



Introduction
Echinolaelaps echidninus is a large worldwide mite belonging to Laelapidae, Gamasida, Parasitiformes, Acari (Deng, 1993). As early as 1929, H.E. Ewing separated the large mites from the genus Laelaps and defined 1 new genus Echinolaelaps (Wang, Reference Wang1963) and then the Latin name for the mite was defined as Echinolaelaps echidninus Berlese, 1887. However, there are 2 Latin names of the mite due to differences in taxonomic opinion. Some researchers have been using Laelaps echidninus Berlese, 1887 (Mishra et al., Reference Mishra, Dhanda and Kulkarni1970; Zhou et al., Reference Zhou, Guo, Song, Zhao, Zhang, Fan, Chen, Lv, Yin and Jin2022), and others were using E. echidninus Berlese, 1887 (Pawar et al., Reference Pawar, Jogdand, Jadhav and Deokar2016) or both (Ugbomoiko and Obiamiwe, Reference Ugbomoiko and Obiamiwe1991). Taxonomic relationships of Gamasida are also confusing, with different taxonomists holding different views. This paper follows the classification system of Krantz, 1978 (Deng,1993).
Echinolaelaps echidninus has a relatively wide range of hosts and can be parasitic on the body surface of most small mammals, among which Rattus tanezemi, Rattus norvegicus and Mus musculus are dominant hosts (Yang, Reference Yang1992; Guo et al., Reference Guo, Ye, Gu and Chen1996). Echinolaelaps echidninus is ovoviviparous and has a 4-stage life history: larva, first nymph, second nymph and adult. The larva does not feed, but the first nymph, second nymph and adult need to feed on blood, tissue fluids, wound exudate and other secretions of their hosts, making them an important medical mite that can directly damage host skin while feeding on animal and human blood, causing pruritus, maculopapular rash and even systemic reactions (Zhao, Reference Zhao2002; Chai et al., Reference Chai, Tao and Li2017). Echinolaelaps echidninus is also an important vector. Researchers have isolated zoonotic pathogens such as Rickettsia tsutsugamushi, Rickettsia Q fever, Rickettsia mooseri, Rickettsia pox pathogens, Corynebacterium pseudotuberculosis and Leptospira in E. echidninus (Deng, 1993; Krantz and Walter, Reference Krantz and Walter2010).
The typical mitochondrial genome of arthropods contains 37 genes, namely 22 tRNA genes, 13 protein-coding genes, 2 rRNA genes (rrnL and rrnS) and a variable length control region (CR) (Gissi et al., Reference Gissi, Iannelli and Pesole2008). Because of its compact structure, smaller molecule (16–19 kb) than nuclear genome, 5–10 times faster evolution rate than nuclear genome and maternal inheritance, mitochondrial genome has become an important molecular marker for studying the origin of species, phylogenetic relationships between related species and within species in recent years (Yang et al., Reference Yang, Chen and Dong2023). In the analysis of the mitochondrial genomes of Gamasida, which have been deposited in Genbank, the mitochondrial genomes of 5 species of mites in the families Diplogyniidae and Parasitidae (Parasitus wangdunqingi, Parasitus fimetorum, Microdiplogynium sp., Quadristernoseta cf. longigynium and Quadristernoseta cf. intermedia) retained the ancestral pattern of mitochondrial gene arrangement of arthropod, while other species show varying degrees of rearrangement (Thomas et al., Reference Thomas, Zalom and Nicola2011; Osuna-Mascaró et al., Reference Osuna-Mascaró, Doña, Johnson, Esteban and de ROJAS2020; Zhang et al., Reference Zhang, Havird, Wang, Lv and Xu2021; Yang et al., Reference Yang, Yang and Dong2022). However, there are multiple gene duplications in the mitochondrial genomes of Euseius nicholsi (2 of trnM and trnF) and Metaseiulus occidentalis (duplicated 18 genes). The genome composition of most arthropod mitochondrial genomes is very stable and the phenomenon of gene duplications or gene loss is rare (Jeyaprakash and Hoy, Reference Jeyaprakash and Hoy2007; Li et al., Reference Li, Shao, Zhang, Deng and Xue2019). In addition, researchers found tRNA truncation in Acariformes and the average tRNA length of 54.8 ± 1 bp in a previous study, and the average length of mitochondrial tRNAs in Parasitiformes is 62.0 ± 1.3 bp (Yuan et al., Reference Yuan, Wei, Wang, Dou and Wang2010). The length of Parasitiformes tRNA genes is longer than that of Acariformes, but shorter than the average length of arthropods tRNA genes (66 bp) (Jeyaprakash and Hoy, Reference Jeyaprakash and Hoy2007), which prevents some tRNA genes from shaping the typical 4-armed cloverleaf secondary structure. This suggests that truncated tRNAs may occur in Parasitiformes.
