


INTRODUCTION
Protozoan parasites, including Plasmodium spp., Trypanosoma spp. and Leishmania spp., are responsible for diseases that cause significant morbidity, mortality and economic burden, predominantly in developing countries. There are currently no effective vaccines available for the prevention of these tropical diseases, including malaria, human African trypanosomiasis (HAT), Chagas disease and leishmaniasis, and therapy relies heavily on antiprotozoal drugs (Pink et al. Reference Pink, Hudson, Mouries and Bendig2005; Anthony et al. Reference Anthony, Burrows, Duparc, Moehrle and Wells2012; Barrett and Croft, Reference Barrett and Croft2012). New antiprotozoals are urgently required, as most existing drugs suffer from one or more liabilities related to poor efficacy, drug resistance, toxicity, high cost or unsuitable pharmacokinetic properties (Wells et al. Reference Wells, Alonso and Gutteridge2009; Phillips, Reference Phillips2012).
The lack of suitable treatments for these tropical diseases is partly due to there having been inadequate pharmaceutical research efforts during the last century (Renslo and McKerrow, Reference Renslo and McKerrow2006; Wells et al. Reference Wells, Alonso and Gutteridge2009). The majority of current antiprotozoal drugs did not arise from programmes common in contemporary pharmaceutical development, and the rigorous pre-clinical and clinical evaluation that is now expected of new drugs did not accompany registrations of most of the antiprotozoals (Renslo and McKerrow, Reference Renslo and McKerrow2006). Indeed, many commonly used antiprotozoal drugs act by unknown mechanisms, which severely impedes optimal clinical utilization and monitoring for efficacy, toxicity and resistance.
Fortunately, the pipeline for new antiprotozoal drugs now looks promising as a result of recent efforts to screen large compound libraries against these pathogenic parasites. Hundreds of ‘hit’ compounds have been identified that inhibit the in vitro proliferation of specific protozoan parasites (Gamo et al. Reference Gamo, Sanz, Vidal, de Cozar, Alvarez, Lavandera, Vanderwall, Green, Kumar, Hasan, Brown, Peishoff, Cardon and Garcia-Bustos2010; Guiguemde et al. Reference Guiguemde, Shelat, Bouck, Duffy, Crowther, Davis, Smithson, Connelly, Clark, Zhu, Jiménez-Díaz, Martinez, Wilson, Tripathi, Gut, Sharlow, Bathurst, Mazouni, Fowble, Forquer, McGinley, Castro, Angulo-Barturen, Ferrer, Rosenthal, DeRisi, Sullivan, Lazo, Roos, Riscoe, Phillips, Rathod, Van Voorhis, Avery and Guy2010; Duffy and Avery, Reference Duffy and Avery2012; Sykes et al. Reference Sykes, Baell, Kaiser, Chatelain, Moawad, Ganame, Ioset and Avery2012). It is clear that screening compound libraries against organisms is superior to primary screens against individual targets, possibly due to the fact that key pharmacological criteria such as membrane-permeability are already built into compounds passing the former type of screen. A major bottleneck in the hit-to-lead optimization pipeline following identification of hits in whole organism screens relates to the lack of mechanistic information concerning their modes of action (Guiguemde et al. Reference Guiguemde, Shelat, Garcia-Bustos, Diagana, Gamo and Guy2012). Identification of target(s) for hit compounds allows rational medicinal chemistry to improve selectivity of binding to the parasite target, and ensures that structural modifications to enhance the pharmacokinetic and toxicity profiles are not likely to compromise activity. The availability of methods to identify modes of action for antiprotozoal compounds in an untargeted manner would greatly enhance the efficiency of drug discovery for parasitic diseases.
Metabolomics is an emerging technology that provides an untargeted overview of cellular metabolism by the simultaneous detection and relative quantification of hundreds of small molecules (<1500 Da) in a biological system (Scalbert et al. Reference Scalbert, Brennan, Fiehn, Hankemeier, Kristal, van Ommen, Pujos-Guillot, Verheij, Wishart and Wopereis2009; Scheltema et al. Reference Scheltema, Decuypere, t'Kindt, Dujardin, Coombs and Breitling2010; Creek et al. Reference Creek, Anderson, McConville and Barrett2012a; Dunn et al. Reference Dunn, Erban, Weber, Creek, Brown, Breitling, Hankemeier, Goodacre, Neumann, Kopka and Viant2012). Metabolomic profiling of protozoan parasites treated with antiprotozoal compounds can detect drug-induced changes to parasite metabolism and identify the individual metabolites and pathways that are directly perturbed. This provides a rapid and unbiased method to discover likely drug targets (Beyoğlu and Idle, Reference Beyoğlu and Idle2013).
