


Cancer-associated malnutrition is diagnosed by the presence of involuntary weight loss, of which the major constituents are skeletal muscle and adipose tissue. Cancer-associated malnutrition leads to poor outcomes: progressive functional impairment, treatment-related complications, reduced quality of life, increased inpatient care, hospitalisation costs and length of stay, burdening the healthcare system. Cancer-associated malnutrition, like other forms of disease-associated malnutrition, differs from a deficiency of nutrients in the absence of underlying disease. Cancer-associated malnutrition is partially, but not completely reversible by conventional nutrition therapy. Underlying metabolic changes in the patients with cancer caused by the tumour or by the cancer therapy alter the ability to utilise nutrients, including inflammation, excess catabolism, futile cycling and anabolic resistance. In this review, we discuss the specific nature of these impairments at the tissue level and their potential for mitigation through nutrition therapy.
Skeletal muscle wasting in cancer-associated malnutrition
Normal and cancer-associated regulation of muscle anabolism/catabolism
In large part, the detrimental outcomes associated with cancer-associated malnutrition are thought to be driven by the depletion of skeletal muscle. It is therefore relevant to appreciate the mechanisms underlying skeletal muscle growth, maintenance or atrophy. A network of signalling pathways serves to control and coordinate muscle protein balance (Fig. 1). This network includes an anabolic arm, reliant on growth factors and nutrient signalling via a pathway involving phosphatidylinositol-3 kinase, serine/threonine protein kinase and the mammalian target of rapamycin (mTOR), which leads to muscle protein synthesis. The catabolic arm is characterised by multiple signalling cascades, connected ultimately to transcriptional control of genes involved in autophagy and to ubiquitin-mediated proteasomal degradation of myofibrils, which leads to poor outcomes to patients with cancer.
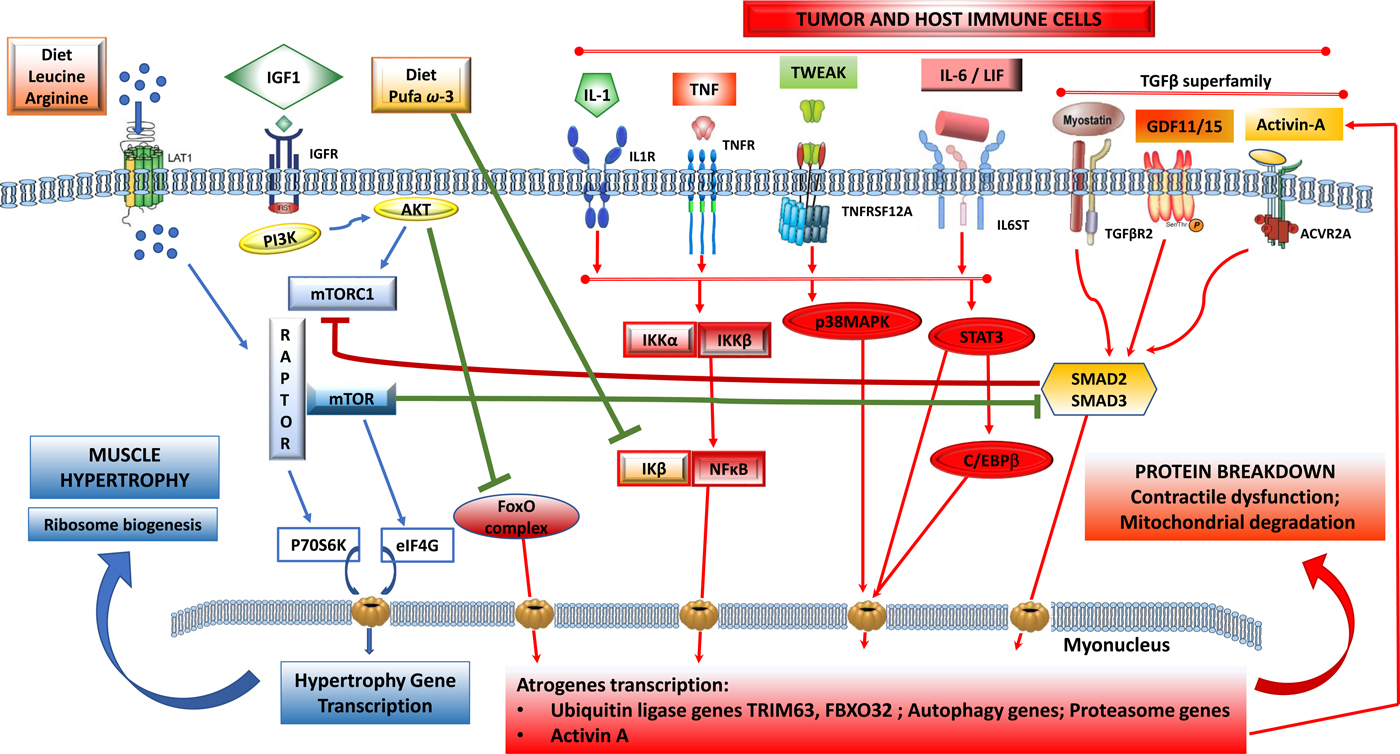
Fig. 1. (Colour online) Tumour-induced signalling pathways involved in the control of skeletal muscle atrophy and hypertrophy: protein synthesis is activated by IGF1 through its receptor IGFR and by downstream elements IRS1/PI3K/AKT/mTOR). TORC1 is a multicomplex protein containing a regulatory protein RAPTOR and mTOR that controls protein synthesis. Phosphorylated by AKT, dissociates RAPTOR and mTOR, and induce translation of p70S6K and eIF4G proteins that induce nuclear transcription of hypertrophy genes. Phosphorylated AKT also blunts catabolic signalling via inhibition of forkhead box O (FoxO) and its downstream signalling to transcription of atrophy genes. Protein breakdown is regulated by signalling pathways that are induced by tumour cells and activated host immune cells: cytokines IL-1, TNF-α, TWEAK acting via downstream signalling that induces a dissociation of NF-κB/IκB complex and NF-κB translocation to