Sulphur amino acids act as nutrient signals controlling oxidative status through their metabolism and synthesis of glutathione and taurine acting as intracellular antioxidants(Reference Schuller-Levis and Park1–Reference Oudit, Trivieni and Khaper4). Inflammation and oxidation are important factors that regulate pro-inflammatory signalling pathways(Reference Pearson, Robinson and Gibson5). The literature supports the notion that dietary antioxidants are useful radioprotectors and play important roles in preventing many diseases such as cancer, atherosclerosis, stroke, neurodegeneration and diabetes in different cell models(Reference Zulli, Lau and Wijaya6, Reference Verzola, Bertolotto and Villaggio7–Reference Sun, Gu and Xu12). The β-amino acid taurine is present in millimolar concentrations in most animal tissues(Reference Schuller-Levis and Park1, Reference Bouckenooghe and Reusens13) including Atlantic salmon (Salmo salar)(Reference Espe, Hevrøy and Liaset14, Reference Espe, Rathore and Du15). Taurine may protect against oxidative stress, neurodegenerative diseases and atherosclerosis, and plays important roles in several physiological processes such as osmoregulation, immunomodulation and bile acid formation(Reference Della Corte, Crichton and Duburs16, Reference Militante and Lomardini17). Atlantic salmon produce taurine from sulphur amino acids through trans-sulfuration(Reference Espe, Hevrøy and Liaset14) and, in addition, taurine is delivered through dietary fishmeal. As marine ingredients are exchanged with plant ingredients, not containing any taurine, a deficiency of taurine may arise using such diets. Deficiencies in taurine may result in increased mitochondrial oxidation and may possibly be linked to the increased lipid accretion reported when dietary taurine is low in rodents(Reference Tsuboyama-Kasaoka, Shozawa and Sano18, Reference Liaset, Madsen and Qin19) and in Atlantic salmon(Reference Espe, Ruohonen and El-Mowafi20, Reference Espe, Ruohonen and El-Mowafi21).
Severe taurine limitation, as observed in taurine transporter knockout mice, may trigger liver disease and apoptosis(Reference Warskulat, Borsch and Reinehr22, Reference Warskulat, Borsch and Reinehr23). Apoptosis is the cellular process in which the cell dies in a controlled fashion either spontaneously or in response to environmental toxicants(Reference Sinha, Manna and Sil24, Reference Das, Ghosh and Manna25). Mitochondria are the cell's sensor to oxidative stress by losing membrane potential, releasing cytochrome c and other pro-apoptotic factors into the cytosol. Mitochondrial permeability is regulated by the Bcl-2 family of proteins. Bcl-2 family proteins regulate apoptosis by controlling mitochondrial permeability and cytochrome c release. Bcl-2 and Bcl-xL inhibit cytochrome c release, thus being anti-apoptotic, while other Bcl-2 family protein members such as Bad, Bid, Bax and Bim are located in the cytosol but may translocate to the mitochondria following death signals and thus promote cytochrome c release(Reference Pradelli, Bénéteau and Ricci26). The mitogen-activating phosphokinase (MAPK) signalling pathway is highly conserved through evolution and transduces a variety of extracellular stress signals such as temperature, irradiation, osmotic shock, cytokines, hormones and inflammation(Reference Brown and Sacks27). When cells become apoptotic, the abundance of phosphorylated MAPK such as p63, p42/44 and p38 has been reported to increase in cells isolated from rodents(Reference Das, Ghosh and Manna25, Reference Xu, Saini and Zhang28–Reference Hou, Shen and Li30). Cytochrome P450 (P450) enzymes are the most important phase I biotransformation enzymes of environmental chemicals(Reference Fent31, Reference Van der Oost, Beyer and Vermaulen32). These enzymes are involved in the detoxification of liver cells in fish(Reference Søfteland, Eide and Olsvik33). As with the abundance of phosphorylated MAPK, P450 also has been found to increase in apoptotic cells(Reference Sinha, Manna and Sil24, Reference Das, Ghosh and Manna29).
It is known that Cd intoxication increases the expression of stress genes(Reference Wang and Templeton34), and modulates antioxidant enzymes(Reference Jay, Zamorano and Munoz35, Reference Watjen, Benters and Haase36). Prophylactic treatments with taurine in rodent models reduce lipid peroxidation following treatments with pro-oxidants such as meththiocarb(Reference Ozden, Catalgol and Gezginci-Oktayoglu37), Cd(Reference Sinha, Manna and Sil24) and Fe overload(Reference Devi and Anuradha38). Hepatotoxicants have been reported to reduce S-adenosylmethionine (SAM) and affect the trans-sulfuration pathway(Reference Schnackenberg, Chen and Sun39). Transmethylation of homocysteine to methionine by the enzyme betaine-homocysteine methyltransferase requires methyl groups from betaine(Reference Li, Mai and Trushenski40). Animals have the capacity of endogenous synthesis of both choline and betaine when intake of these metabolites is low. Choline is synthesised from ethanolamine through three successive transmethylation reactions consuming SAM(Reference Vance and Vance41–Reference Brosnan, Jacobs and Stead43), and choline can be metabolised to betaine within the mitochondrion(Reference Obeid and Herrmann44). Glutathione is an important detoxifying molecule for reactive oxygen species and other reactive metabolites acting as a free radical scavenger, and SAM is the primary precursor for glutathione; thus, the depletion of cellular SAM has been associated with liver toxicity(Reference Sun, Schnackenberg and Holland45). As the endogenous synthesis of taurine might be too low to protect the cells against oxidative stress and apoptosis, the present study aimed to assess whether taurine supplementation could improve cell viability and methylation capacity (i.e. the SAM:S-adenosylhomocysteine (SAH) ratio) in primary liver cells isolated from Atlantic salmon. To maximise any protective effects of taurine administration(Reference Sinha, Manna and Sil24), cells were also stressed with CdCl2 for 24 h and compared with unstressed cells.
