


The resident microbiota that colonises the intestinal tract influences our health and nutrition via multiple routes. Most obvious is that it can act as a reservoir for infectious pathogens, but we also know that the more numerous non-pathogenic gut micro-organisms exert a sustained influence on the host's immune system and metabolism through interactions of microbial cell components and gene products (e.g. extracellular polysaccharides and flagella). Just as significant, however, is the combined metabolic activity of the resident gut microbiota, especially that arising from the very dense microbial community that occupies the large intestine. Here the largely fermentative metabolism of undigested food and of host-derived products creates a microbial metabolome (Fig. 1) that in turn impacts on the host metabolome that is detected in faeces, urine and blood( Reference Martin, Dumas and Wang 1 ). It is increasingly recognised that microbial metabolites have a major influence on host physiology. The major products of carbohydrate fermentation, the SCFA acetate, propionate and butyrate, provide energy sources for host tissues, but also exert anti-inflammatory and anti-apoptotic effects that may be important for the prevention of colorectal cancer and colitis. SCFA may influence the regulation of lipogenesis, whereas their interactions with host receptors have been linked to regulation of hormones that affect satiety( Reference Sleeth, Thompson and Ford 2 ). Furthermore, SCFA also play a major role in determining the gut environment, influencing pH, gut transit, nutrient uptake and microbial balance within the large intestine. Apart from SCFA there is a vast array of other potentially bioactive compounds that are generated through microbial activity in the gut, for example from protein, dietary phytochemicals and xenobiotics (including drugs).
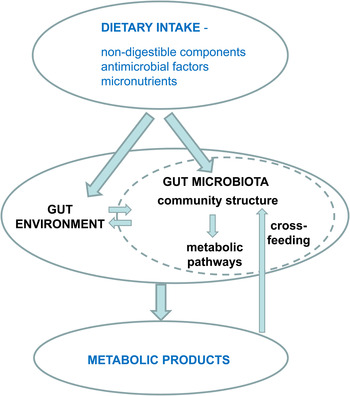
Fig. 1. Impact of diet on microbial metabolites.
The formation of microbial metabolites is strongly influenced by dietary intake, particularly of non-digestible dietary carbohydrates, protein and fat. This is dictated primarily by the chemical structures of the substrates themselves and the microbial pathways by which they are processed( Reference Macfarlane, Gibson, Mackie and White 3 ). It is becoming apparent however that many pathways are limited in their distribution among the various phylogenetic groups of bacteria that dominate the gut microbiota. This has the important implication that changes in the species composition of the gut microbiota, which could potentially come about as a result of individual variation or differences in diet or medication, may change the metabolic profile of the community. The composition of the gut microbiota changes and becomes increasingly complex following birth to adulthood. Within an individual adult, the intestinal microbiota is reasonably stable in composition, but there is potential for change in response to diet change, during periods of gastrointestinal disease and during antibiotic treatment. We will start here by considering recent evidence for the impact of diet upon microbiota composition.
Dietary modulation of gut microbiota composition
Microbial utilisation of dietary polysaccharides
Based on 16S rRNA-targeted molecular analyses, the majority of bacteria detected in faecal samples from healthy human volunteers belong to two phyla, Bacteroidetes and Firmicutes( Reference Suau, Bonnet and Sutren 4 – Reference Eckburg, Bik and Bernstein 6 ). The Gram-negative Bacteroidetes phylum includes the genera Bacteroides, Prevotella, Parabacteroides and Alistipes. Detailed investigations on human colonic Bacteroides isolates show that these organisms possess the capability to utilise a very wide range of substrates of both host and dietary origin( Reference Martens, Koropatkin and Smith 7 – Reference Kaoutari, Armougom and Gordon 9 ). Genes encoding polysaccharide utilisation functions in Bacteroides spp. are organised into clusters (polysaccharide utilisation loci) that include genes encoding hydrolases and also outer membrane proteins responsible for the initial binding of the soluble polysaccharide and its transfer into the periplasmic space (the region between the outer and cytoplasmic membranes in Gram-negative bacteria)( Reference Martens, Koropatkin and Smith 7 ). Much of the hydrolysis of the polysaccharide fragments occurs in this periplasmic space in Bacteroides spp. Functional analysis of the original starch-utilisation polysaccharide utilisation loci by Salyers et al. ( Reference