Currently, the study of Gamasida is hampered by extreme body miniaturization, which has likely stifled the interest of taxonomists in this group and delayed the application of new sequencing technologies. This study filled a gap of the complete mitochondrial genome of the genus Echinolaelaps, and some novel insights into the rearrangement patterns and phylogeny of Gamasida are provided by the unique mitochondrial genome presented by E. echidninus.
Materials and methods
Specimen collection, morphological identification, DNA extraction and PCR amplification
Echinolaelaps echidninus were collected from the body surface of Rattus tanezumi (Muridae, Rodentia) in Yunnan Province, and all samples were stored at –80°C in a refrigerator for backup. The mite specimens and R. tanezumi were deposited in the Institute of Pathogens and Vectors, Dali University, China. The main reference for morphological identification of E. echidninus is the volume 40 of the Economic insect fauna of China Fasc (Deng, 1993). The essential distinguishing features of E. echidninus were that the 2 sides of the genito-ventral plate are extremely enlarged after VI1, the posterior margin was deeply concave and the distance between the genito-ventral plate and anal plate is small, showing a narrow groove. The anal plate is inverted pear-shaped, the front part is blunt and round and the rear end is sharp and narrow. The adanal setae lie behind the level of the back end of the anus and reach the base of the postanal seta. The postanal seta is thicker and longer than the adanal setae (Fig. 1).

Figure 1. Morphological characteristics of E. echidinus from China.
Total DNA was extracted from individual mite with DNeasy Tissue Kit (QIAGEN, Germany). Ex Taq (Takara) and the conserved universal primers: bcdF01: 5′-CATTTTCHACTAAYCATAARGATATTGG-3′, bcdR04: 5′-TATAAACYTCDGGATGNCCAAAAAA-3′ (Dabert et al., Reference Dabert, Witalinski, Kazmierski, Olszanowski and Dabert2010), 16Sbr: 5′-CCGGTCTGAACTCAGATCACGT-3′ and 16Sar: 5′-CGCCTGTTTAACAAAAACAT-3′ (Simon et al., Reference Simon, Frati, Beckenbach, Crespi, Liu and Floork1994) were used to amplify 421 bp cox1 gene and 300 bp rrnL gene of E. echidninus. The polymerase chain reaction (PCR) cycling conditions were 94°C for 3 min; 37 cycles of 94°C for 1 min, 46–54°C for 1 min, 72°C for 1 min; 72°C for 10 min. The long fragment-specific primers for E. echidninus, 43 cox1-rrnL-F2: 5′-CTGTTTATCCACCTTTGGCTGGAAG-3′ and 43 cox1-rrnL-R1: 5′-GCGACCTCGATGTTGAATTAATGAACC-3′, were designed with the sequences of cox1 and rrnL short fragments to amplify the complete mitochondrial genome of E. echidninus. Amplification conditions were 98°C for 2 min; 34–40 cycles of 98°C for 10 s, 46–56°C for 30 s, 68°C for 5 min; 68°C for 10 min.