Materials and methods
Isolation of liver cells
Liver cells were isolated from four Atlantic salmon (S. salar) with an average body weight of 1443 (se 71) g, two males and two females. The fish were anaesthetised by metocaine (MS222, 5–8 g/l) and the liver was perfused with a 0·09 m-HEPES buffer containing 1·4 m-NaCl, 0·067 m-KCl and 0·03 m-EDTA, pH 7·4, at a flow of 4 ml/min until free of blood. Thereafter, the liver was digested with collagenase (0·1 % collagenase type IV was dissolved in the 0·9 m-HEPES buffer as used for perfusion). The isolated cells were harvested in 10 ml of 10 % PBS buffer (0·002 m-KH2PO4, 0·02 m-Na2HPO4, 0·03 m-KCl and 0·14 m-NaCl, pH 7·4), filtered (100 μm filter) and washed twice in the PBS buffer, resuspended in the respective test media before the viability of the isolated cells was assessed. All centrifugations were done by 50 g for 5 min. The viability of the cells was examined with the Trypan Blue exclusion test in accordance with the protocol supplied by the manufacturer (Lonzo; Medprobe). The viability of the isolated primary liver cells was above 90 (range 90·8–94·4) %. The isolations of the cells were done with sterile equipment and buffers. The experimental protocol was approved by the Norwegian Board of Experiments with Living Animals.
In vitro study
Cells were grown in Leibovitz's-15 medium (catalogue no. L1518; Sigma-Aldrich) supplemented with 1 % of 2 mm-glutamax 100 × (Gibco Life Technologies), 1 % antibiotics (penicillin, 10 000 U/ml, streptomycin, 10 000 μg/ml and amphotericin B, 25 μg/ml; Lonzo Medprobe) and 10 % fetal calf serum (Sigma-Aldrich) to which taurine was added or not added (Sigma-Aldrich). The mean analysed taurine concentration in Leibovitz's-15 medium not supplemented with taurine was 14 (se 5) μm (n 4), while the corresponding taurine-supplemented media contained 25 (se 0·5) mm-taurine close to the physiological concentration in the liver of Atlantic salmon fed adequate methionine and cyst(e)ine (26 mm) calculated from Espe et al. (Reference Espe, Hevrøy and Liaset14). Cells were grown for 3 d on laminin-coated (1·8 μg/cm2; Sigma-Aldrich) flasks and cover slides before the addition of 100 μm-CdCl2 (prepared in sterile water as described elsewhere(Reference Søfteland, Holen and Olsvik46)) to half of the cells and cells were allowed to grow for 24 h until harvested at day 4. Cells were grown at a density of 0·5 mill cells/cm2 and a temperature of 9 ± 0·5°C in the dark in a normal atmosphere incubator (Sanyo Incubator model MIR-253). The cell study design is illustrated in Fig. 1.

Fig. 1 Schematic illustrating the design of the cell study conducted. Primary liver cells isolated from Atlantic salmon (Salmo salar) were grown in the respective media containing 14 μm- or 25 mm-taurine for 3 d before supplemented or unsupplemented with 100 μm-CdCl2. Following CdCl2 supplementation, cells were grown for 24 h before being harvested for imaging and analyses. Each cell study was repeated with cells isolated from four fish and at each trial, the four treatments were assessed.
Chemical analyses
Amino acid composition in deproteinised cells was determined on the Biochrom 20 plus Amino Acid Analyzer (Amersham Pharmacia Biotech) equipped with a lithium column using post-column derivatisation with ninhydrin as described previously(Reference Espe, Lemme and Petri47). Standards for both amino acids and N-metabolites were from Sigma. SAM and SAH were analysed as described in Wang et al. (Reference Wang, Kramer and Yang48). Total homocysteine was determined as described previously(Reference Espe, Hevrøy and Liaset14).
Cells were harvested and washed three times in 10 ml PBS buffer. Thereafter, cells were lysed in 1 ml lysis buffer (50 mm-Tris–HCl, pH 8·0, 150 mm-NaCl and 1 % NP-40 containing protease inhibitor cocktail) on ice for 30 min. The lysed cells were spun at 10 000 g for 15 min and the supernatant was aliquoted and stored at − 80°C until analysed. Betaine and choline were analysed in the lysed supernatants by LC MS/MS (Bevital, www.bevital.no).
Assessment of cell viability
Viability of the cells was estimated by the In vitro Toxicology assay kit, MTT (3-(4,5-dimethylthizol-2yl)-2,5-diphenyltetrazolium bromide) following the instructions from the supplier using a dual wavelength (570 and 690 nm, catalogue no. M5655, M8910; Sigma-Aldrich). The assay is based on the fact that the yellow-coloured MTT will be cleaved by mitochondrial dehydrogenases present in viable cells yielding a purple colour, while dead cells will remain yellow. Thus, the higher the absorbance, the less is the viability. Change in absorbance was calculated relative to the absorbance of dead cells, which was set equal to 100.
