarly, following oral administration of the same dose, a marked and rapid decline in plasma concentrations of sulforaphane is evident between 1 h after administration, when peak concentrations are achieved, and 2 h after administration, followed by a prolonged terminal phase (Fig. 3). At the higher doses, however, the decline in plasma concentrations, after maxima have been attained, is more gradual.

Fig. 3 Plasma concentration v. time curves in rats following oral and intravenous exposure to sulforaphane. Blood samples were withdrawn 0·25, 0·5, 1, 2, 4, 6, 8 and 24 h after administration. Bars represent standard deviations where n 4. For details of procedures, see Experimental methods.
The pharmacokinetic parameters of sulforaphane following oral and intravenous administration are shown in Table 1. The C max and AUC0-24 values in orally treated rats increased with dose, but not proportionately; the rise in AUC0-24 and C max values was lower than would be anticipated. Comparison of AUC0-24 values between the intravenously and orally treated groups, at the 0·5 mg/kg dose, indicates an absolute bioavailability of 82 %, which, however, decreased at the higher doses. Finally, the rate of absorption constant k ab, biological half-life t ½ and apparent volume of distribution decreased at the highest dose used.
Table 1 Absolute bioavailability and pharmacokinetic parameters of sulforaphane following administration of dietary doses†
(Values are means and standard deviation for four animals)
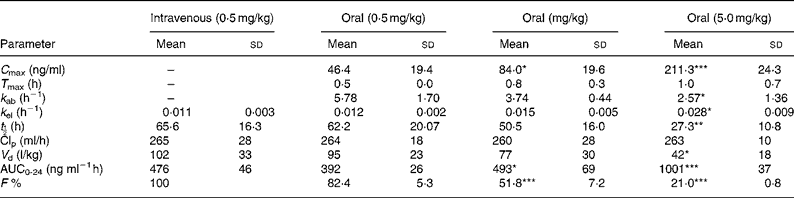
*P < 0·05; **P < 0·01; ***P < 0·001; when compared with the 0·5 mg/kg oral dose.
† For details of procedures, see Experimental methods.
AUC, area under the curve.
Discussion
An analytical method, utilising liquid chromatography-MS/MS, which allows the determination of sulforaphane in small volumes of rat plasma following exposure to low dietary doses, was developed and validated, and employed to determine its absolute bioavailability and pharmacokinetic characteristics in the rat following intravenous and oral administration. A 150 g serving of fresh broccoli will release 56–112 mg sulforaphaneReference Howard, Jeffery, Wallig and Klein19, so that the intake for a 70 kg individual would be 0·8–1·6 mg/kg. The doses employed in the present study of 0·5–5·0 mg/kg were chosen to represent such concentrations of intake.
In the current studies it was considered prudent not to deprive the animals of food prior to administration of sulforaphane, as it could have an impact on its metabolic clearance. The principal pathway of metabolism of this compound involves conjugation with glutathione, followed by further processing of the conjugate to the mercapturateReference Kassahun, Davis, Hu, Martin and Baillie20. Withdrawal of food can result in a decline in the cellular concentrations of glutathioneReference Pessayre, Dolder, Artigou, Wandscheer, Descatoire, Degott and Benhamou21.
Following oral administration, sulforaphane peak plasma concentrations were attained at about 1 h, indicating rapid absorption, compatible with its lipophilicity and small molecular size. Similarly, in studies conducted in human volunteers, isothiocyanates, measured as total dithiocarbamates, were rapidly absorbed, reaching peak plasma concentrations 1 h after ingestion of broccoli sprout preparationsReference Ye, Dinkova-Kostova, Wade, Zhang, Shapiro and Talalay22. In a recent study, where sulforaphane levels were determined in volunteers consuming a broccoli soup, maxima were attained 1 h after intake, in concordance with the present findings in ratsReference Al Janobi, Mitchen, Gasper, Shaw, Middleton, Ortori and Barrett23. At the highest dose only, the absorption rate constant decreased and this may explain in part the fact that C max values did not rise proportionately to the dose. This observation raises the possibility that sulforaphane may to some extent be absorbed by a carrier-mediated transport mechanism that is saturated at this dose level.
Oral absolute bioavailability of sulforaphane in rats was over 80 % at the lowest oral dose studied. It is likely that sulforaphane is subjected to modest first-pass metabolism as glutathione and mercapturate metabolites of this isothiocyanate can be generated by intestinal as well as hepatic enzymes, or even possibly in the blood as it contains low concentrations of glutathione that can interact chemically with the isothiocyanate. Studies using human jejunum in situ have established that sulforaphane is well absorbed by enterocytes where it is conjugated with glutathione during absorption and secreted back into the lumenReference Petri, Tannergen and Holst24.
In the present study, the oral bioavailability of sulforaphane was dose-dependent, being only about 20 % at the highest dose studied of 28 μmol/kg, i.e. a quarter of that observed at a dose of 2·8 μmol/kg. These observations imply that intake of sulforaphane supplements may not be as effective as envisaged in achieving high plasma concentrations of the compound. Isothiocyanates display high protein bindingReference Ji, Kuo and Morris25, Reference Kassie and Knasmuller26, presumably because of their facile interaction with –SH groups, and it is conceivable that at the higher doses protein-binding sites are saturated so that sulforaphane remains free and available for metabolism and excretion. It is relevant to point out that albumin contains only a single residue of free cysteine (Cys34). The observed dose-dependent decrease in biological half-life values concord with such a mechanism of action. Non-linear pharmacokinetics in rats have also been reported for phenethyl isothiocyanate, having an aromatic substituentReference Ji, Kuo and Morris25 and collectively these observations indicate that the isothiocyanate group is more likely to be responsible for this effect rather than the substituent.
Following intravenous and oral dosing, a rapid marked drop is observed in the plasma concentrations of sulforaphane and this most likely reflects cellular uptake. Isothiocyanates, such as sulforaphane, attain very high intracellular concentrations as a result of their interaction with glutathioneReference Zhang and Talalay16, Reference Ye and Zhang27, Reference Zhang and Callaway28. As the absorbed sulforaphane is readily conjugated with glutathione and possibly other thiols, the concentration gradient drives the further cellular uptake of the isothiocyanate, which can achieve mm concentrations, and is accompanied by a marked drop in glutathione levelsReference Conaway, Wang, Pittman, Yang, Schwarz, Tian, McIntee, Hecht and Chung17. The glutathione and cysteinylglycine conjugates are exported through membrane transporters such as P-glycoproteinReference Zhang29, Reference Callaway, Zhang, Chew and Sherry Chow30. It has been demonstrated in in vitro studies that peak intracellular concentrations of isothiocyanates are attained within 3 h of exposure and intracellular concentration may be as much as 200-fold higher than extracellular concentrationReference Zhang3. Such extensive intracellular localisation would explain the large apparent volume of distribution. Elimination of sulforaphane was characterised by a long terminal phase; in fact, no major difference was evident in plasma concentrations between 6 and 24 h following intravenous administration or oral administration at the lower doses. Most likely, this is a consequence of protein binding, rendering the isothiocyanate unavailable for elimination through metabolism and excretion.
In summary, the present paper demonstrates that in the rat, following oral administration of dietary doses, sulforaphane is rapidly absorbed, achieving high absolute bioavailability at low doses. However, dose-dependent pharmacokinetics was evident, with bioavailability decreasing with increasing dosage.
Acknowledgements
The authors acknowledge with gratitude the financial support of the Association for International Cancer Research.