the nucleus. IL-6 and LIF induce STAT3 and C/EBPβ signalling pathways and TGF-β superfamily members (myostatin, activin-A, GDF11, GDF15) activate SMAD2/SMAD3 downstream. All these pathways induce transcription of ubiquitin, proteasome and autophagy genes involved in myofibrillar protein breakdown, mitochondrial degradation and contractile dysfunction. Nutrients, including amino acids (leu, arg) induce dissociation of TORC1 and activate mTOR pathway and muscle synthesis. n-3 PUFA (EPA, DHA) suppress the dissociation of NF-κB/IκB and decrease the translation of atrogenes in the nucleus induced by NF-κB. ACVR2A, activin receptor type 2A; AKT, serine/threonine protein kinase; C/EBPβ, CCAAT/enhancer-binding protein-β; FoxO, forkhead box protein O complex; GDF, growth differentiation factor; IκB, inhibitory subunit of NF-κB; IGF1, insulin-like growth factor-1; IGFR, IGF receptor; IKK, IκB kinase; IL-1R, IL 1 receptor; IL-6ST, IL-6 receptor subunit β; IRS1, insulin receptor substrate 1; FXBO32, F-box protein 32; LAT1, L-type amino acid transporter 1; LIF, leukaemia inhibitor factor; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol-3 kinase; RAPTOR, regulatory-associated protein of mTOR; STAT3, signal transducer and activator of transcription 3; TGFBR2, TGF-β receptor type 2; TNFR, TNF receptor; TNFRSF12A, TNF receptor superfamily member 12A; TORC1, target of rapamycin complex 1; TRIM63, tripartite motif containing 63; TWEAK, TNF-related weak inducer of apoptosis.
Muscle anabolic signalling
Insulin, insulin-like growth factor and growth factor receptor-mediated signalling cause activation of the canonical phosphatidylinositol-3 kinase/serine/threonine protein kinase pathway and stimulation of mTORC1 to induce muscle hypertrophy(Reference Adegoke, Abdullahi and Tavajohi-Fini1, Reference Egerman and Glass2). Effects of activation of mTORC1 include mRNA translation, inhibition of apoptosis causing an increase in cell size and number, implicated in the regulation of myogenesis(Reference Yecies and Manning3). mTORC1 also activate ribosome biogenesis and also suppresses autophagy. Insulin resistance and/or reduced levels of growth factors(Reference Barazzoni, Short and Asmann4) in muscle are associated with reduced phosphorylation of insulin receptor substrate 1 and reduction in its signalling to phosphatidylinositol-3 kinase(Reference Guertin and Sabatini5).
Amino acids per se have pronounced effects in stimulating anabolism and reducing breakdown of protein in muscle(Reference Egerman and Glass2, Reference Barazzoni, Short and Asmann4, Reference Guertin and Sabatini5). The branched-chain amino acid leucine, as well as arginine, can activate downstream signalling to mTORC1 by independent mechanisms that are linked with increasing muscle hypertrophy(Reference Adegoke, Abdullahi and Tavajohi-Fini1). Low plasma concentrations of amino acids are permissive for activation of the ubiquitin system, autophagy and apoptosis(Reference Adegoke, Abdullahi and Tavajohi-Fini1, Reference Barazzoni, Short and Asmann4).