In addition, viable or dead cells were assessed by the Viability/Cytotoxicity Assay Kit for Animal Live and Dead Cells (catalogue no. 30 002; Biotum, Inc.). The EthD-III component binds to nucleic acids and produces a red fluorescence in dead cells. EthD-III is excluded by intact membranes and live cells are stained green by calcein AM. The assay followed the procedure given by the supplier. Cells were investigated for fluorescence using a Confocal Microscope (Leica TCS SP2 AOBS; Molecular Imagine Center, University of Bergen). Results are given as calcein AM-positive cells as a percentage of total cells present on each image.
Transmission electron microscopy
Cell cultures were prepared for transmission electron microscopy (Jeol JEM-1230; Molecular Imaging Center, University of Bergen). Electron microscopy was used to evaluate any structural changes in the primary liver cells following the treatments.
DNA fragmentation
Monolayer cells (without the supernatants) were lysed in liquid N2 before purified for total DNA using the DNeasy® Blood & Tissue Kit (catalogue no. 69 504), following the supplier's instructions (Qiagen), and frozen at − 80°C until analysed. Samples were dissolved in loading buffer (1:6, v/v, 0·25 % bromophenol blue, 0·25 % xylene cyanol and 15 % Ficoll in distilled water). DNA fragmentation was analysed on 1 % agarose gels, stained with GelRed Nucleic Acid Stain (Biotium) and run with 1 × Tris–acetate–EDTA buffer at 70 V for 1 h. Molecular weight ladder (2log DNA ladder 0·1-10 kb, catalogue no. NEBFN3200; BioLab, Inc.) was used. DNA fragmentation was visualised using a Chemi Chemiluminescence Image Capture (Syngene).
Western blot
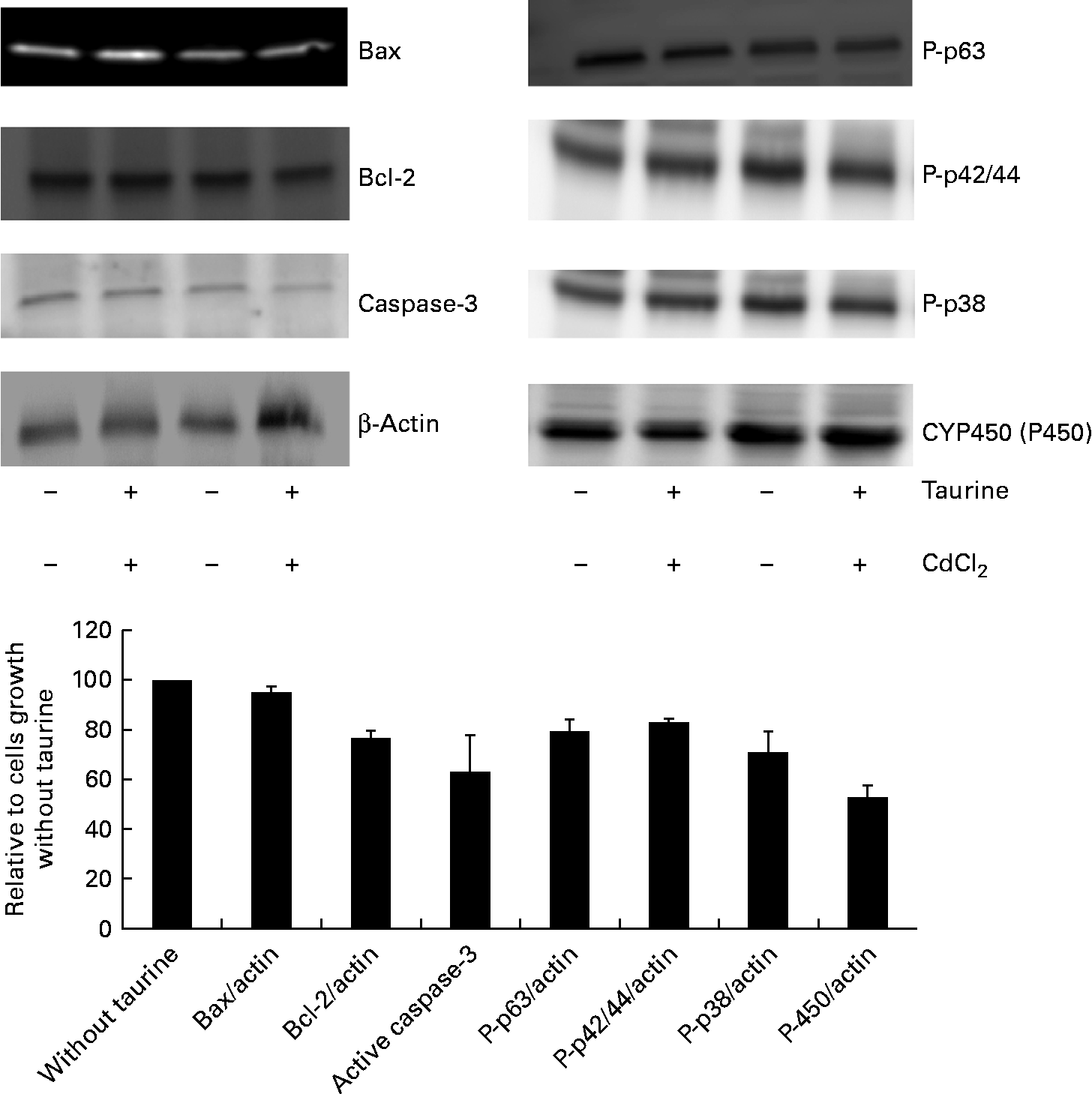