The untargeted nature of metabolomics has allowed rapid and unbiased classification of numerous antimicrobial compounds according to their modes of action (Gao et al. Reference Gao, Shi, Tian, Shi, Yuan, Lu and Xu2007; Liu et al. Reference Liu, Wen, Wang, Li and Xu2009; Halouska et al. Reference Halouska, Fenton, Barletta and Powers2012). Investigation of the metabolic response of Staphylococcus aureus to triphenylbismuthdichloride revealed pyruvate dehydrogenase as a target for this novel antibiotic (Birkenstock et al. Reference Birkenstock, Liebeke, Winstel, Krismer, Gekeler, Niemiec, Bisswanger, Lalk and Peschel2012). A more detailed elucidation of the mechanism of antibacterial action was demonstrated by isotope-labelled metabolic flux profiling of Escherichia coli following antifolate exposure (Kwon et al. Reference Kwon, Lu, Melamud, Khanam, Bognar and Rabinowitz2008). Trimethoprim is a known inhibitor of dihydrofolate reductase (DHFR), which was confirmed by accumulation of oxidized folates in the treated cells. However, an unexpected inhibiton of folylpoly-gamma-glutamate-synthetase was also observed; a response to dihydrofolate accumulation (Kwon et al. Reference Kwon, Lu, Melamud, Khanam, Bognar and Rabinowitz2008). This systems-based approach allows observation of pharmacological effects that often go unnoticed in classical biochemical or genetic approaches based on individual candidate drug targets. Here we discuss the development of platforms that have enabled measurement of hundreds of metabolites in protozoa and their application to drug-target identification.
METABOLOMICS METHODOLOGY
Metabolomics studies fall into two broad categories, targeted and untargeted. Targeted studies involve hypothesis-driven experiments that aim to provide accurate quantification of a subset of known metabolites, usually restricted to a particular metabolic pathway or chemical class. Untargeted studies by contrast are usually hypothesis-generating experiments that provide relative quantification of all detectable metabolite signals, with subsequent identification of the most significant metabolites (Dunn et al. Reference Dunn, Erban, Weber, Creek, Brown, Breitling, Hankemeier, Goodacre, Neumann, Kopka and Viant2012). Untargeted studies are particularly attractive for the investigation of drugs of unknown mode of action. Targeted studies inherently provide less coverage of the metabolome, but have the advantage of enabling accurate quantification as well as flux analysis to determine dynamic responses in candidate pathways (Kwon et al. Reference Kwon, Lu, Melamud, Khanam, Bognar and Rabinowitz2008).
Sample preparation
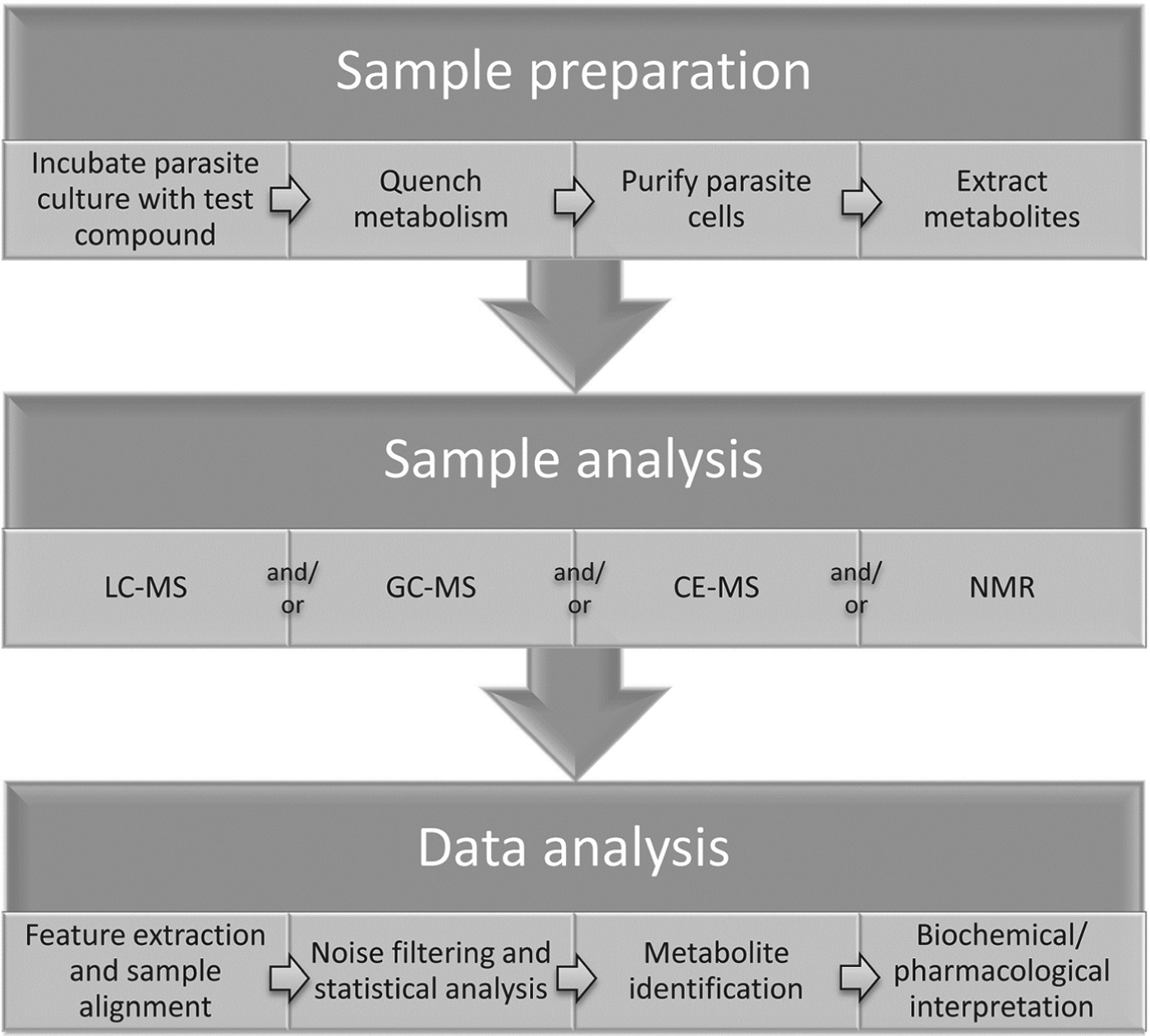
Measurements of metabolic responses to drugs are often obtained from in vitro cell culture systems (Fig. 1). This approach allows maximum control over genetic and environmental variables, such as temperature, pH, atmosphere, extracellular environment and cell density, which can contribute to unwanted metabolic alterations and obscure the desired observation of drug-specific effects (Cuperlovic-Culf et al. Reference Cuperlovic-Culf, Barnett, Culf and Chute2010). The choice of culture media may interfere with drug action and/or metabolic responses, particularly when a standard culture medium does not mimic the in vivo environment of the