Muscle catabolic signalling
Inflammation and glucocorticoids are physiological signals that induce muscle protein degradation(Reference Egerman and Glass2) by their individual and combined actions. Pro-inflammatory cytokines activate the hypothalamic–pituitary–adrenal axis, leading to the production of catabolic stress hormones (adrenalin, cortisol, glucagon) resulting in reduced muscle sensitivity to insulin, increased proteolysis and reduced protein anabolism(Reference Rittig, Bach and Thomsen6). The two most important cellular degradation systems in muscle are the autophagy and ubiquitin–proteasome systems(Reference Egerman and Glass2, Reference Sandri7). Autophagy is a non-selective catabolic pathway through which damaged organelles and macromolecules are degraded and recycled within the cell. Autophagy genes are up-regulated at transcriptional and protein level in the tumour-bearing host. Autophagy is induced in the muscle by tumours, reduced nutrient supply, inflammation and several chemotherapy agents (see later). Autophagy is not only related to muscle loss and weakness but is involved in mitochondrial dysfunction, oxidative stress and associates with mortality(Reference Egerman and Glass2, Reference Sandri7). In the ubiquitin–proteasome system, proteins are targeted for degradation by the 26S proteasome. Ubiquitin ligases confer specificity in protein ubiquitination and play a key role in muscle loss. Tripartite motif containing 63 (TRIM63; also known as muscle-specific ring finger protein 1) and F-box protein 32 (FBOX32; also known as atrogin-1 or muscle atrophy F-box protein) were the first described muscle-specific ubiquitin protein ligases and are considered the main enzymes of this class responsible for targeting degradation of muscle structural and contractile proteins(Reference Egerman and Glass2, Reference Sandri7).
Muscle protein catabolism is a transcriptionally regulated process(Reference Lecker, Jagoe and Gilbert8). A cluster of atrophy genes is strongly up-regulated in animal models of cancer with multiple upstream signals (Fig. 1) generating a series of transcription factors converging on the expression of TRIM63 and FBOX32. Forkhead box protein O complex are important activators of the ubiquitin–proteasome system, whose activity is normally repressed by insulin/growth factor signalling(Reference Sandri7).
Tumour-derived catabolic mediators
A complex tumour secretome contributes prominently to cancer-associated muscle wasting (Fig. 1, Table 1). Tumour cells, as well as the enhanced inflammation elicited by the tumour, generate factors that directly elicit muscle catabolism(Reference Baracos, Martin and Korc9). These factors can also act in the central nervous system where they prompt catabolic neural outputs as well as neuroendocrine outputs (such as the release of adrenal corticosteroids)(Reference Braun, Zhu and Szumowski10) which activate proteolysis in skeletal muscle.
Table 1. Catabolic mediators of proteolysis in skeletal muscle and lipolysis in adipose tissue related to cancer-associated cachexia

BAT, brown adipose tissue; ERK1/2, extracellular signal-regulated protein kinase; GDF, growth differentiation factor; HSL, hormone-sensitive lipase; LIF, leukemia inhibitory factor; MAPK, mitogen-activated protein kinase; TWEAK, TNF-related weak inducer of apoptosis; UCP, uncoupling protein; WAT, white adipose tissue.
Multiple pro-inflammatory factors expressing catabolic actions on muscle have been described. PGE2 is a known agent of tumour-induced bone resorption(Reference Tashjian11) and has similarly been documented as a mediator of protein catabolism in skeletal muscle(Reference Strelkov, Fields and Baracos12). Additional inflammatory mediators of muscle catabolism include IL-6, IL-1, TNF-α, interferon γ, leukaemia inhibitory factor and TNF ligand superfamily member 12 (TNF-related weak inducer of apoptosis). These factors signal through their respective cell surface receptors and activate selective transcription factors, which in turn promote the transcription of ubiquitin–proteasome and autophagy genes. TNF-α, IL-6 and IL-1 activate downstream pathways to induce TRIM63 and FBOX32(Reference Egerman and Glass2, Reference Sandri7) and muscle atrophy. Signal transducer and activator of transcription 3 (STAT 3) signalling downstream of IL-6 activates the ubiquitin–proteasome pathway, overexpression of signal transducer and activator of transcription 3 is sufficient to induce muscle atrophy and to up-regulate TRIM63 as well as autophagy(Reference Bodine and Baehr13, Reference Yuan, Han and Meng14).
Members of the transforming growth factor (TGF)-β superfamily are produced by both tumours and immune cells(Reference Loomans, Andl and Andl15). Myostatin, a major autocrine inhibitor of muscle growth, binds to its receptor in skeletal muscle. Myostatin-dependent catabolic signaling involves SMAD transcription factors, resulting in the transcriptional activation of the ubiquitin–proteasome system as well as inhibition of the mTOR pathway(Reference Egerman and Glass2, Reference Cohen, Nathan and Goldberg16) (Fig. 1). Additional TGF-β family members, including TGF-β1, activin-A, growth differentiation factor (GDF)-11 and GDF-15 stimulate SMAD(Reference Egerman and Glass2, Reference Chen, Walton and Qian17, Reference Lerner, Tao and Liu18) and inhibit muscle differentiation with similar or even greater potencies than myostatin. The mechanisms that regulate myostatin expression locally in muscle are still mostly unknown, although glucocorticoids, forkhead box protein O complex, NF-κB and SMAD can all enhance myostatin expression in muscle(Reference Loumaye, De Barsy and Nachit19, Reference Togashi, Kogita and Sakamoto20). This generates an autocrine contribution, in addition to the effects of tumour-derived myostatin (Fig. 1).