Cells (22·5 million) were harvested and washed three times in 10 ml PBS buffer. Thereafter, cells were lysed in 1 ml lysis buffer (50 mm-Tris–HCl, pH 8·0, 150 mm-NaCl and 1 % NP-40 containing a protease inhibitor cocktail tablet, 11697498001; Roche) on ice for 30 min. The lysed cells were centrifuged at 10 000 g for 15 min and the supernatants were stored at − 80°C until analysed. Samples were prepared for SDS gel by mixing equal amounts of sample and Laemmli sample buffer (catalogue no. 161-0710; BioRad) before heating the samples for 5 min at 95°C. Samples and molecular weight marker (Precision Plus Protein™ WesternC™ Standards, catalogue no. 161-0376; BioRad) were loaded into precast 10 % SDS gels (catalogue no. 456-1033; BioRad) using a BioRad Mini Protean® Cell according to the manufacturer's instructions. Proteins were blotted onto a polyvinylidene difluouride membrane (catalogue no. 162-0176; BioRad) for 1 h at 100 V, using the BioRad Criterion™ Blotter System. The membranes were washed five times with 20 ml TPBS buffer (PBS/0·1 % Tween-20), shaking, before blocking for 1 h with blocking buffer (catalogue no. RPN2135; GE Healthcare) or 5 % bovine serum albumin/TPBS, shaking. Primary antibodies were added directly into the blocking solution (1:1000) and incubated overnight at 4°C, shaking. The following primary rabbit antibodies from Cell Signaling (MedProbe) were as follows: β-actin (no. 4967), P-p63 (S160/162, no. 4981), P-p42/44 MAPK (T202/Y204, no. 4370), Bax (no. 2772), Bcl-2 (50E3, no. 2870), P-p38 (Thr180/Tyr182, no. 9215), while active caspase-3 (ab13847) and P450 (A1+A2 cytochrome, no. ab37131) were purchased from AbCam. The membranes were washed as described above. Then, horseradish peroxidase-linked secondary anti-rabbit IgG (1:500, no. 7074; Cell Signaling) and