parasite. For example, the high folate levels present in standard Trypanosoma brucei culture medium, HMI11 (Hirumi and Hirumi, Reference Hirumi and Hirumi1989), inhibits the in vitro activity of antifolate compounds that demonstrate trypanocidal activity in vivo (Sienkiewicz et al. 2008), suggesting that culture media should be modified to more closely reflect in vivo conditions wherever possible. In addition, the impact of drug treatment on parasite growth and viability can often alter metabolite abundances, independent of the molecular target of the drug. Multiple controls, time courses and drug concentrations are often necessary to delineate mechanism-related metabolic alterations from non-specific stress responses (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012).

Fig. 1. General outline of methodology for metabolomics studies of protozoan parasites in cell culture.
Intracellular parasites present unique challenges, and isolation of parasites from uninfected host cells should be performed to avoid interference from host cell metabolites (e.g. saponin lysis or MACS purification for Plasmodium falciparum) (Zhang et al. Reference Zhang, Watts, Hodge, Kemp, Hunstad, Hicks and Odom2011; Biagini et al. Reference Biagini, Fisher, Shone, Mubaraki, Srivastava, Hill, Antoine, Warman, Davies, Pidathala, Amewu, Leung, Sharma, Gibbons, Hong, Pacorel, Lawrenson, Charoensutthivarakul, Taylor, Berger, Mbekeani, Stocks, Nixon, Chadwick, Hemingway, Delves, Sinden, Zeeman, Kocken, Berry, O'Neill and Ward2012), although it is intriguing to consider how impacts on host cell metabolism can contribute to the pharmacological effect of a drug. For example, human cytomegalovirus was shown to lead to up-regulation of host cell fatty acid biosynthesis and inhibitors of fatty acid biosynthesis could inhibit viral growth (Munger et al. Reference Munger, Bennett, Parikh, Feng, McArdle, Rabitz, Shenk and Rabinowitz2008). Metabolomic analysis of uninfected host-cell response to drug treatment is also recommended to confirm the parasite-specificity of drug-induced metabolic changes. Care must be taken to avoid metabolic perturbation during any purification step, and metabolic quenching is often achieved by rapid cooling to 0 °C (Saunders et al. Reference Saunders, Ng, Chambers, Ng, Naderer, Kramer, Likic and McConville2011). Following quenching and isolation of cells, metabolites are generally extracted with organic solvent mixtures such as chloroform/methanol/water (1:3:1) (Scheltema et al. Reference Scheltema, Decuypere, t'Kindt, Dujardin, Coombs and Breitling2010; t'Kindt et al. Reference t'Kindt, Jankevics, Scheltema, Zheng, Watson, Dujardin, Breitling, Coombs and Decuypere2010b).
Sample analysis
Metabolomics analysis is generally performed with mass spectrometry or NMR spectroscopy. Mass spectrometry is usually coupled to chromatographic separation such as gas chromatography (GC-MS), capillary electrophoresis (CE-MS) or liquid chromatography (LC-MS) (Dunn et al. Reference Dunn, Bailey and Johnson2005; Scalbert et al. Reference Scalbert, Brennan, Fiehn, Hankemeier, Kristal, van Ommen, Pujos-Guillot, Verheij, Wishart and Wopereis2009; Scheltema et al. Reference Scheltema, Decuypere, t'Kindt, Dujardin, Coombs and Breitling2010; Creek et al. Reference Creek, Anderson, McConville and Barrett2012a).