Chemotherapy effects on muscle anabolism/catabolism
Catabolic sequelae of cancer chemotherapy add substantially to overall weight loss and muscle catabolism. Common side effects of cytotoxic chemotherapy include anorexia, nausea and vomiting, which are nutritionally impactful symptoms that contribute to reduced food intake and hence to weight loss. These effects can be substantial. Mean weight loss in patients receiving neoadjuvant chemotherapy (various combinations of epirubicin, cisplatinum, fluorouracil, capecitabine or oxaliplatin) for oesophago-gastric cancer was 4·2 kg(Reference Awad, Tan and Cui21) and during platinum-based chemoradiotherapy for head and neck cancer, 11·4 kg(Reference Kubrak, Olson and Jha22). Diagnostic imaging i.e. dual-energy X-ray absorptiometry or computed tomography has been used to determine that these losses are often composed mostly of muscle(Reference Awad, Tan and Cui21, Reference Silver, Dietrich and Murphy23). Patients receiving neoadjuvant chemoradiotherapy (gemcitabine + cisplatin) with pancreatic cancer were evaluated using their abdominal computed tomography scan and these lost 8 % of their pretreatment adipose tissue and 2·5 % of skeletal muscle mass (P ≤ 0·01)(Reference Cooper, Slack and Fogelman24).
While some of the chemotherapy-induced weight loss might be due to non-specific effects such as loss of appetite, there is also a mechanistic basis for direct effects of cytotoxic and targeted cancer therapies on muscle cells, including altered contractile properties, insulin resistance and atrophy(Reference Sorensen, Cheregi and Timpani25). Since many cancers show aberrant activation in pathways upstream of mTORC1, targeted cancer therapies are designed to inhibit at the mTORC1 complex (e.g. rapamycin derivatives: sirolimus, everolimus and ridaforolimus). These therapies may interfere with the mTOR-dependent pathways by which muscle protein synthesis is activated by insulin and amino acids(Reference Adegoke, Abdullahi and Tavajohi-Fini1). A drug of this class, sorafenib, intensified muscle loss during 6 and 12 months of treatment in a clinical trial of patients with renal cell carcinoma(Reference Antoun, Birdsell and Sawyer26).
Several cytotoxic agents (e.g. oxaliplatin(Reference Sorensen, Petersen and Timpani27), cisplatin(Reference Sirago, Conte and Fracasso28), anthracyclines, 5-fluorouracil, irinotecan) appear to be taken up by muscle cells and induce atrophy as well as mitochondrial dysfunction (mitophagy), oxidative damage, cellular energy depletion and apoptotic or necrotic cell death(Reference Sorensen, Cheregi and Timpani25, Reference Sorensen, Petersen and Timpani27, Reference Sirago, Conte and Fracasso28). Doxorubicin suppresses protein synthesis and activates proteolytic and apoptotic signalling(Reference Nissinen, Degerman and Räsänen29). Cisplatin reduced serine/threonine protein kinase activation, up-regulated TRIM63 and FBOX32, decreased levels of phosphorylated forkhead box protein O complex, and also increased Beclin1, a protein involved in the initiation of autophagy and activation of LC3 (LC3AII), a molecule essential for autophagosome formation(Reference Sirago, Conte and Fracasso28, Reference Conte, Camerino and Mele30). In the muscles of mice, gemcitabine + cisplatin therapy induced myostatin, activin-A, TNF-α, IL-6, IL-1β, forkhead box protein O complex expression and activation, NF-κB activation, TRIM63 and FBOX32 expression and proteasome activity(Reference Chen, Hsu and Hwang31).
Adipose tissue wasting in cancer-associated malnutrition
Normal and cancer-associated regulation of lipogenesis and lipolysis
Precise physiological controls on lipogenic and lipolytic pathways allow for an overall state of neutral fat balance in healthy individuals(Reference Ratnayake and Galli32). Lipogenesis is activated by insulin during feeding, inducing glucose uptake and activation of lipogenic enzymes. Adipocyte lipoprotein lipase releases fatty acids for adipocyte uptake from circulating lipoproteins such as chylomicrons and VLDL(Reference Luo and Liu33). During fasting, decreased insulin levels suppress lipogenesis and induce lipolysis. TAG in adipocytes are converted to NEFA and glycerol by adipocyte triacylglycerol lipase and hormone-sensitive lipase (HSL). Glycerol is a substrate for hepatic gluconeogenesis and NEFA released are used for oxidation according to energy needs in other organs(Reference Voss, Vendelbo and Kampmann34). Additional physiological lipolytic stimuli include glucagon as well as catecholamines (released by sympathetic neurons innervating adipocytes); both activate 3′,5′cyclic AMP-dependent protein kinase A pathway and are responsible for the induction of lipolysis during fasting. Catecholamines and natriuretic peptides system stimulate adipocyte lipolysis is mediated through different signal pathways by either 3′,5′cyclic AMP and 3′,5′cyclic GMP pathway, stimulating protein kinase G and activating HSL(Reference Sorensen, Cheregi and Timpani25, Reference Agustsson, Rydén and Hoffstedt35).