Precision Protein™ StrepTactin-HRP conjugate (catalogue no. 161-0380; BioRad) were added to freshly made blocking buffer (catalogue no. RPN2135; GE Healthcare) and incubated for 2 h, shaking, before washing (six times, 20 ml) with PBS buffer. The Amersham ECL-Advance™ Western Blotting detection kit (GE Healthcare, catalogue no. RPN2135) and the Chemi Chemiluminescence Image capture (Syngene) were used to detect the proteins.
Statistical analyses
Values are reported as means with their standard errors, based on the liver cells isolated from four individual fish (n 4). Results of taurine supplementation were calculated relative to unsupplemented taurine cells and assessed by the Mann–Whitney U test. P values less than 0·05 were considered as statistically different. All statistical analyses were performed using the Statistica program (version 9; Statsoft, Inc.).
Results
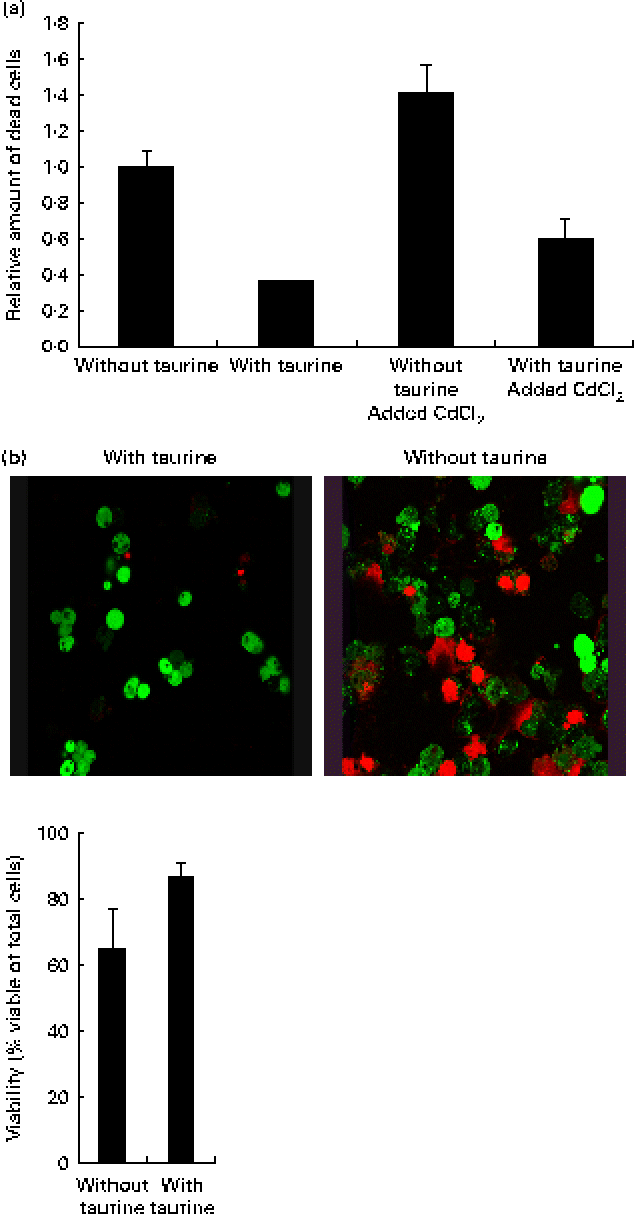
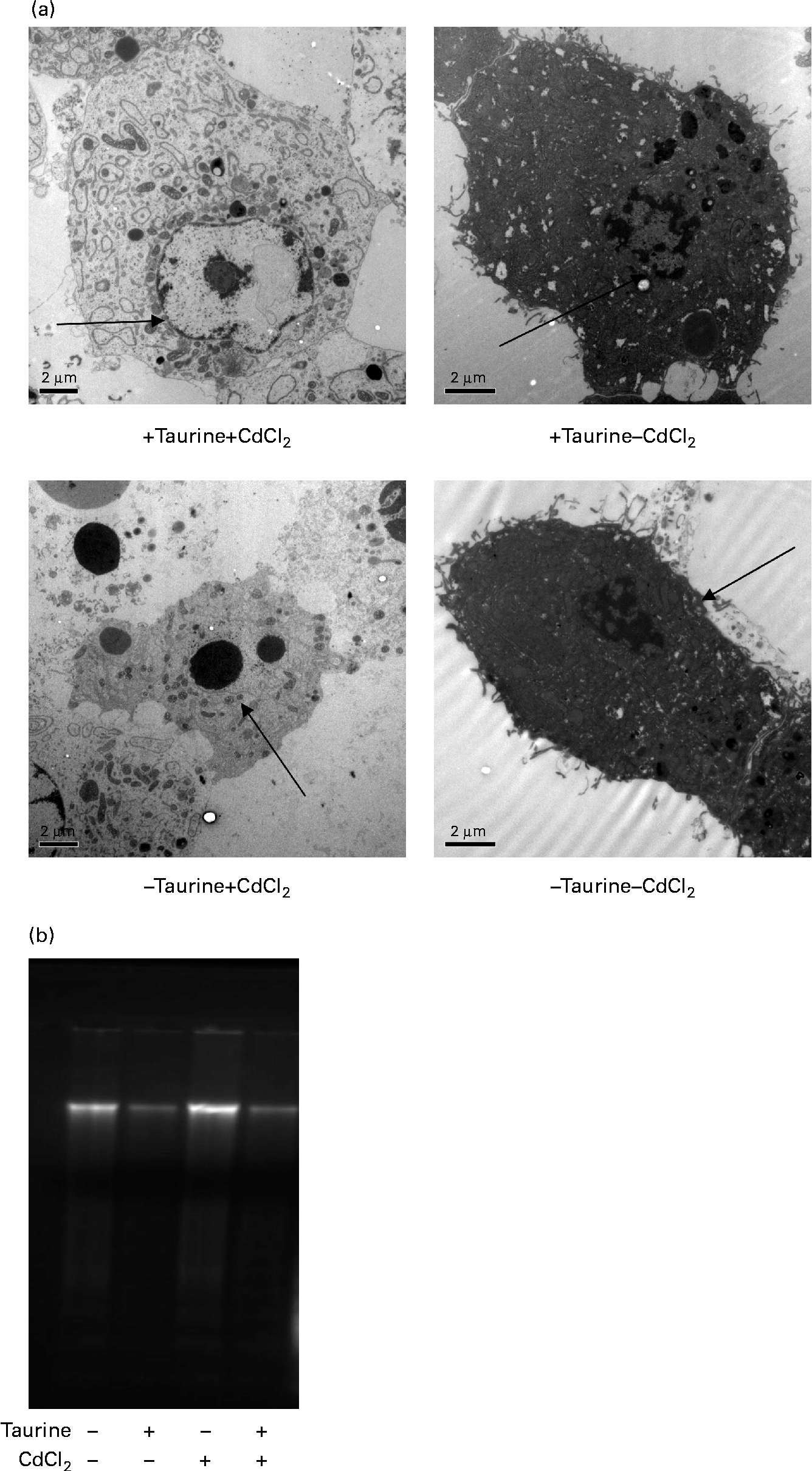
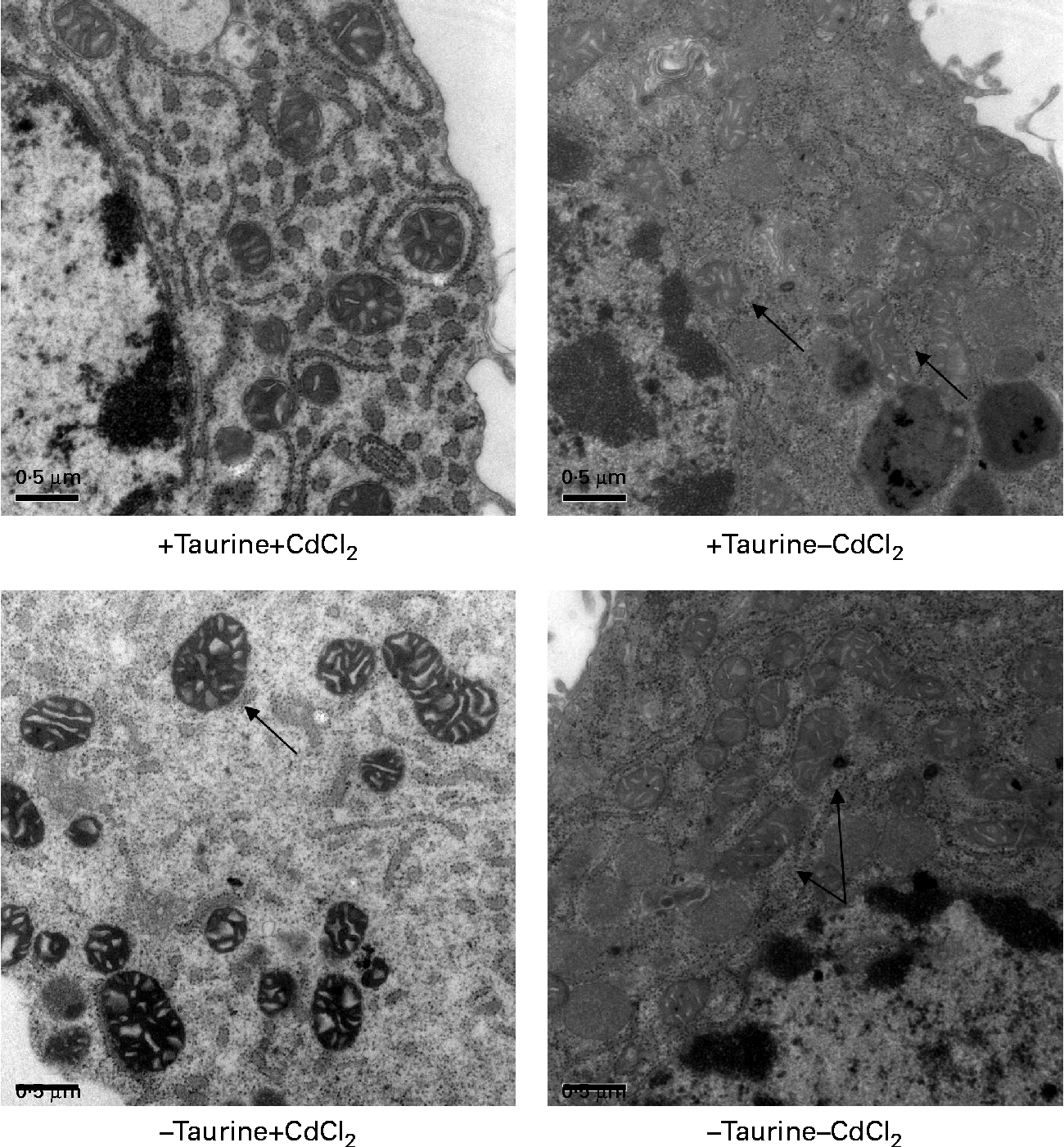
Supplementation of taurine to the media improved the viability of the cells (Fig. 2). The addition of CdCl2 strengthened this effect of taurine supplementation on cell survival without showing any interaction between CdCl2 and taurine. Cells grown in media without taurine supplementation had a more condensed nucleus, while taurine supplementation seemed to counteract this even in the cells stressed with CdCl2 (Fig. 3(a)). Further, the cells with the more condensed nuclei had more smeared DNA when compared with cells grown in taurine-supplemented media (Fig. 3(b)). As expected, the mitochondrial membranes in cells grown in media supplemented with CdCl2 were more damaged when compared with cells grown in media unsupplemented with CdCl2. However, taurine supplementation by itself did not seem to affect the mitochondrial membranes (Fig. 4).