Sequencing, assembly and annotation of mitochondrial DNA sequence
PCR products were purified with the Wizard SV Gel and PCR clean-up system (Promega) kit according to the manufacturer's instructions. Purified PCR products were sequenced directly with Illumina Hiseq X-Ten platform at Winner Biotech in Shanghai, China. The clean sequencing data of E. echidninus libraries were assembled with Geneious Prime 11.0 software (Kearse et al., Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012), tRNA genes were searched with tRNAscan SE (Chan et al., Reference Chan, Lin, Mak and Lowe2021) and ARWEN (Laslett and Canback, Reference Laslett and Canback2008), and protein-coding and rRNA genes were identified with Geneious Prime 11.0 software, BLAST and MITOS (Altschul et al., Reference Altschul, Madden, Schaffer, Zhang, Zhang, Miller and Lipman1997; Bernt et al., Reference Bernt, Donath, Juhling, Externbrink, Florentz, Fritzsch, Putz, Middendorf and Stadler2013). The annotated mitochondrial genome sequence of E. echidninus was deposited in GenBank (accession number: OP954302).
Rearrangement and phylogenetic analysis of the mitochondrial genome
The breakpoint distances between each of 2 taxa of Gamasida were compared using the CREx web server (Bernt et al., Reference Bernt, Merkle, Ramsch, Fritzsch, Perseke, Bernhard, Schlegel, Stadler and Middendorf2007) to analyse the extent of rearrangement of the mitochondrial genome (M. occidentalis and E. nicholsi were not calculated due to duplicated mitochondrial genes). Thirteen protein-coding genes of 24 complete mitochondrial genome sequences of 17 genera and 10 families of Gamasida, currently published in GenBank, were aligned using Mega 11 (Tamura et al., Reference Tamura, Stecher and Kumar2021). Tachypleus tridentatus, Carcinoscorpius rotundicauda and Limulus polyphemus were used as outgroups for phylogenetic analysis using the maximum likelihood (ML) method (Stamatakis, Reference Stamatakis2006) and Bayesian inference (BI) method (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003). In ML analysis, the GTR + I + G model constructs ML trees with 1000 bootstrap replicates. For BI analysis, 4 simultaneous Markov chains were run for 1 million generations, with tree sampling occurring every 1000 generations, and a burn-in of 25% of the trees in Mrbayes to phylogenetic analysis. Finally, the Figtree 1.4.4 program (http://tree.bio.ed.ac.uk/software/figtree/) was used to embellish the evolutionary tree.
Result
Base composition and genes distribution of the mitochondrial genome of E. echidninus
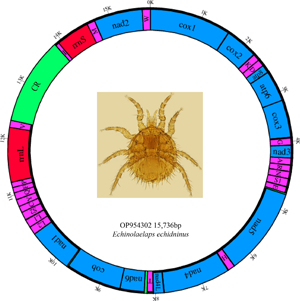
The complete mitochondrial genome of E. echidninus is 15 736 bp in length, was assembled by sequencing and contains 37 genes, namely 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes (rrnL and rrnS) and a CR of 1561 bp in length. Base composition of the entire mitochondrial genome is A: 40.8%, C: 7.7%, G: 10.7%, T: 40.8%, with an AT content of 81.6%, AT-skew is 0 and GC-skew is 0.163. The lengths of rrnL and rrnS are 1079 and 714 bp, respectively, with rrnL on the N strand and rrnS on the J strand. Protein-coding and tRNA genes are on the J strand except for nad5, nad4, nad4L, nad1, trnL1, trnL2, trnS2, trnH, trnC, trnQ, trnY, trnF, trnP and trnV, which were located in the N strand (Table 1).
Table 1. Distribution of the E. echidninus complete mitochondrial genome

Analysis of tRNA and protein-coding genes in the mitochondrial genome of E. echidninus
The average length of 22 mitochondrial tRNA genes of E. echidninus was 62 ± 0.8 bp, of which trnC gene (50 bp in length) was the shortest, trnQ and trnI were the longest (68 bp in length). Except for trnC and trnS1, which are missing the D-arm, all the tRNAs have the typical 4-armed cloverleaf secondary structure (Fig. 2). The anticodon of trnK is CUU, which does not preserve the universal anticodon of the arachnid mitochondrial tRNA (trnK: UUU), while the rest of tRNAs preserve the universal anticodon of the arachnid mitochondrial tRNA. The length of 13 protein-coding genes in order is nad5>cox1>nad4>cob>nad2>nad1>cox3>cox2>atp6>nad6>nad3>nad4L>atp8. There were 3632 amino acids encoded in the complete mitochondrial genome of E. echidninus. Cox1, atp6, nad3, nad6, nad2 have ATA as the start codon, cox2, cox3, nad4, cob have ATG as the start codon and atp8, nad5, nad4, nad1 have ATT as the start codon. All protein-coding genes have complete stop codons, except atp8 which uses TAG as a stop codon, and the remaining 12 protein-coding genes use TAA as a stop codon (Table 1).