Chemotherapy effects on fat lipolysis and lipogenesis
Decreased de novo lipogenesis and increased lipolysis by the action of agents used in cancer chemotherapy have been postulated to contribute to weight loss; however, there are only a few observations available concerning direct effects of chemotherapy drugs on adipocytes and lipid metabolism. Cisplatin and doxorubicin suppress the expression of genes associated with de novo lipogenesis (fatty acid synthase, acetyl-CoA carboxylase), biosynthesis of PUFA (stearoyl coenzyme A desaturase1) and fatty acid uptake (lipoprotein lipase)(Reference Garcia, Scherer and Chen36, Reference Biondo, Lima and Souza37). Cisplatin additionally increased the expression of carnitine palmitoyl transferase-1α (a regulatory enzyme for fatty acid β-oxidation), adipocyte TAG lipase, HSL and fatty-acid-binding protein-4(Reference Garcia, Scherer and Chen36, Reference Klose, Krzywinska and Castells38).
Tumour-derived mediators of excess lipolysis in adipose tissue
The balance of lipogenesis and lipolysis is profoundly altered in the tumour-bearing state (Fig. 2). The response of lipolysis to its normal physiological regulators is altered, with loss of the inhibitory action of insulin with concurrent hypersensitivity to the lipolytic activation by catecholamines and natriuretic peptides and resulting in overexpression and activation of HSL(Reference Agustsson, Rydén and Hoffstedt35). Multiple elements of the tumour secretome are potent lipolysis, inducing factors, including IL-6, TNF-α, zinc-α2-glycoprotein (ZAG), adrenomedullin and parathyroid hormone-related protein (PTHrP; Table 1, Fig. 2).
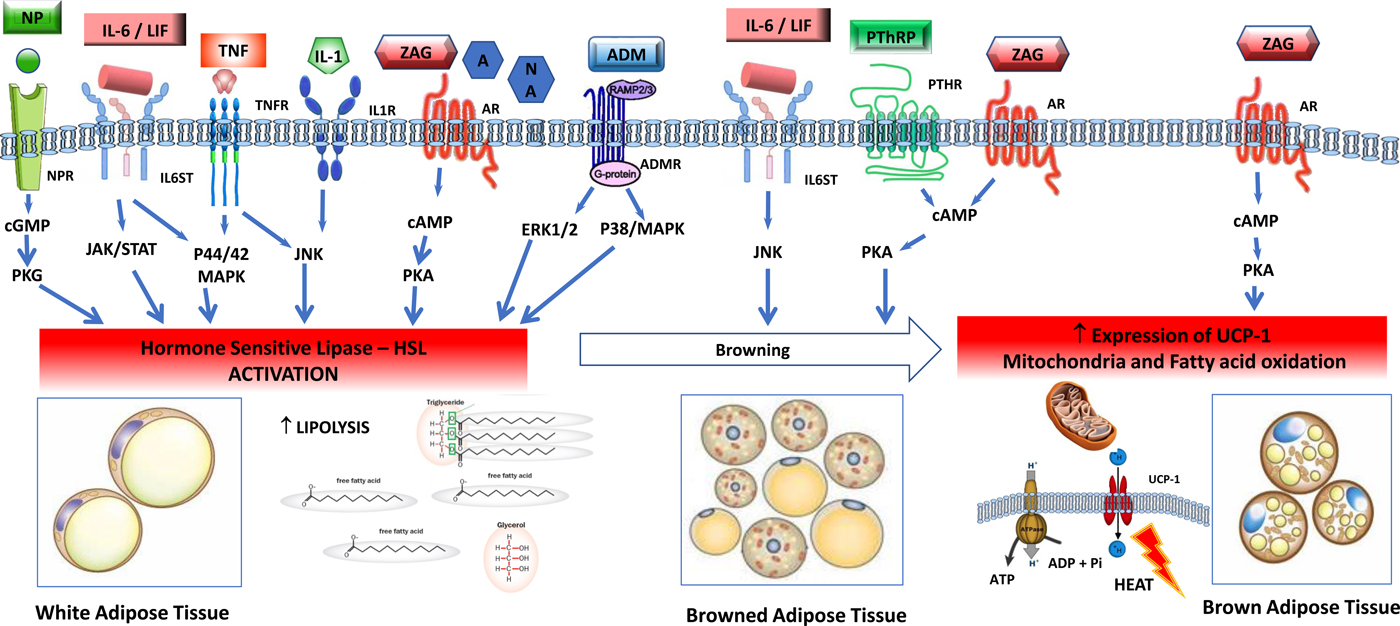
Fig. 2. (Colour online) Tumour-induced signalling pathways involved in the control of adipose tissue lipolysis. Signals released from tumour and activated immune cells, sympathetic nervous system, cardiovascular system and adrenal gland activate lipolysis in white adipose cells through increased gene expression and activation of hormone-sensitive lipase. Lipolytic stimuli include cytokines (IL-1, TNF-α, IL-6, leukaemia inhibitor factor (LIF)), zinc-α2-glycoprotein (ZAG), adrenomedullin (ADM), natriuretic peptides (NP) and catecholamines such as adrenaline (A) and noradrenaline (NA). In healthy individuals, lipolysis in white adipose tissue is normally regulated by adrenal catecholamines, sympathetic input and natriuretic peptides, and in the tumour-bearing state white adipose is abnormally sensitive to these stimuli. Several factors overproduced by tumours, including IL-6, LIF, ZAG and parathyroid hormone-related protein (PTHrP) induce browning of white adipose tissue, characterised by increased expression of uncoupling protein (UCP)-1 and energy wastage via futile cycling. In brown adipocytes, ZAG also increases expression of UCP-1, elevated NEFA oxidation and futile cycling. AM, adrenomedullin receptor; AR, adrenergic receptor; BAT brown adipose tissue, cAMP, 3′,5′cyclic AMP; cGMP, 3′, 5′cyclic GMP; CREB, factor cAMP response element-binding protein; ERK1/2 (p44/42 MAPK), extracellular signal-regulated protein kinase, HLS, hormone-sensitive lipase; IL-1R, IL 1 receptor; IL-6ST, IL-6 receptor subunit β; JAK/STAT, Janus kinase/signal transducer and activator of transcription; JNK, Jun Nterminal kinase; MAPK, mitogen-activated protein kinase; NPR, natriuretic peptide receptor; TG, TAG; TNFR, TNF receptor.