Fig. 2 (a) Taurine supplementation improved viability (P= 0·03), while the addition of CdCl2 reduced the viability (P= 0·001) and no interaction between taurine and cadmium occurred (P= 0·47). Values are means of four repeated treatments, with their standard errors represented by vertical bars. (b) Representative images of cells grown in media supplemented or unsupplemented with taurine stained for viable cells. The red-coloured cells are dead while the green-coloured cells are alive. Cells grown in media supplemented with taurine had better viability (P= 0·052; Mann–Whitney U test).
Fig. 3 Representative transmission electron microscopy images of liver cells isolated from Atlantic salmon (Salmo salar) (n 4) and grown in media supplemented or unsupplemented with taurine and CdCl2. Cells grown in media without taurine supplementation had a more condensed chromatin in the nucleus ( → ) when compared with cells grown in media supplemented with taurine. (a) Addition of CdCl2 to the media increased the abundance of apoptotic cells. When cells were grown without taurine supplementation, a higher DNA smearing occurred. (b) Representative image showing the DNA smearing occurring in cells grown in the four different media. Scale bar = 2·0 μm.
Fig. 4 Representative transmission electron microscopy images of the mitochondrion in liver cells isolated from Atlantic salmon (Salmo salar) (n 4) and grown in media supplemented or unsupplemented with taurine and CdCl2. Cells supplemented with CdCl2 had more swollen mitochondrial membranes ( → ) in line with the lower viability of these cells. Taurine did not seem to affect this swelling ( → ). Scale bar = 0·5 μm.