Salyers, Vercellotti and West 8 ) showed that this type of organisation results in the sequestration of soluble substrates from the gut lumen, which is assumed to offer a competitive advantage within the very dense microbial community of the large intestine( Reference Martens, Koropatkin and Smith 7 , Reference Flint, Bayer and Rincon 10 ). The Firmicutes have received considerably less study although they account for about 70 % of the species diversity within the human colonic microbiota( Reference Eckburg, Bik and Bernstein 6 ). However, we know that the Firmicutes include several highly abundant species such as Faecalibacterium prausnitzii, Eubacterium rectale and Eubacterium hallii that have been identified as the dominant producers of butyrate in the colon( Reference Louis, Young and Holtrop 11 ). They also include species that convert lactate to butyrate or propionate and may help to stabilise the colonic microbiota by preventing lactate accumulation and excess acidity( Reference Belenguer, Duncan and Holtrop 12 ). With regard to substrate utilisation, evidence is emerging that the Firmicutes include some highly specialised degraders of non-digestible polysaccharides that may be regarded as ‘keystone’ species within the microbiota( Reference Ze, Duncan and Louis 13 ). Firmicutes typically carry fewer genes concerned with polysaccharide degradation than Bacteroides spp.( Reference Kaoutari, Armougom and Gordon 9 ), and this is likely to reflect both their smaller genome sizes and their greater nutritional specialisation. Human colonic bacteria that became associated with insoluble starch particles or wheat bran in in vitro fermentors included mostly Firmicutes rather than Bacteroides spp.( Reference Leitch, Walker and Duncan 14 ) and Firmicutes (specifically Ruminococcaceae) were also enriched among particle-associated bacteria from human stool samples( Reference Walker, Duncan and Harmsen 15 ). The species Ruminococcus bromii has recently been shown to play a keystone role in the degradation of resistant starch, as individuals who lack R. bromii in their microbiota are unable to degrade resistant starch which is then detectable in their faeces( Reference Ze, Duncan and Louis 13 ). The enzyme systems involved in polysaccharide degradation by human colonic Firmicutes are less well studied than those of Bacteroides spp., but comprise extracellular degradative enzymes and enzyme complexes ( Reference Flint, Bayer and Rincon 10 , Reference Ramsay, Scott and Martin 16 , Reference Flint, Scott and Louis 17 ). These polysaccharide-degrading Gram-positive anaerobes often produce large multi-domain enzymes that are attached to the cell wall either directly( Reference Ramsay, Scott and Martin 16 ) or via extracellular multi-enzyme complexes( Reference Rincon, Dassa and Flint 18 ).
Other bacterial phyla within the healthy colonic microbiota include Actinobacteria (that include Bifidobacterium spp.), Proteobacteria (including Escherichia coli) and Verrucomicrobia (including Akkermansia muciniphila). These are typically present in smaller numbers than the dominant Bacteroidetes and Firmicutes, but have considerable potential to influence health outcomes. Although only about 30 % of human intestinal species are currently represented by cultured isolates, the most abundant species appear to be well represented( Reference Walker, Ince and Duncan 19 ) and most of the remaining organisms are probably capable of being cultured under appropriate conditions( Reference Lagier, Armougom and Million 20 , Reference Goodman, Kallstrom and Faith 21 ).
Impact of diet on microbiota composition
Two major mechanisms seem likely to result in shifts in the species composition of the intestinal microbiota. First, different microbial species vary in their capabilities, determined by their genomes, for utilising substrates of dietary and host origin. This is expected to determine the outcome of competition for substrates available in the large intestine, with certain species favoured over others when particular substrates are in greater abundance. This is the principle behind manipulation of the microbiota using prebiotics. Secondly, different species may vary in their tolerance of a wide range of factors in the gut environment, such as high or low pH, high bile salt concentrations or low micronutrient (e.g. Fe) concentrations that tend to limit microbial growth. Recent work with human volunteers and animal models indicates that both of these mechanisms are likely to play an important role in defining species composition.