Activation of thermogenesis appears to be an additional mechanism that contributes to a hypercatabolic state in tumour-bearing animals(Reference Shellock, Riedinger and Fishbein39). Brown adipose tissue has a characteristic thermogenic mechanism. This tissue expresses uncoupling protein (UCP)-1, a transmembrane protein which increases the permeability of the inner mitochondrial membrane, uncoupling mitochondrial oxidative phosphorylation. This is a futile cycle, a means of heat production and is characteristic of brown adipose tissue. Browning of white adipose tissue (WAT) consists of a differentiation of the white adipocytes towards a brown adipose tissue-like phenotype, also characterised by the expression of UCP-1 as well as a suite of brown-selective genes, e.g. Dio2, Pgc1α, PPARγ, Cidea (cell death activator), Prdm16 and mitochondrial protein NDUFS1(Reference Petruzzelli, Schweiger and Schreiber40, Reference Kir, White and Kleiner41). Browning of WAT switches mitochondrial activity to thermogenesis instead of ATP synthesis, causing lipid mobilisation and energy expenditure(Reference Petruzzelli, Schweiger and Schreiber40, Reference Tsoli, Moore and Burg42). A variety of physiological and tumour-derived effectors induce browning of WAT (Table 1). For example, IL-6 induces lipolysis in WAT as well as browning of WAT(Reference Han, Meng and Shen43). In an animal tumour model, a neutralising antibody to IL-6 abolished these effects, and in patients IL-6 levels in blood were correlated with circulating NEFA levels(Reference Petruzzelli, Schweiger and Schreiber40).
ZAG is a molecule encoded by the AZGP1 gene in human subjects, which is produced by secretory epithelial cells, adipocytes and tumour cells. ZAG is overexpressed in cancer patients and can be detected in plasma(Reference Xue, Yu and Yan44) up to 7-fold above normal concentrations(Reference Cabassi and Tedeschi45). ZAG is an adipokine, involved in the regulation of lipid and glucose metabolism, and control of fat mass and energy expenditure. ZAG activates β1 and β2-adrenergic receptors in WAT, and acts in adipocyte intracellular pathways by signalling adenylyl cyclase, 3′,5′cyclic AMP cascade involved in lipolysis(Reference Cabassi and Tedeschi45). To increase energy expenditure, ZAG is reported to raise fatty acid oxidation and expression of UCP-1 in brown adipose tissue. ZAG causes a reduction of mRNA and proteins of lipogenic transcriptional factors: CCAAT/enhancer-binding protein, PPARγ, sterol regulatory element binding protein-1c, increased levels of UCP-1 causing shrinkage of adipocytes(Reference Cabassi and Tedeschi45).
Like all cells, cancer cells secrete exosomes, small membranous vesicles containing bioactive molecules, which can transfer their contents to other cells, locally and systemically. Human pancreatic cancer-secreted exosomes were shown to induce lipolysis in both murine and human white adipocytes(Reference Sagar, Sah and Javeed46). The exosome factor responsible was identified as adrenomedullin, a ubiquitously expressed peptide originally isolated from pheochromocytoma, which has a number of biological activities. A specific adrenomedullin receptor found in adipocytes, activates HSL via extracellular signal-regulated protein kinase and p38 mitogen-activated protein kinase signalling(Reference Sagar, Sah and Javeed46).