LC-MS with high resolution mass spectrometry is becoming the most widely used metabolomics platform due to its high sensitivity, broad specificity and the ability to annotate or identify hundreds of polar and non-polar metabolites from a wide range of metabolic classes (Theodoridis et al. Reference Theodoridis, Gika, Want and Wilson2012). Reversed phase chromatography is well established, and is particularly suitable for analysis of lipids and other non-polar metabolites (Theodoridis et al. Reference Theodoridis, Gika, Want and Wilson2012). Hydrophilic interaction chromatography (HILIC) is finding increasing application in metabolomics, with particular advantages for the separation and identification of polar metabolites (Cubbon et al. Reference Cubbon, Antonio, Wilson and Thomas-Oates2009; Zhang et al. Reference Zhang, Creek, Barrett, Blackburn and Watson2012). High resolution mass spectrometry is critical for the detection and identification of metabolites in untargeted studies. Recent advances in mass spectrometry have enabled widespread application of untargeted metabolomics, as most new time-of-flight (TOF) and Fourier transform (Orbitrap and FT-ICR) mass spectrometers routinely achieve the required resolution (>30 000) and mass accuracy (<5 ppm) (Dunn et al. Reference Dunn, Erban, Weber, Creek, Brown, Breitling, Hankemeier, Goodacre, Neumann, Kopka and Viant2012; Theodoridis et al. Reference Theodoridis, Gika, Want and Wilson2012).
Data analysis
Computational approaches to metabolomics data analysis are essential, as modern metabolomics platforms routinely detect tens of thousands of ion signals per sample. Many detected peaks arise from noise, contaminants and LC-MS artefacts, and extensive data processing is necessary to allow meaningful interpretation of results (Jankevics et al. Reference Jankevics, Merlo, de Vries, Vonk, Takano and Breitling2011; Kuhl et al. Reference Kuhl, Tautenhahn, Böttcher, Larson and Neumann2011; Creek et al. Reference Creek, Jankevics, Burgess, Breitling and Barrett2012b; Weber et al. Reference Weber, Li, Bruty, He and Viant2012). Numerous open-source software packages are available to provide high-throughput detection, alignment, quantification, filtering and/or identification of metabolite signals, including XCMS, Metalign, MZmine, mzMatch and IDEOM (Smith et al. Reference Smith, Want, O'Maille, Abagyan and Siuzdak2006; Lommen, Reference Lommen2009; Pluskal et al. Reference Pluskal, Castillo, Villar-Briones and Oresic2010; Scheltema et al. Reference Scheltema, Jankevics, Jansen, Swertz and Breitling2011; Creek et al. Reference Creek, Jankevics, Burgess, Breitling and Barrett2012b). Pre-processed metabolomics data provide relative metabolite abundances for hundreds to thousands of putative metabolites for interrogation by univariate or multivariate statistics (Liland, Reference Liland2011; Vinaixa et al. Reference Vinaixa, Samino, Saez, Duran, Guinovart and Yanes2012).
The major bottleneck for interpretation of untargeted metabolomics data is metabolite identification (Dunn et al. Reference Dunn, Erban, Weber, Creek, Brown, Breitling, Hankemeier, Goodacre, Neumann, Kopka and Viant2012). Many metabolites can be putatively annotated by matching accurate masses to the exact mass of metabolites in biochemical databases. However, accurate metabolite identification is often hampered by metabolites with the same (isomers) or similar (within the error margin of the MS) mass (Scheltema et al. Reference Scheltema, Decuypere, t'Kindt, Dujardin, Coombs and Breitling2010). Confirmation of identification requires orthogonal data such as fragmentation spectra (MS/MS) and chromatographic retention time, and should be compared to authentic standards (Sumner et al. Reference Sumner, Amberg, Barrett, Beale, Beger, Daykin, Fan, Fiehn, Goodacre, Griffin, Hankemeier, Hardy, Harnly, Higashi, Kopka, Lane, Lindon, Marriott, Nicholls, Reily, Thaden and Viant2007). Unfortunately, many metabolite standards are not easily accessible, and detected metabolites are often reported as putative, or unknown, identities. Isolation and characterization of unidentified metabolites is labour intensive, and is generally only performed for a few metabolites that show statistical significance in a particular study.