Human volunteer studies that involve precise control over dietary intake have been used to investigate the impact of different dietary non-digestible carbohydrates on the species composition of the gut microbiota over short time intervals. Changing the major non-digestible carbohydrate from wheat bran to resistant starch (and vice versa) in diets delivering the same intake of protein, fat and total carbohydrate, led to rapid and reversible shifts in the representation of certain bacterial groups within a few days in overweight volunteers( Reference Walker, Ince and Duncan 19 ). Changes affecting multiple species were detected by 16S rRNA gene sequencing, qPCR and HitChip microarray analysis( Reference Walker, Ince and Duncan 19 , Reference Salonen, Lahti and Salojärvi 22 ). On the other hand, global analyses of sequence and HitChip data indicated that inter-individual variation played a more major role than dietary change in determining the overall species composition of the microbiota( Reference Walker, Ince and Duncan 19 , Reference Salonen, Lahti and Salojärvi 22 ). The explanation for this apparent contradiction is twofold. Firstly, many species (especially the less abundant ones) occur only in one or a few individuals. Secondly, it appears that within the microbiota only certain species are responsive to the particular dietary switches, in this case including those bacteria that are specialists at utilising resistant starch or wheat bran. In the present study, many of the diet-responsive species were Firmicutes, notably Ruminoccocus bromii in the case of resistant starch diets( Reference Walker, Ince and Duncan 19 ) and various Lachnospiraceae for the wheat bran diet( Reference Salonen, Lahti and Salojärvi 22 ). Significant shifts in faecal microbiota composition resulting in decreased butyrate-producing Firmicutes have also been observed with weight loss diets that include decreased amounts of fibre and total carbohydrate( Reference Walker, Ince and Duncan 19 , Reference Duncan, Belenguer and Holtrop 23 ). In contrast, many dominant species showed limited responses to such dietary switches. These may be considered generalists that have a greater capacity to switch between dietary energy sources, or between host- and diet-derived substrates, as has been elegantly demonstrated using animal models, for example, for Bacteroides spp.( Reference Sonnenburg, Xu and Leip 24 ).
Rapid changes in microbiota were also reported recently in another human intervention study involving more extreme diets( Reference David, Maurice and Carmody 25 ). A switch between a plant-based diet (high in fibre, low in fat and protein) and an animal-based diet (70 % of energies from fat and 30 % from protein) resulted in an increased proportion of Bacteroides spp. and a decreased proportion of many Firmicutes on the animal-based diet. In this case, the change in fat content was proposed to be a major factor( Reference David, Maurice and Carmody 25 ), mediated via the effect on bile acids, which are known from animal studies to exert an important selective influence upon gut microbiota composition( Reference Islam, Fukiya and Hagio 26 ).
There is also indirect evidence that variations in habitual dietary intake are responsible for differences in gut microbiota profiles. A survey of ninety-eight US adults( Reference Wu, Chen and Hoffmann 27 ) showed that individuals whose faecal microbiota is high in Prevotella tend to consume more fibre, whereas those high in Bacteroides tend to consume more protein and fat, indicating that there is a strong influence of long-term dietary intake upon the relative abundance of these two genera of Bacteroidetes within the gut microbiota. This is in agreement with a previous study in which a group of Italian children showed higher Bacteroides and a group of rural African children higher Prevotella, with the difference being ascribed mainly to the very different habitual diets of each group( Reference De Filippo, Cavalieri and Di Paola 28 ). Meanwhile, metagenomic sequence analysis of DNA extracted from faecal samples has revealed a bimodal distribution of low gene count (LGC) and high gene count (HGC) individuals within the general population( Reference Le Chatelier, Nielsen and Qin 29 ). The microbiota of LGC individuals tends to be Bacteroides-dominated and phylogenetically less diverse than that of HGC individuals, and has lower representation of Firmicutes, including butyrate-producers. Remarkably, the LGC group of individuals was reported to show a higher incidence of obesity and metabolic syndrome than the HGC group( Reference Le Chatelier, Nielsen and Qin 29 , Reference Cotillard, Kennedy and Kong 30 ). When obese LGC volunteers were put onto a controlled weight loss diet, their microbiota diversity increased towards that of the HGC individuals, while the symptoms of metabolic syndrome improved in both HGC and LGC groups( Reference Cotillard, Kennedy and Kong 30 ). Again, it seems likely that the LGC state is a consequence of dietary intake. The composition of the gut microbiota present in the colon of each individual along with different dietary intakes is likely to impact on microbial metabolic output including the formation of SCFA.