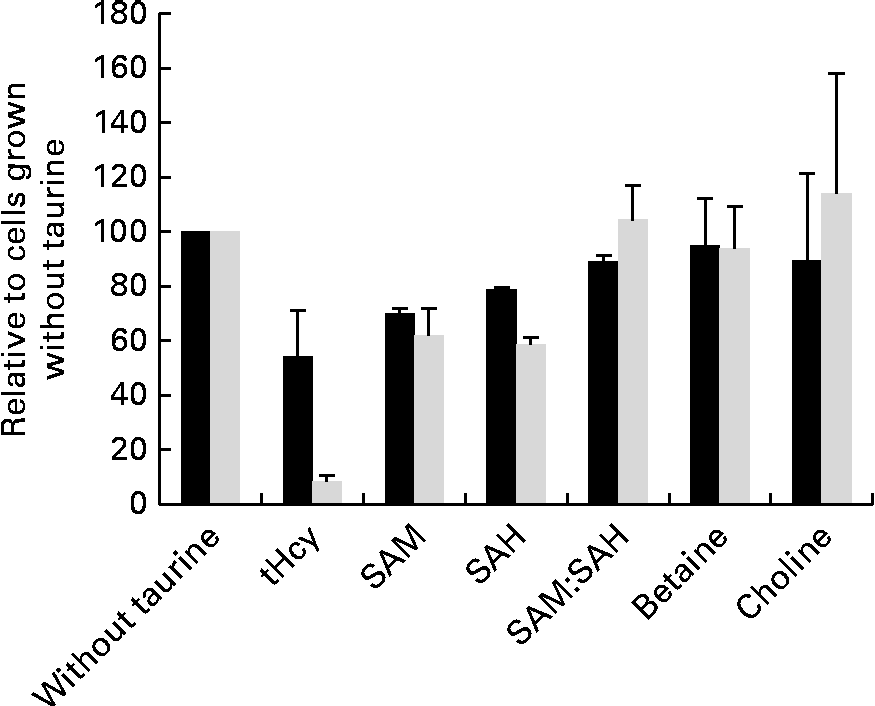
Taurine supplementation increased the intracellular taurine concentration approximately ten times (from 1093 to 10 982 μm per million cells), but none of the other free amino acids was affected by taurine supplementation (data not shown). SAM, SAH and total homocysteine were less in cells grown in media supplemented with taurine, but the capacity of methylation analysed as the SAM:SAH ratio was unaffected by taurine supplementation (Fig. 5). Neither choline nor betaine was affected by taurine supplementation (Fig. 5).
Fig. 5 Metabolites in sulphur amino acid metabolism analysed in free monolayer liver cells isolated from Atlantic salmon (Salmo salar) and grown for 4 d in media supplemented or unsupplemented with taurine and CdCl2 (n 4). Values are relative means, as cells grown without taurine supplementation are set equal to 100, with their standard errors represented by vertical bars. S-adenosylmethionine (SAM), S-adenosyl homocysteine (SAH) and total homocysteine (tHcy) all reduced (P= 0·03, 0·03 and 0·1, respectively, without CdCl2 and P= 0·02. 0·02 and 0·03 with CdCl2 supplementation; Mann–Whitney U test) by taurine supplementation. Capacity of methylation (SAM:SAH ratio) and methyl donor concentration (choline and betaine) was unaffected by the treatment (P>0·05). ■, Without CdCl2; ![]() , with CdCl2.
, with CdCl2.
As CdCl2 increased the effects of taurine supplementation without showing any interaction with taurine on either the viability or the concentration of metabolites in sulphur amino acid metabolism, the abundance of proteins associated with apoptosis focused on taurine supplementation only (i.e. without taurine was set equal to 100 when independent of CdCl2 administration). The abundance of active cleaved caspase-3 (P= 0·08) was reduced when cells were grown in media supplemented with taurine, while Bax (P= 0·39) and Bcl-2 (P= 1·0) were unaffected by the treatment (Fig. 6). Furthermore, taurine supplementation decreased the protein abundance of the phosphorylated MAPK (P-p63, P-p42/44, P-p38, P= 0·02, P= 0·001 and P= 0·063, respectively). Additionally, taurine supplementation reduced the abundance of P450 (P= 0·02; Fig. 6).
Fig. 6 Abundance of proteins associated with apoptosis as affected by taurine supplementation. Cytochrome P450 (P= 0·02) and the phosphorylated mitogen-activating phosphokinase decreased following taurine supplementation (P-p63 and P-p42/44, P= 0·02 and P= 0·001, respectively; Mann–Whitney U test). Also, active caspase-3 abundance and P-p38 decreased when taurine was supplemented, but did not reach significant difference (P= 0·08 and P= 0·06, respectively). Bax (P= 0·39) and Bcl-2 (P= 1·0) abundance was not affected by taurine supplementation. The proteins present were calculated relative to actin present in each cell lysate, and presented as relative means, as cells grown in media without taurine supplementation is set equal to 100, with their standard errors represented by vertical bars. Representative images from the Western blot of liver cells isolated from four fish (n 4).
Discussion
Here, we report that taurine supplementation improves the viability of primary liver cells and reduces the abundance of some proteins associated with apoptosis (caspase-3, MAPK and P450), while others were unaffected by the treatment (Bax and Bcl-2). Risso-de Faverney et al. (Reference Risso-de Faverney, Orsini and de Sousa49) analysed Bcl-2 family proteins and caspase activation following Cd-induced apoptosis in rainbow trout. Cd increased the abundance of caspase-3, 9 and 8, and increased cytosolic cytochrome c and decreased mitochondrial cytochrome c. Similarly, Bax increased in the mitochondria and decreased in the cytosol. In accordance with the results reported by Risso-de Faverney et al. (Reference Risso-de Faverney, Orsini and de Sousa49), the present study found that supplementation of CdCl2 reduced viability. The lack of significant changes in Bax and Bcl-2 in the present experiment may be due to the fact that whole cells were analysed and not fractionised into mitochondrial and cytosolic fractions.