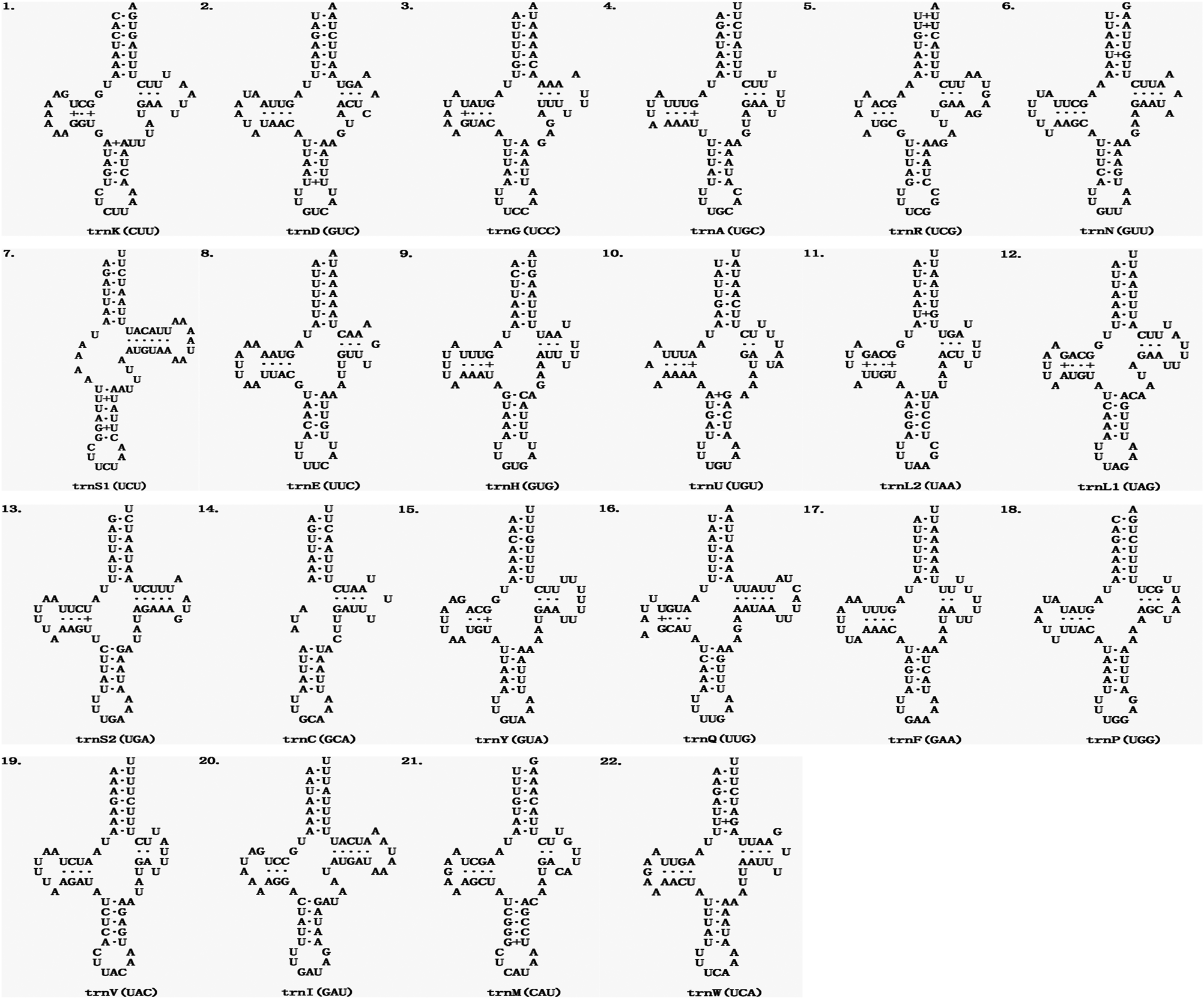
Figure 2. Secondary structure of the 22 mitochondrial tRNAs of E. echidninus.