Metabolic activities of the gut microbiota
Formation of propionate and butyrate
The main fermentation products of gut microbial metabolism are the SCFA acetate, propionate and butyrate plus gases. SCFA are absorbed by the host and utilised as an energy source, with butyrate acting as the main fuel for the colonic wall( Reference Sleeth, Thompson and Ford 2 ). Butyrate has received much attention for its anti-inflammatory and anti-carcinogenic effects( Reference Canani, Costanzo and Leone 31 ) and more recently SCFA have been studied for their effects on systemic host metabolism, particularly as satiety-inducing agents( Reference Arora, Sharma and Frost 32 , Reference Frost, Sleeth and Sahuri-Arisoylu 33 ). In order to increase the concentration of propionate and butyrate in the lower gut by dietary means, it is important to understand which bacteria form these SCFA. Much progress has been made in the identification of butyrate-producing bacteria, revealing that they are present in several different classes within the Firmicutes( Reference Louis, Young and Holtrop 11 , Reference Louis and Flint 34 ). E. rectale/Roseburia spp. is an abundant group of butyrate producers (estimated at 2–15 % of the total bacteria( Reference Louis and Flint 34 )) within the Lachnospiraceae (clostridial cluster XIVa). These bacteria showed a marked drop in abundance in human volunteers on a high-protein, low-carbohydrate diet and their abundance correlated significantly with faecal butyrate levels( Reference Duncan, Belenguer and Holtrop 23 ). F. prausnitzii, another abundant butyrate-producing species within the Rumincoccaceae (clostridial cluster IV)( Reference Lopez-Siles, Khan and Duncan 35 ), has been linked to anti-inflammatory effects( Reference Sokol, Pigneur and Watterlot 36 , Reference Martín, Chain and Miquel 37 ). Interestingly, F. prausnitzii has been shown to use an extracellular electron shuttle to transfer electrons to oxygen, which enables it to grow in the presence of low levels of oxygen( Reference Khan, Duncan and Stams 38 ). Many other genera within several clostridial clusters produce butyrate, some of which are known to contribute specialist functions including species belonging to Lachnospiraceae that can convert lactate into butyrate( Reference Duncan, Louis and Flint 39 ).
Butyrate is formed from two molecules of acetyl-CoA and two different types of reaction contribute to the final step, the liberation of butyrate from butyryl-CoA (Fig. 2). Few bacteria in the gut use the butyrate kinase pathway, whereas the butyryl-CoA:acetate CoA-transferase pathway is utilised by the majority of known butyrate-producing gut strains( Reference Louis, Young and Holtrop 11 , Reference Louis, Duncan and McCrae 40 ). Butyrate producers are phylogenetically interspersed with non-butyrate producers, making it difficult to quantify them with molecular techniques targeting the 16S rRNA gene. Degenerate primers specifically amplifying the butyryl-CoA:acetate CoA-transferase gene have proven valuable in specifically targeting the butyrate-producing community. This approach currently allows a more in-depth analysis compared with metagenomic mining( Reference Louis, Young and Holtrop 11 ), however, with improved depth and coverage of metagenomic datasets, metagenomic mining will become increasingly attractive to investigate specific functions within the microbiota. It has to be kept in mind however that genes closely related to those under study are often present that may perform a different function. Therefore results based on molecular analysis of genes have to be evaluated critically and backed up by biochemical analysis for less closely related genes. On the other hand, different genes may have evolved to perform the same function. Thus, it appears that a CoA-transferase gene more closely related to propionate CoA-transferase carries out the last step of butyrate formation in butyrate-producing bacteria within the Erysipelotrichaceae (clostridial cluster XVI)( Reference Eeckhaut, van Immerseel and Croubels 41 ).

Fig. 2. Metabolic routes for butyrate and propionate formation by representative bacterial genera and species from the human colon. Species shown in purple can utilise lactate to form butyrate; species shown in blue can utilise lactate, and those shown in green succinate to produce proprionate. DHAP, dihydroxyacetonephosphate; PEP, phosphoenolpyruvate.
Three different biochemical pathways for propionate formation exist within the gut microbiota( Reference Reichardt, Duncan and Young 42 ) (Fig. 2). The succinate pathway is present in Bacteroidetes and some Firmicutes bacteria within the Negativicutes (Veillonellaceae). Bacteroidetes are the second most abundant phylum in healthy human subjects and faecal propionate levels (as a percentage of all fermentation acids) have been found to be significantly correlated with their relative abundance( Reference Salonen, Lahti and Salojärvi 22 ). The level of propionate formation is dependent on the environmental conditions, as succinate may become a major fermentation product instead of propionate. Thus, carbon limitation results in higher propionate production in Bacteroides ovatus ( Reference Macfarlane and Macfarlane 43 ) and vitamin B12 is required for propionate formation in Prevotella ruminicola ( Reference Strobel 44 ). Some Negativicutes bacteria, however, can utilise succinate as a substrate for propionate formation, either in conjunction with other substrates or as the sole energy source( Reference Reichardt, Duncan and Young 42 ).