Cells grown in the media without taurine supplementation tended to have a higher abundance of active caspase-3 and showed more DNA fragmentation, indicative of increased apoptosis when compared with cells grown in media supplemented with taurine. Recently, Ghosh et al. (Reference Ghosh, Sulistyoningrum and Glier50) reported that active caspase-3 increased in the heart of transgenic mice with reduced cystathionine β-synthase activity, the rate-limiting enzyme for trans-sulfuration(Reference Mato, Corrales and Lu51), especially when fed high-fat diets. Also, these mice had less total glutathione and oxidised glutathione in the heart, but no differences in liver glutathione status were observed. We did not analyse glutathione status in the isolated liver cells, but neither catalase nor glutathione peroxidase gene expression was affected (data not shown) by the treatment in the present study. Reduced cystathionine β-synthase activity not only decreases taurine and glutathione synthesis but may also increase homocysteine(Reference Mato, Corrales and Lu51, Reference Finkelstein52), especially so if betaine is limiting. In the present study, neither betaine nor choline was affected by taurine supplementation, but SAM, SAH and homocysteine all reduced following taurine supplementation. Zulli et al. (Reference Zulli, Lau and Wijaya6) reported that elevated homocysteine present in rabbits fed an atherogenic diet (high cholesterol and methionine) normalised after taurine administration. However, taurine supplementation failed to improve hyperlipidaemia or the oxidative stress system(Reference Zulli, Lau and Wijaya6) in rabbits. Thus, the elevated total homocysteine present in cells grown in media without taurine supplementation also may have contributed to apoptosis in the present study.
Previous studies have shown that phosphorylated MAPK such as P-p63, P-p42/44 and P-p38 increase during apoptosis(Reference Das, Ghosh and Manna25, Reference Xu, Saini and Zhang28–Reference Hou, Shen and Li30, Reference Meng, Pei and Zhang53). P450 has also been reported to increase in apoptotic cells(Reference Sinha, Manna and Sil24, Reference Das, Ghosh and Manna29). The data from the present study thus support increased apoptosis in cells grown in media without taurine supplementation. P-p38 may activate the PPARα pathway(Reference Tsuboyama-Kasaoka, Shozawa and Sano18, Reference Hou, Shen and Li30, Reference Meng, Pei and Zhang53), thus affecting lipid mobilisation. As cells grown in media supplemented with taurine had less P-p38, this suggests the involvement of MAPK signalling pathways for the reduced fat accretion reported when Atlantic salmon were fed diets supplemented with taurine(Reference Espe, Ruohonen and El-Mowafi20, Reference Espe, Ruohonen and El-Mowafi21). Unfortunately, this could not be answered in the present study as the gene expression of PPARα was unaffected by the treatment (results not shown). However, cytokine activation and lipid mobilisation deserve to be studied in more detail both in vitro and in vivo to unravel cell signalling linking fuel sensing, mobilisation of lipids and MAPK signalling.
Taurine also functions as an osmolyte; therefore supplementation of taurine to the media may have reduced cell shrinkage that acts as a signal for programmed cell death(Reference Militante and Lomardini17, Reference Li, Mai and Trushenski40, Reference Yu, Chen and Wei54). This may also add to the increased apoptosis observed in the present study.
Several investigators have reported that oxidation is a major determinant for apoptosis and cell dysfunction(Reference Sinha, Manna and Sil24, Reference Hou, Shen and Li30, Reference Sun, Schnackenberg and Holland45, Reference Giris, Depooylu and Dogru-Abbasoglu55). Even though taurine administration has been reported to prevent apoptosis in experimental inflammatory cell models, taurine treatment alone did not affect either Bax or Bcl-2 expression, oxidative stress or apoptosis in normal rats(Reference Giris, Depooylu and Dogru-Abbasoglu55). On the contrary, we here report that taurine administration by itself significantly improved cell viability and reduced the phosphorylated MAPK. As both too little(Reference Ronchi, Giudici and Mendieta10) and too much(Reference Gomez, Caro and Sanchez56) of the sulphur amino acids have been reported to affect the amount of reactive oxygen species and apoptosis, further studies are required to assess the minimum requirement of taurine supplementation to attenuate apoptosis including possible interactions with lipid mobilisation. In doing so, the primary liver cells should be fractionised to enable study of the localisation of apoptotic proteins(Reference Risso-de Faverney, Orsini and de Sousa49).
Conclusion
Taurine may be conditionally indispensable in Atlantic salmon, as it attenuates apoptosis and protects nuclear DNA in primary liver cells isolated from Atlantic salmon. Neither the capacity of methylation nor betaine or choline availability was affected by taurine treatment. More research is needed to determine the minimum concentration of taurine necessary to attenuate apoptosis including cell localisation and mobility of anti-apoptotic and pro-apoptotic proteins.
Acknowledgements
This study was financed by the Norwegian Ministry of Fisheries and Coastal Affairs. Assistance in the isolation of primary cells by Synnøve Wintertun at the NIFES is highly appreciated. The confocal imaging/electron microscopy was performed at the Molecular Imaging Center (Fuge, Norwegian Research Council), University of Bergen. Both authors contributed equally to the planning and analysis of the cell trials. They participated in the interpretation of the results as well as in the writing of the manuscript. There is no conflict of interest.