Advanced biochemical interpretation of metabolomics studies can be achieved by software solutions that allow mapping of metabolite levels on to metabolic pathways (Paley and Karp, Reference Paley and Karp2006; Jourdan et al. Reference Jourdan, Cottret, Huc, Wildridge, Scheltema, Hillenweck, Barrett, Zalko, Watson and Debrauwer2010; Leader et al. Reference Leader, Burgess, Creek and Barrett2011; Yamada et al. Reference Yamada, Letunic, Okuda, Kanehisa and Bork2011). The bottlenecks associated with metabolite identification limit the usefulness of these approaches, and pathway-based analyses are best suited to targeted metabolomics studies. A compromise approach, which combines untargeted metabolomics with retention time data from authentic standards to improve the confidence of putative metabolite identifications (Creek et al. Reference Creek, Jankevics, Breitling, Watson, Barrett and Burgess2011), is supported by the IDEOM software (Creek et al. Reference Creek, Jankevics, Burgess, Breitling and Barrett2012b). Interpretation of data in the context of metabolic pathways may also improve the accuracy of metabolite identification (Rogers et al. Reference Rogers, Scheltema, Girolami and Breitling2009; Weber and Viant, Reference Weber and Viant2010).
METABOLOMICS FOR INVESTIGATING THE MODE OF ACTION OF ANTIPROTOZOAL DRUGS
Trypanocidal compounds
T. brucei is the causative agent of HAT, otherwise known as sleeping sickness. Infection occurs following the bite of a Tsetse fly, and parasites remain in the bloodstream during stage 1 disease, where symptoms are minor and non-specific (Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007). Stage 2 disease occurs after parasites enter the central nervous system, leading to neurological complications including disturbed sleep-wake patterns. The infection is inevitably fatal if untreated. Five compounds are approved for the treatment of HAT, but each has serious limitations (Brun et al. Reference Brun, Don, Jacobs, Wang and Barrett2011; Barrett and Croft, Reference Barrett and Croft2012). Suramin and pentamidine must be given by injection and are only effective during stage 1 disease because they do not penetrate the blood-brain barrier. Melarsoprol is active against both stage 1 and stage 2 disease, but this arsenical compound is particularly toxic, with 5–10% of patients suffering from reactive encephalopathy, which is often fatal. Eflornithine alone, or in combination with nifurtimox is also active against stage 2 CNS disease (Barrett et al. Reference Barrett, Boykin, Brun and Tidwell2007; Phillips, Reference Phillips2012). Clinical utilization of eflornithine may be limited by the requirement for administration of large doses by intravenous infusion, and there is concern that resistance to these drugs may develop rapidly (Vincent et al. Reference Vincent, Creek, Watson, Kamleh, Woods, Wong, Burchmore and Barrett2010). Eflornithine is the only compound among the five approved drugs that has a well-defined mode of action, which involves inhibition of ornithine decarboxylase (ODC) and subsequent depletion of polyamines (Grishin et al. Reference Grishin, Osterman, Brooks, Phillips and Goldsmith1999).
The mode of action of eflornithine was recently confirmed using untargeted metabolomics, providing a proof of concept that untargeted metabolomic profiling of protozoan cell cultures is a powerful tool for the unbiased determination of mode of action for antiprotozoal compounds (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012). Eflornithine induced significant accumulation of ornithine (the substrate for ODC), and depletion of putrescine (the product of ODC), consistent with direct inhibition of ODC by the drug (Fig. 2). The only other metabolites to consistently show marked changes in abundance were the acetylated forms of ornithine and putrescine. This study also demonstrated a significant depletion of spermidine, the downstream product of the polyamine pathway. Interestingly, only minor down-regulation was observed for trypanothione, the spermidine-bis-glutathione conjugate responsible for antioxidant activity in trypanosomatids, suggesting that the trypanocidal action of eflornithine is more likely related to polyamine depletion than disruption of the antioxidant defence system. Additional metabolite changes were detected after 48 h incubation with higher drug concentrations, highlighting the importance of metabolomic measurements across a dose-range and time-course to delineate direct drug mechanisms from downstream metabolic responses that may include non-specific cell death pathways (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012).
Fig. 2. Metabolomic response of Trypanosoma brucei to eflornithine over a 72 h incubation time-course (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012). (A) Heat map shows the relative change in abundance of all putative metabolites following incubation with eflornithine. Putative metabolites are ranked according to fold-change at 72 h. The most significant increases and decreases correspond to metabolites of the polyamine pathway. (B) Metabolite abundance profiles of polyamine pathway metabolites showing accumulation of metabolites upstream of ornithine decarboxylase (ODC), and depletion of downstream metabolites. Y-axes represent peak heights from LC-MS data.
Metabolomic analysis of nifurtimox-treated cells revealed a complex pattern of metabolic perturbations, which could not be interpreted as inhibition of a single metabolic enzyme (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012). Altered abundances of nucleotide and glycolytic metabolites were consistent with previous hypotheses that implicate nucleic acid interactions and oxidative stress as likely mediators of nifurtimox activity. One advantage of the untargeted approach was the detection of nifurtimox metabolites that support the likely activation mechanism by T. brucei type-1 nitroreductase (Hall et al. Reference Hall, Bot and Wilkinson2011). In contrast to previous hypotheses, nifurtimox was found to lack synergistic activity with eflornithine, and metabolomics data support this lack of synergism (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012).