The acrylate pathway (Fig. 2) is utilised for the conversion of lactate to propionate. Based on genomic and metagenomic analyses, this pathway appears to be limited to very few different bacterial genera within the Firmicutes, including Megasphaera spp. (Veillonellaceae), Coprococcus catus and uncultured clones related to Clostridium lactatifermentans (both Lachnospiraceae)( Reference Reichardt, Duncan and Young 42 ).
The propanediol pathway (Fig. 2) operates specifically from deoxy-sugar substrates (fucose, rhamnose) and has been identified in the Proteobacterium Salmonella enterica serovar Typhimurium( Reference Bobik, Havemann and Busch 45 ) and Lachnospiraceae bacteria related to Blautia spp. and Ruminococcus obeum as well as Roseburia inulinivorans ( Reference Scott, Martin and Campbell 46 , Reference Reichardt, Duncan and Young 42 ). The full conversion of the C5 ring deoxy-sugars to propionate involves a toxic intermediate and thus occurs within polyhedral bodies, the synthesis of which requires approximately ten independent genes encoded on a complex operon. Transcriptional analysis of R. inulinivorans revealed that these genes were strongly up-regulated during growth on fucose compared with glucose( Reference Scott, Martin and Campbell 46 ). Bacteroides thetaiotaomicron, which is able to grow on fucose although lacking the genes required for 1,2-propanediol utilisation, has been shown to signal to the host resulting in more fucose production( Reference Xu, Bjursell and Himrod 47 ). Phylogenetic distribution of the acrylate and propanediol pathways suggests that they may have been acquired by horizontal gene transfer by some bacteria, providing a competitive advantage by enabling recipients to utilise additional substrates. For instance, many of the glycoconjugates lining the gut epithelium are fucosylated, which may provide an alternative energy source in times of dietary starvation. The succinate pathway for propionate formation and the two butyrate production pathways have not been found to be present within the same species and consequently the ability to produce butyrate and propionate is rarely found within the same organism( Reference Reichardt, Duncan and Young 42 ). Several bacteria carrying either the acrylate or the propanediol pathway, however, are also able to produce butyrate from non-deoxy-sugars, and consequently change their fermentation product profile in response to the growth substrate available. For example, R. inulinivorans produces butyrate when grown on glucose, whereas propionate, propanol and butyrate are produced when it is grown on fucose( Reference Scott, Martin and Campbell 46 ).
Lactate formation and utilisation
Although normally detected at low levels in adult faecal samples, lactate is a common end product of bacterial fermentation and is produced, among others, by lactic acid bacteria of the genera Lactobacillus and Bifidobacterium. These two genera are considered as key members of the gut microbiota due to their perceived health-promoting effects( Reference Metchnikoff 48 ). Lactobacilli are usually detected in lower abundance than bifidobacteria in stool samples but may be more abundant in the small intestine( Reference Mitsuoka 49 ). Lactate is a major bacterial fermentation product in breast-fed babies that are mainly colonised by Bifidobacterium ( Reference Favier, Vaughan and De Vos 50 – Reference Turroni, Peano and Pass 52 ), whereas the intestinal microbiota of formula-fed infants is more complex, with a lower abundance of bifidobacteria( Reference Bezirtzoglou, Tsiotsias and Welling 53 ). Breast-fed babies have been reported to show significantly higher levels of lactic acid (mean 8·4 mm) compared with formula-fed babies (mean 1·7 mm) at age 1 month, with the latter indicating a much more mixed acid fermentation. These differences affected the relative faecal pH, with the former pH 5·8 and the latter pH 7·1. The increased faecal lactate and lower pH values were also found in slightly older (age 2–5 months) babies( Reference Ogawa, Ben and Pons 54 ).
Lactobacillus species, which are widely used as probiotics, use either the homo-fermentative pathway, yielding 2 mol lactate per mol glucose, or the hetero-fermentative pathway that yields lactate, ethanol and carbon dioxide. Bifidobacterium species ferment sugars using the bifid shunt route that yields acetate and lactate in the molar ratios of 3:2. However, it is less well recognised that many Bacteroidetes and Firmicutes that are dominant inhabitants in the large intestine can also form lactate in combination with other fermentation acids (Table 1).