The relative synonymous codon usage (RSCU) was analysed for the mitochondrial genome of E. echidninus (Table 2). UUU, UUA, AUU, AUA and AAU were the most frequently used (>150 times). Twenty-eight codons of UUA, AGA, UCU, CCU, GCU, etc., are preference codons, and the RSCU was >1. The codons of Ile, Leu, Phe and Ser were the most frequently used (1780 times in total), accounting for 49.0% of the total number of codons for uses. Using T. tridentatus as an outgroup, the evolutionary rate of 13 protein-coding genes was calculated and analysed with DnaSP6 (Rozas et al., Reference Rozas, Ferrer-Mata, Sanchez-Delbarrio, Guirao-Rico, Librado, Ramos-Onsins and Sanchez-Gracia2017). Among the 13 protein-coding genes, the ratio of non-synonymous evolutionary rate to synonymous evolutionary rate (Ka/Ks) for cox2, atp6, nad1, cox3, nad4 and nad5 was >1, while the ratio of non-synonymous evolutionary rate to synonymous evolutionary rate (Ka/Ks) for the rest of the protein-coding genes was <1. The evolutionary rates of 13 protein-coding genes were cox2>atp6>nad1>cox3>nad4>nad5>nad2>nad3>atp8>mad4L>nad6>cox1>cob (Fig. 3).

Figure 3. Comparison of non-synonymous and synonymous evolutionary rates (Ka/Ks) of 13 protein-coding genes in E. echidninus using the Tachypleus tridentatus as outgroup.
Table 2. Codon usage in protein-coding genes in the E. echidninus complete mitochondrial genome

Highlight codons with RSCU > 1 in bold.
Arrangement pattern and rearrangement degree of the mitochondrial genome of E. echidninus
Among the complete mitochondrial genomes of Gamasida that have been studied so far, the complete mitochondrial genome of E. echidninus shows a novel arrangement pattern. Except that trnC, trnY, trnQ, trnF, trnP and trnI were translocated, and trnS2 and rrnS were translocated and inverted, the remaining 13 protein-coding genes and tRNA genes still retained the ancestral pattern of mitochondrial gene arrangement of arthropods. Because of the rearrangement of rrnS gene, the ancestral arthropod ‘rrnL-trnV-rrnS’ gene cluster is no longer retained by E. echidninus. By breakpoint distance analysis (Table 3), comparing with that of the ancestral arthropod (as the presentative of T. tridentatus), the breakpoint distance of the mitochondrial genome of E. echidninus is 13; the breakpoint distances of Amblyseius tsugawai and Amblyseius swirskii are the highest (33); the breakpoint distances of Macrocheles muscaedomesticae and Stylochyrus rarior are the lowest (7). The breakpoints of species in the same family are similar, with certain differences between different families.
Table 3. Breakpoint distance analysis between species of Gamasida

a“1.Tachypleus tridentatus” (as the presentative of ancestral arthropod) retained the ancestral pattern of mitochondrial gene arrangement of arthropod.
bThe mitochondrial gene arrangement patterns of Ptilonyssus chloris and Tinaminyssus melloi are consistent. Two species are represented by “17.Ptilonyssus chloris” in the table.
cThe mitochondrial gene of 5 species of mites in Diplogyniidae and Parasitidae (Parasitus wangdunqingi, Parasitus fimetorum, Microdiplogynium sp., Quadristernoseta cf. longigynium and Quadristernoseta cf. intermedia) retained the ancestral pattern of mitochondrial gene arrangement of arthropod. Five species are represented by “1.Tachypleus tridentatus” in the table.