The application of metabolomics to experimental trypanocidal compounds can provide a method to elucidate attractive drug targets for future drug discovery. The impact of fluorinated pyrimidines on T. brucei metabolism was recently described (Ali et al. Reference Ali, Creek, Burgess, Allison, Field, Mäser and De Koning2013). 5-fluoro-2′-deoxyuridine resulted in specific accumulation of dUMP in wild-type cells, and minimal change in resistant cells, demonstrating inhibition of thymidylate synthase, most likely by the active metabolite, 5-fluoro-dUMP. In contrast, 5-fluorouracil and 5-fluoroorotic acid were incorporated into the uracil nucleotide pool and then into parasite RNA, revealing a distinct mode of action for the fluoropyrimidines compared to the fluorodeoxynucleoside (Ali et al. Reference Ali, Creek, Burgess, Allison, Field, Mäser and De Koning2013). The metabolomics platform has recently been applied to a variety of currently used and experimental trypanocidal drugs and work is following up on potential modes of action identified in this way.
Antileishmania compounds
Leishmania spp. are trypanosomatid parasites responsible for a spectrum of diseases that include visceral, cutaneous and mucocutaneous manifestations of leishmaniasis. The infection is transmitted by sand-fly bite, and parasites develop mostly within macrophages of the mammalian host (Naderer and McConville, Reference Naderer and McConville2008). Treatment options for leishmaniasis are severely limited, and the commonly used treatments, pentavalent antimonials, amphotericin B, miltefosine and paromomycin, have poor safety, efficacy and pharmacokinetic profiles (Barrett and Croft, Reference Barrett and Croft2012). The mode of action of paromomycin has been investigated by proteomics and, like other aminoglycosides, appears to act by inhibition of protein synthesis (Chawla et al. Reference Chawla, Jhingran, Panigrahi, Stuart and Madhubala2011). Amphotericin B and miltefosine are thought to interact with sterols and phospholipid metabolism, respectively (Lux et al. Reference Lux, Heise, Klenner, Hart and Opperdoes2000; Ouellette et al. Reference Ouellette, Drummelsmith and Papadopoulou2004; Singh et al. Reference Singh, Kumar and Singh2012). It is expected that metabolomic or lipidomic studies would be able to confirm the impact of these drugs on lipid metabolism pathways to further characterize the modes of action and resistance.
The mode of action of antimonial compounds is a matter of debate, and is thought to be mediated primarily by oxidative stress. A metabolomics approach using CE-MS demonstrated significant perturbation of sulphur-containing amino acids and polyamine pathway metabolites following treatment of Leishmania infantum promastigotes with Sb(III) (Canuto et al. Reference Canuto, Castilho-Martins, Tavares, López-Gonzálvez, Rivas and Barbas2012). This finding is consistent with depletion of trypanothione, the main metabolite responsible for redox homeostasis in trypanosomatids. The mode of resistance to antimonials has also been studied by metabolomics with LC-MS, which revealed significant alterations in the membrane lipid composition of resistant L. donovani field isolates (t'Kindt et al. Reference t'Kindt, Scheltema, Jankevics, Brunker, Rijal, Dujardin, Breitling, Watson, Coombs and Decuypere2010a).
Antimalarial compounds
Plasmodium spp. are the infectious agents responsible for malaria, with P. falciparum and P. vivax the most common causative agents of human disease. Plasmodium parasites undergo a complex life-cycle, and are spread by the bite of an infectious mosquito, with the symptomatic phase of disease occurring during replication of asexual parasites within red blood cells of the human host. Numerous drugs are approved for treatment of malaria, although resistance has emerged to most antimalarials (Dondorp et al. Reference Dondorp, Nosten, Yi, Das, Phyo, Tarning, Lwin, Ariey, Hanpithakpong, Lee, Ringwald, Silamut, Imwong, Chotivanich, Lim, Herdman, An, Yeung, Singhasivanon, Day, Lindegardh, Socheat and White2009; White, Reference White2012). Artemisinin combination therapies (ACTs) are currently recommended due to their potent activity and limited evidence of clinical resistance to date (Anthony et al. Reference Anthony, Burrows, Duparc, Moehrle and Wells2012; Meshnick, Reference Meshnick2012). Nevertheless, new antimalarials are urgently required, as ACTs suffer from emerging resistance, high cost and a pharmacokinetic mismatch between short-acting artemisinins and long-acting partner drugs, usually quinolines (Nosten and White, Reference Nosten and White2007; Anthony et al. Reference Anthony, Burrows, Duparc, Moehrle and Wells2012). Artemisinins are thought to act by production of free radicals following exposure of the peroxide pharmacophore to Fe(II) iron and/or haem, which is released as a by-product of haemoglobin digestion within the parasite (O'Neill et al. Reference O'Neill, Barton and Ward2010). The quinoline antimalarials are also thought to act by interaction with intraparasitic haem, and preventing its detoxification (Loria et al. Reference Loria, Miller, Foley and Tilley1999). However, the precise mode of action of most antimalarials remains a matter of debate (Olliaro, Reference Olliaro2001; O'Neill et al. Reference O'Neill, Barton and Ward2010). The antifolate antimalarials are known to act by inhibition of DHFR and dihydropteroate synthase (DHPS), but the clinical application of these antifolates is limited by widespread resistance. Nevertheless, characterization of the target enzyme, DHFR, from wild-type and resistant parasites, has lead to the design of novel antimalarial leads that exhibit potent activity against the resistant enzyme (Yuthavong et al. Reference Yuthavong, Tarnchompoo, Vilaivan, Chitnumsub, Kamchonwongpaisan, Charman, McLennan, White, Vivas, Bongard, Thongphanchang, Taweechai, Vanichtanankul, Rattanajak, Arwon, Fantauzzi, Yuvaniyama, Charman and Matthews2012). This demonstrates the potential benefit of understanding the targets of current drugs as a paradigm for new drug discovery.