PTHrP is a member of the parathyroid hormone family, whose normal physiological functions are exerted at an autocrine or paracrine level; in healthy human subjects, PTHrP is undetectable in peripheral blood. Mutations that amplify PTHrP expression in tumours(Reference Sidler, Alpert and Henderson47) can promote high systemic concentrations in patients with cancer and this has been associated with malignant hypercalcaemia and with poor prognosis. PTHrP production also appears to be activated in fibroblasts that exist within the tumour stroma(Reference Kong, Lei and Du48). In tumour-bearing animals, PTHrP was demonstrated to induce thermogenesis in WAT via a G-protein-coupled receptor, resulting in browning(Reference Kir, White and Kleiner41). UCP-1 expression in WAT has been shown in human subjects with cancer cachexia(Reference Petruzzelli, Schweiger and Schreiber40).
Potential for nutritional modulation of excess catabolism
In the preceding sections, we have described numerous catabolic stimuli capable of provoking lipolysis and proteolysis as well as increased energy expenditure, in the tumour-bearing host. The efficacy of nutritional intervention as a sole therapy is clearly a challenge in this context for several reasons. Malnutrition is generally more prominent in cancers of advanced stage and therefore the context of dietary intervention is concurrent chemotherapy or chemoradiotherapy, which have their own catalogue of catabolic actions. Low levels of dietary intake pose another challenge. Taking several randomised clinical trials of cancer nutrition therapy as examples(Reference Bourdel-Marchasson, Blanc-Bisson and Doussau49–Reference Ovesen, Allingstrup and Hannibal51), intakes achieved by the participants (7113–8368 kJ (1700–2000 kcal)) typically fall well short of target intakes (8786–11715 kJ (2100–2800 kcal)), and therefore energy requirements were not met by the intervention. Reaching nutritional targets based on volitional food intake is limited, even in the protected arena of a clinical trial. While further studies are needed to enhance dietary intake, some approaches might include ensuring that pain and symptom management addresses all modifiable nutrition impact symptoms, proposing higher nutritional goals between cycles when patients are experiencing fewer side effects, improving patient education and awareness, increasing the energy density and/or improved palatability of supplements, and eventually an earlier consideration of escalation to non-volitional modalities of nutritional support.
An anabolic intervention may be required to support nutrition therapy, and it seems promising that robust anabolic responses have been demonstrated to specific drugs. Selumetinib treatment was associated with an increase in computed tomography-defined skeletal muscle mass in patients with cholangiocarcinoma(Reference Macdonald, Johns and Stephens52). Anamorelin, a growth hormone secretagogue receptor type 1 (ghrelin receptor) agonist, increased dual-energy X-ray absorptiometry-defined muscle and fat mass in a large randomised clinical trial in patients with non-small-cell lung cancer(Reference Temel, Abernethy and Currow53). These studies show that there is a strong anabolic potential lurking in the background that responds remarkably to these stimuli; patients added kilogram quantities of muscle even though they had advanced disease and inflammation.
Finally, whatever supply of nutrients is provided, the provision of food or nutritional supplements may or may not alter the crescendo of catabolic stimuli and the alterations of metabolism that they provoke. As described later, leucine and n-3 fatty acids appear to intercept catabolic signalling pathways, so these nutrients have the possibility to correct the tumour-induced alterations in metabolism. This is quite exciting and more work to optimise the nutrient mixtures and combine them with anabolic agents may provide even stronger responses.