Phylogenetic relationships of E. echidninus
For the phylogenetic analysis of Gamasida using the ML (Stamatakis, Reference Stamatakis2006) and BI methods (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003), T. tridentatus, C. rotundicauda and L. polyphemus were used as the outgroup and the 13 protein-coding genes of 24 species in 17 genera and 10 families of the Gamasida as the ingroup. The phylogenetic tree of Gamasida is divided into 2 main branches (Fig. 4). The first branch consists of 3 species of 2 genera in the family Diplogyniidae located at the base of the phylogenetic tree, and the second branch contains the largest number of species, which consist of 21 species of 15 genera and 9 families. Echinolaelaps echidninus is located in the second branch, clustered with the species of Dermanyssoidea and formed a more supportive sister group with Varroa destructor in this study.

Figure 4. Phylogenetic relationships of Gamasida were inferred from the nucleotide sequences of 13 protein-coding genes using the Bayesian inference (BI) and maximum likelihood (ML) methods. The posterior probabilities for BI (left) and bootstrap support values for ML (right) are shown on the corresponding nodes in the identical topology of BI and ML tree. On the right side of the picture are the mitochondrial genomic arrangement patterns of the species, the different colours have different meanings (yellow: translocations; blue: inversions; green: translocations and inversions; red marks the ‘rrnL-V-rrnS’ gene cluster; pink marks the atp8 and atp6 genes; grey marks the control region and the 18 duplicated genes in the Metaseiulus occidentalis).
Discussion
The mitochondrial genome of E. echidninus was reported for the first time in this study. It is 15 736 bp in length and contains 37 genes and 1 CR typical of mitochondrial genomes. AT average content of the E. echidninus mitochondrial genome was 81.6%, with AT-skew of 0 and GC-skew of 0.163. The AT-skew of the mite is 0, which is more special in the mitochondrial genome of Acari. In general, due to the directional mutation pressure and asymmetric replication process, the AT skew of the J strand of the mitochondrial genome of most mites is positive, while the GC skew is negative (Hassanin et al., Reference Hassanin, Leger and Deutsch2005). Does a zero value for the J strand AT skew of E. echidninus predict low directional mutational pressure on its mitochondrial genome and/or a relatively short period of time for the mitochondrial double strand to remain in a single strand? Among the other species of Gamasida, we found that V. destructor, E. nicholsi, Phytoseiulus persimilis, A. tsugawai and A. swirskii showed negative AT-skew and positive GC-skew, while Coleolaelaps cf. liui, Blattisocius keegani and Hypoaspis linteyini showed negative AT-skew and GC-skew. The remaining mites maintained base skew characteristic of the mitochondrial genome of Acari. Analysing from parasitic host, living environment, life history and other factors, AT skew value does not seem to show significant species specificity, so the mechanisms responsible for differences in AT skew between the same genera need to be further explored in the next studies.
The 2 rRNAs of the ancestral arthropods are both located in the N strand and contain an rrnL-trnV-rrnS gene cluster, but rrnS is located in the J strand while rrnL is located in the N strand in the E. echidninus mitochondrial genome, due to a translocation and inversion of rrnS. This phenomenon is relatively rare in the mitochondrial genomes of Gamasida, and is currently only found in V. destructor from Japan (Harada et al., Reference Harada, Yoshioka, Okuyama, Kato, Martin and Takahashi2020) and E. echidninus studied here. The average length of mitochondrial tRNAs was 54.8 ± 1 bp in the Acariformes and 62.0 ± 1.3 bp in the Parasitiformes (Yuan et al., Reference Yuan, Wei, Wang, Dou and Wang2010). The average length of the 22 tRNA genes of E. echidninus is 62 ± 0.8 bp, with trnC (50 bp) being the shortest and trnQ (68 bp) and trnI (68 bp) being the longest. All tRNAs have typical cloverleaf secondary structure, with the exception of trnC and trnS1, which lack the D-arm (Fig. 2). The absence of the D-arm of trnS1 is an ancestral feature of the Acari mitochondrial genome (Wolstenholme, Reference Wolstenholme1992). The missing D-arm of trnC is reported for the first time in Echinolaelaps and may be of some reference value for succeeding studies. Typically, mitochondrial tRNAs in mites retain the universal anticodons of Arachnida mitochondrial tRNAs, but E. echidninus studied here show trnK (UUU to CUU) and most of the species of Gamasida show trnK (UUU to CUU) and/or trnS1 (UCU to GCU), which may be a synapomorphy of Gamasida.