Atovaquone is another antimalarial with a known mode of action, inhibition of the cytochrome bc1 complex in the mitochondrial electron transport chain. This results in loss of the mitochondrial membrane potential and subsequent disruption of pyrimidine synthesis by indirect inhibition of the essential enzyme dihydroorotate dehydrogenase (DHODH). The mode of action of atovaquone has been confirmed by a targeted metabolomics study of P. falciparum, which demonstrated significant accumulation of dihydroorotate, the substrate of DHODH, and its precursor, carbamoyl-L-aspartate (Biagini et al. Reference Biagini, Fisher, Shone, Mubaraki, Srivastava, Hill, Antoine, Warman, Davies, Pidathala, Amewu, Leung, Sharma, Gibbons, Hong, Pacorel, Lawrenson, Charoensutthivarakul, Taylor, Berger, Mbekeani, Stocks, Nixon, Chadwick, Hemingway, Delves, Sinden, Zeeman, Kocken, Berry, O'Neill and Ward2012). The mode of action for the novel quinolone, CK-2-68, was also confirmed by observation of the same metabolic response (Biagini et al. Reference Biagini, Fisher, Shone, Mubaraki, Srivastava, Hill, Antoine, Warman, Davies, Pidathala, Amewu, Leung, Sharma, Gibbons, Hong, Pacorel, Lawrenson, Charoensutthivarakul, Taylor, Berger, Mbekeani, Stocks, Nixon, Chadwick, Hemingway, Delves, Sinden, Zeeman, Kocken, Berry, O'Neill and Ward2012).
Several compounds are currently under development for the treatment of malaria, and metabolomics studies may play an important role in defining the mode of action of new antimalarials. Fosmidomycin is an antibiotic that acts on Plasmodium parasites by inhibition of the essential non-mevalonate pathway of isoprenoid biosynthesis. A targeted metabolic profiling study of intermediates in the non-mevalonate isoprenoid synthesis pathway confirmed deoxyxylulose phosphate reductoisomerase (DXR) as the molecular target of the drug, and surprisingly, identified a second target in the pathway, methylerythritol phosphate cytidyltransferase (IspD) (Zhang et al. Reference Zhang, Watts, Hodge, Kemp, Hunstad, Hicks and Odom2011). The accumulation of all metabolites upstream of IspD suggests that inhibition of this enzyme, either directly by fosmidomycin or by accumulation of 2-C-methylerythrose 4-phosphate, is the primary mode of isoprenoid depletion in P. falciparum (Zhang et al. Reference Zhang, Watts, Hodge, Kemp, Hunstad, Hicks and Odom2011).
The response of P. falciparum in vitro cultures to polyamine pathway inhibitors, eflornithine and MDL73811, was investigated by targeted metabolomics combined with transcriptomics and proteomics. Polyamine depletion was confirmed as the primary mode of action, and unexpected accumulation of glutamate metabolites could be explained by transcriptional upregulation of ornithine aminotransferase in response to ornithine accumulation (van Brummelen et al. Reference van Brummelen, Olszewski, Wilinski, Llinas, Louw and Birkholtz2009).