Protein and amino acid nutrition
There is some encouraging evidence that loss of muscle mass and reduced rates of muscle protein synthesis are at least partially reversible in patients with cancer. Protein synthesis is evidently not shut down or impaired. Several studies suggest that this process is responsive to the supply of amino acids, albeit a somewhat higher quantity than in younger, healthy individuals(Reference Acharyya, Butchbach and Sahenk54–Reference Baracos57). Winter et al. (Reference Winter, MacAdams and Chevalier56) demonstrated a normal whole-body anabolic response to infusion of amino acids in non-small-cell lung cancer patients, even though they were insulin-resistant for glucose uptake. Macdonald et al. (Reference Macdonald, Johns and Stephens52) showed that myofibrillar protein synthesis is normal in patients with cancers of the upper gastrointestinal tract. Activation of protein synthesis has been demonstrated in patients with advanced disease and inflammation after oral intake of high-quality proteins(Reference Deutz, Safar and Schutzler55) (whey protein + added leucine). It is of interest that leucine supplementation reduces body and muscle protein loss(Reference Macdonald, Johns and Stephens52, Reference Deutz, Safar and Schutzler55, Reference Winter, MacAdams and Chevalier56), even in the presence of acute inflammation, as has been shown after endotoxin injection in human subjects(Reference Rittig, Bach and Thomsen6). Since leucine activates muscle protein synthesis at the level of mTORC1, it is possible that leucine may bypass the effect of catabolic stimuli that act upstream of mTORC1, or act by an alternative mechanism(Reference Egerman and Glass2, Reference Barazzoni, Short and Asmann4, Reference Guertin and Sabatini5, Reference Macdonald, Johns and Stephens52, Reference Deutz, Safar and Schutzler55–Reference Baracos57). Targeting protein anabolism in cancer patients requires optimisation. Since the most favourable stimulation of protein synthesis and suppression of protein breakdown is coordinated by insulin and amino acids, it may be helpful to ensure optimal conditions of muscle insulin sensitivity. Anabolic deficit may be partly addressed by the maintenance of physical activity, a notion that is endorsed within current oncology nutrition clinical practice guidelines(Reference Arends, Bachmann and Baracos58) as well as in the design of clinical trials of multimodal intervention (see, e.g. NCT02330926 in which protein- and energy-dense nutrition is combined with exercise). Amino acids are not the only nutrients specifically required for muscle building and others, including n-3 PUFA, vitamin D, creatine and carnitine, may need to be added(Reference Chanet, Verlaan and Salles59). Muscle protein synthesis was stimulated in catabolic cancer patients by a formulated medical food, in which whey protein, free amino acid leucine, specific oligosaccharides and fish oil were combined(Reference Deutz, Safar and Schutzler55). Optimal conditions for exploiting this anabolic potential are currently under study, with the overall aim of net improvements in muscle mass, functionality, performance status and treatment tolerance.
While muscle protein synthesis can clearly be activated in patients with cancer under controlled conditions, their day-to-day protein intake may be suboptimal. Reported dietary intakes of protein are often below levels recommended in the European Society for Clinical Nutrition and metabolism guidelines on nutrition in cancer patients(Reference Arends, Bachmann and Baracos58), i.e. daily protein intake >1 g/kg and if possible up to 1·5 g/kg. Available commercial nutritional supplements for cancer patients are typically energy-dense, but standard formulations may have an inadequate amount of high-quality protein(Reference Engelen, van der Meij and Deutz60). The protein levels required to maximally support muscle mass and anabolism in patients with cancer requires further study, but there are data from non-malignant disease to suggest that these may be closer to the upper end of the range of current recommendations. Mitchell et al. randomised men aged >70 years to a complete diet containing either 0·8 (RDA) or 1·6 (2RDA) g protein/kg daily. Whole-body lean mass was increased, mainly accounted for by an increase in lean mass of the trunk and preservation of appendicular lean mass in the 2RDA group compared with the RDA group, which lost lean mass, suggested that higher protein intake prevents atrophy(Reference Mitchell, Milan and Mitchell61).
n-3 PUFA nutrition
The n-3 fatty acids are potentially useful therapeutic agents for treatment and prevention of muscle loss(Reference Pappalardo, Almeida and Ravasco62). The participation of PGE2 as a catabolic mediator in muscle raised(Reference Strelkov, Fields and Baracos12) interest in the possible utility of n-3 PUFA (EPA and DHA) in cancer nutrition therapy. The main studied effect of n-3 fatty acids is to down-regulate the synthesis of catabolic pro-inflammatory eicosanoids (PGE2), cytokines (TNF-α, IL-6, IL-1β) and their downstream effectors such as NF-κB that induce muscle proteolysis(Reference Pappalardo, Almeida and Ravasco62–Reference Schiessel, Yamazaki and Kryczyk64). The n-3 fatty acids may suppress cytokine transcription in muscle via regulation of transmembrane toll-like receptors and repression of NF-κB activation and its downstream catabolic signalling(Reference Liu, Chen and Odle65). It has been postulated that n-3 fatty acids induce activation (phosphorylation) of anabolic signalling proteins in the muscle tissue(Reference Smith, Atherton and Reed66). While this has not been studied in patients with cancer, older subjects after 8 weeks of dietary supplementation with n-3 fatty acids, showed enhancements in the fractional rate of skeletal muscle protein synthesis, and increases in muscle mTOR-p70s6k (ribosomal protein kinase S6) signalling pathway(Reference Smith, Atherton and Reed66).
Conclusion
Cancer nutrition therapy often is of limited success and we suggest that it may be seriously undermined by a barrage of catabolic stimuli generated by the tumour, activation of the host immune system and cancer therapy. Research is required to determine how to optimise nutrient mixtures so that they not only provide energy fuels and biosynthetic building blocks, but they also modulate altered metabolism and catabolic drive. It also seems likely that overexpression of some specific mediators by tumours will require targeted intervention to mitigate their catabolic influence so that nutrition therapy can be fully utilised.
Acknowledgements
The authors thank L Martin RD, MSc for useful discussion of this manuscript.
Financial support
V. E. B. receives financial support from the Canadian Institutes of Health Research and the Alberta Cancer Foundation.
Conflicts of interest
None.
Authorship
Both authors contributed equally to the conception and writing of this paper.