The start codons of 13 protein-coding genes are ATN and the stop codons are TAA or TAG, encode a total of 3632 amino acids. The frequency of UUU, UUA, AUU, AUA and AAU was considered to be high (>150 times) according to the analysis of synonymous codon usage (Table 2). Twenty-eight codons, including UUA, AGA, UCU, CCU and GCU, were preferred codons, with RSCU >1. The codons for Ile, Leu, Phe and Ser were the most frequently used (1780 times in total), accounting for 49.0% of the total codon usage. Codon preference can reveal the proximity of species relatedness and is used in the taxonomic study of species (Sharp and Li, Reference Sharp and Li1986; Kokate et al., Reference Kokate, Techtmann and Werner2021), so to a certain extent codon preference can reveal the taxonomic relationships of species. Based on the analysis of evolutionary rates of 13 protein-coding genes in T. tridentatus (Fig. 3), cox2, atp6, nad1, cox3, nad4 and nad5, the ratio of non-synonymous evolutionary rate to synonymous evolutionary rate (Ka/Ks) was >1, demonstrating that these 6 protein-coding genes are subject to positive selection, and because of this positive selection, the species are able to continuously improve their adaptability to the environment. The ratio of non-synonymous to synonymous evolutionary rates (Ka/Ks) for the remaining protein-coding genes is <1, suggesting that these protein-coding genes are subject to negative selection, and the effect of negative selection is to restrict mutations in mitochondrial genes so that oxidative phosphorylation-related protein functions in mitochondria are performing their functions in a consistent manner (Hurst, Reference Hurst2002; Wang et al., Reference Wang, Zhong, Fu, Dou and Bian2014).
In Gamasida, with the exception of Parasitidae and Diplogyniidae, which retain the ancestral arthropod mitochondrial genome arrangement pattern, other species show rearrangements with varying degrees. The breakpoint distance analysis (Table 3) (M. occidentalis and E. nicholsi were not calculated due to duplicated mitochondrial genes) revealed that the breakpoint distances of A. tsugawai and A. swirskii are the highest (33) compared to the ancestral arthropods, while the breakpoint distances of M. muscaedomesticae and S. rarior are the lowest (7). The phylogenetic analysis of Gamasida (Fig. 4) revealed that E. echidninus and V. destructor, A. tsugawai and A. swirskii, Blattisocius tarsalis and B. keegani, C. cf. liui and H. linteyini, Ptilonyssus chloris and Tinaminyssus mello, Macrocheles glaber and M. muscaedomesticae, P. wangdunqingi and P. fimetorum, Quadristernoseta cf. longigynium and Quadristernoseta cf. intermedia formed sister branches and the breakpoint distances between each sister branch were 13, 15, 11, 6, 0, 2, 0, 0, respectively. This suggests a different degree of mitochondrial genome rearrangement within species that form sister branches, with more closely related species maintaining a relatively constant pattern of gene arrangement (e.g. P. chloris and T. mello, M. glaber and M. muscaedomesticae, P. wangdunqingi and P. fimetorum, Q. cf. longigynium and Q. cf. intermedia), but also rapid gene rearrangements in a relatively short evolutionary time (e.g. E. echidninus and V. destructor, A. tsugawai and A. swirskii, B. tarsalis and B. keegani, C. cf. liui and H. linteyini). However, the breakpoint distances of mitochondrial genome between species that form sister branches are smaller (<15) compared to other species of Gamasida. The similarity of breakpoint distances between species of the same family or genus may be a typical feature of specific taxa (e.g. Phytoseiidae, Macrochelidae). Phylogenetic analysis supported gene rearrangement breakpoint analyses within Gamasida: E. echidninus was sister of V. destructor and showed a moderate number of breakpoints (13). Therefore, mitochondrial genome rearrangements may occur regularly with evolutionary divergence in Gamasida. There are certain similarities in rearrangement patterns and breakpoints distance in closely related taxonomic level.