FUTURE DIRECTIONS
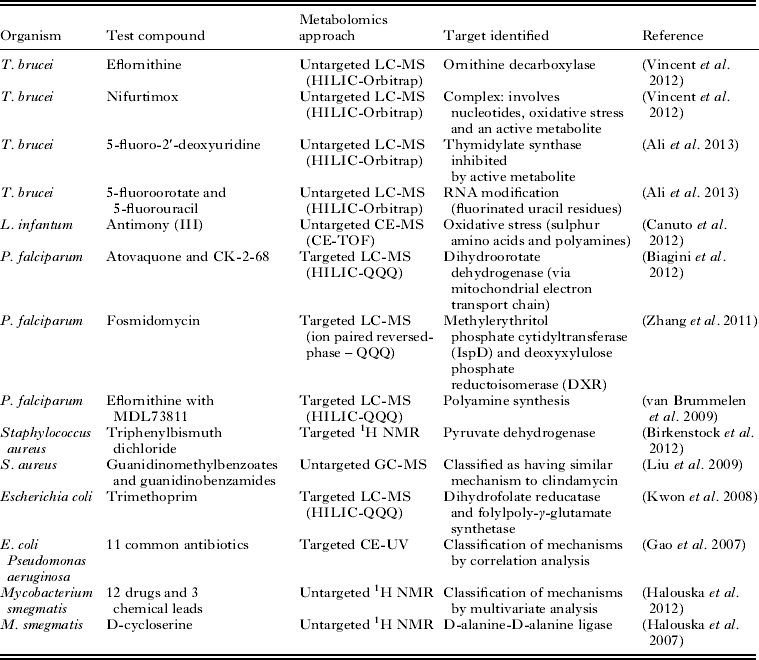
The suitability of metabolomics technology for determination of the mode of action of antiprotozoal compounds has been clearly demonstrated (Table 1). Further developments in technology will be necessary in order to expand the application of this approach efficiently to all the currently available antiprotozoal drugs, and the hundreds of compounds that display antiprotozoal activity in vitro. Whilst improvements in analytical hardware are expected to continue to expand metabolite coverage and increase sensitivity, the major issue limiting the accessibility of high-throughput metabolomics is data analysis. This situation is rapidly improving as evidenced by many recent advances in freely available data analysis software and web servers. However, it is expected that automated identification of metabolites will remain a significant bottleneck for untargeted metabolomics for the foreseeable future (Dunn et al. Reference Dunn, Erban, Weber, Creek, Brown, Breitling, Hankemeier, Goodacre, Neumann, Kopka and Viant2012).
Table 1. Examples of applications of metabolomics to determine the mode of action of drugs and other compounds with antimicrobial activity
The ability to relate changes in metabolite abundance to drug action remains largely dependent on a priori knowledge of metabolic pathways within the parasite. Unfortunately, many aspects of parasite metabolism have not been fully elucidated, and often rely on bioinformatic reconstructions of metabolic pathways based on genomic homology with other organisms. Many parasite genes are not annotated, or annotated as hypothetical proteins, and it is likely that some drugs will cause perturbations in metabolites from uncharacterized pathways. In these cases, untargeted metabolomics studies of drug action are likely to provide significant new discoveries in basic parasite biochemistry, as has already been demonstrated for polyamine acetylation (Vincent et al. Reference Vincent, Creek, Burgess, Woods, Burchmore and Barrett2012) and pyrimidine metabolism (Ali et al. Reference Ali, Creek, Burgess, Allison, Field, Mäser and De Koning2013) in T. brucei, and the two-step DXR reaction in P. falciparum isoprenoid biosynthesis (Zhang et al. Reference Zhang, Watts, Hodge, Kemp, Hunstad, Hicks and Odom2011).
The major advantage of untargeted metabolomics is the ability to identify a drug-mediated response at the biochemical level with no prior knowledge of the mode of action of a particular antiprotozoal compound. This method can be used to classify compounds according to their mode of action based on a metabolic signature and, in many cases, highlight the specific metabolic enzyme or pathway responsible for antiprotozoal activity. The primary limitation of this approach is that some drugs do not act by disruption of metabolic pathways. In addition, drugs may induce non-specific metabolic changes in response to stress, or as secondary responses to the initial drug action. Targeted metabolomics approaches are often required in these circumstances to provide a detailed characterization of the temporal and dose-dependent biochemical effects of drugs. Additional studies, such as transcriptomics, may also be required to delineate primary and secondary metabolic responses to drug treatment (van Brummelen et al. Reference van Brummelen, Olszewski, Wilinski, Llinas, Louw and Birkholtz2009). Generally, measurements of RNA or protein levels are unlikely to reveal the primary targets of drugs that inhibit metabolic enzymes, particularly in trypanosomatid parasites that exhibit polycistronic transcription. Nevertheless, transcriptomic and proteomic approaches are likely to detect the secondary cellular responses to drug treatment, which may be relevant to determination of potential resistance mechanisms. A systems biology approach, that combines metabolomics with other functional genomics tools, is a promising avenue to obtain comprehensive descriptions of the modes of action and resistance for antimicrobial drugs. Metabolomics may also be combined with chemical biology (e.g. protein or ligand microarrays (MacBeath et al. Reference MacBeath, Koehler and Schreiber1999) and chemoinformatic methods (e.g. similarity ensemble approach - Keiser et al. Reference Keiser, Irwin and Shoichet2010) to improve identification of unknown drug targets in an untargeted manner.
Metabolomics methods offer effective tools for the identification and characterization of mechanisms of drug action, which should optimize drug discovery by allowing detailed pre-clinical pharmacokinetic/pharmocodynamic descriptions of antiprotozoal drugs, and have the potential to translate into clinical studies to monitor both host and parasite responses to drug treatment (Beyoğlu and Idle, Reference Beyoğlu and Idle2013).
FINANCIAL SUPPORT
D. J. C. is supported by an NHMRC postdoctoral training fellowship. M. P. B. is partly supported by the Wellcome Trust through The Wellcome Trust Centre for Molecular Parasitology, which is supported by core funding from the Wellcome Trust (Grant 085349).


 Open access
Open access