Echinolaelaps echidninus belongs to Dermanyssoidea, so their breakpoint distances and mitochondrial genome arrangement patterns are similar to those of other species in Dermanyssoidea. Whilst 13 protein-coding genes retained the ancestral arthropod arrangement order, tRNA and rRNA show variant degrees of rearrangement. The analysis of the phylogenetic tree (Fig. 4) revealed that E. echidninus clustered into a taxon with other species of Dermanyssoidea (H. linteyini, C. cf. liui, V. destructor, Dermanyssus gallinae, P. chloris, T. mello), but did not form a sister branch with other species of Laelapidae (H. linteyini, C. cf. liui) that were more closely related morphologically, instead of forming a sister group with V. destructor, indicating a close kinship between V. destructor and E. echidninus at the molecular level. This is not only the concentration of the complete mitochondrial genome arrangement pattern, but also is a further confirmation at the phylogenetic level. In terms of morphological classification, both E. echidninus and V. destructor belong to the large gamasid mite, but they belong to different families, with morphological characteristics of E. echidninus being more similar to those of species in Laelapidae. However, phylogenetic analyses at the molecular level have shown that E. echidninus is more closely related to V. destructor. Casanueva suggested that Varroidae was synonymized into Laelapidae (Casanueva, Reference Casanueva1993), which is consistent with the results of the phylogenetic tree in this paper. It seems to suggest that a single morphological classification is not sufficient in the taxonomic study of Acari and that strong molecular evidence is needed to provide a more complete explanation of their taxonomic and phylogenetic relationships.
Conclusion
Structure and evolution of the E. echidninus complete mitochondrial genome were reported for the first time in this study. It provides novel insights into rearrangement patterns and evolution of the complete mitochondrial genomes of Gamasida. The complete mitochondrial genome of the mite has 37 genes typical of metazoans, including 13 protein-coding genes, 22 tRNA genes and 2 rRNA genes. It is unusual for the E. echidninus complete mitochondrial genome structure with zero AT-skew and positive GC-skew, unlike the complete mitochondrial genome structure of Gamasida. The mitochondrial gene arrangement patterns and breakpoint distances indicated that the complete mitochondrial genomes of E. echidninus show a novel arrangement pattern. Phylogenetic relationships analysis of Gamasida showed that E. echidninus is located in the second branch and clustered into a taxon with other species of Dermanyssoidea (H. linteyini, C. cf. liui, V. destructor, D. gallinae, P. chloris, T. mello), the close kinship between V. destructor and E. echidninus. To obtain a more reliable phylogenetic tree, we still need to collect more representative species of Gamasida, to sequence the complete mitochondrial genomes of more species, and to study evolutionary mechanisms and rearrangement patterns of Gamasida in depth.
Data availability
All data generated or used during the study appear in the submitted article.
Acknowledgements
We are grateful to Professor Guo Xian-Guo for supporting this project by providing us specimens.
Author's contribution
Wenge Dong and Bili Yuan designed and conducted the research. Wenge Dong and Bili Yuan contributed reagents and materials. Wenge Dong, Bili Yuan and Gangxian He analysed the data. Bili Yuan and Wenge Dong wrote the manuscript. All the authors have read and approved the final manuscript.
Financial support
We acknowledge the funding support from the National Natural Science Foundation of China (No. 32060143 to Wenge Dong) and the Expert work station for Dao-Chao Jin in Dali Prefecture.
Conflict of interest
None.
Ethical standards
All methods and procedures used in the capture of rodent process were in accordance with the guidelines and regulations approved by the Animal Ethics Committees at Dali University. Approval ID is MECDU-201806-11.








 Open access
Open access