Table 1. Major fermentation products of representative dominant species of human colonic bacteria
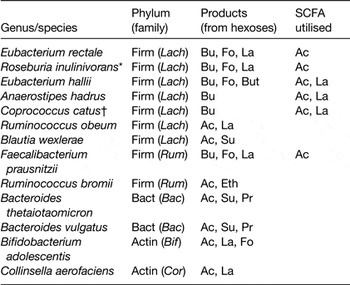
Firm, Firmicutes; Lach, Lachnospiraceae; Rum, Ruminococaceae; Bact, Bacteroidetes; Bac, Bacteroidaceae; Actin, Actinobacteria; Bif, Bifidobacteriaceae; Cor, Coriobacteriaceae; Bu, butyrate; Fo, formate; La, lactate; But, butanol; Ac, acetate; Eth, ethanol; Su, succinate.
* Roseburia inulinivorans forms propionate and propanol from deoxy-sugars.
† Coprococcus catus forms propionate from lactate.
Belenguer et al. ( Reference Belenguer, Duncan and Holtrop 12 ) reported that lactate formation occurred across a pH range between 5·2 and 6·4 in in vitro incubations with mixed faecal microbiota, with the highest production occurring under slightly acidic conditions (pH 5·9). At low pH (<5·2) lactate formation was slightly increased but lactate utilisation was strongly inhibited, resulting in lactate accumulation. Normal colonic pH values range between pH 5·5 and 7·5( Reference Nugent, Kumar and Rampton 55 ) although lower pH values have been reported in severe bowel disease( Reference Fallingborg, Christensen and Jacobsen 56 ) and low pH values in combination with high colonic lactate concentrations (up to about 100 mm) have been associated with ulcerative colitis( Reference Vernia, Caprilli and Latella 57 , Reference Hove, Nordgaard-Andersen and Brobech Mortensen 58 ). Faecal lactate concentrations clearly depend on the balance between production by bacteria and by host tissues, microbial utilisation and host absorption. In contrast to the reported beneficial effects of butyrate on gut health, lactate accumulation may be detrimental by causing acidosis and d-lactate can result in neurological problems. In the healthy colon, much of the lactate formed must be utilised by other bacteria via metabolite cross-feeding as faecal concentrations are generally low. Bacteria isolated from the human colon that have been reported to utilise lactate include the acetate producer Desulfovibrio piger, which concomitantly forms hydrogen sulphide( Reference Marquet, Duncan and Chassard 59 ). Separately, some Firmicutes can form propionate from lactate( Reference Bourriaud, Robins and Martin 60 ), including C. catus ( Reference Reichardt, Duncan and Young 42 ) as discussed earlier, while lactate is also utilised together with acetate by certain Lachnospiraceae including Eubacterium hallii and Anaerostipes species to form butyrate( Reference Belenguer, Duncan and Holtrop 12 , Reference Duncan, Louis and Flint 39 , Reference Morrison, Mackay and Edwards 61 ). Thus a number of bacterial groups are likely to compete for lactate; in terms of bacterial energy gain, acetate formation from lactate is the least favourable( Reference Thauer, Jungermann and Decker 62 ).
U13C-labelled dl-lactate provided as the sole added energy source to in vitro incubations with mixed faecal microbiota, gave rise to more propionate, formed via the acrylate pathway, than butyrate( Reference Belenguer, Duncan and Holtrop 12 ). In the presence of polysaccharide substrates, however, the reverse was true, with more butyrate than propionate formed from lactate, and this was accompanied by a marked increase in the population of E. hallii within the community( Reference Belenguer, Duncan and Holtrop 12 ). In a recent fermentor study, lactate cross-feeding was estimated to contribute to approximately 20 % of the butyrate pool( Reference Belenguer, Holtrop and Duncan 63 ). It is clear that lactate-utilising bacteria, including those producing butyrate, play a key role in modulating the colonic metabolome.
Dietary fibre and phytochemicals
Consumption of fibre-enriched diets is considered to offer several health benefits( Reference Scott, Duncan and Flint 64 – Reference Russell, Hoyles and Flint 66 ). In addition to increasing faecal bulking and transit rates along the large intestine, fibre delivers a wide range of phytochemicals and bacterially transformed metabolites within the human colon( Reference Russell, Gratz and Duncan 67 ) with many of these compounds possessing anti-inflammatory activity( Reference Russell, Scobbie and Chesson 68 ). Free phytochemicals are likely to be taken up in the small intestine whereas those that complex with plant polymers will be released and metabolised by the gut microbiota in the large intestine. In contrast, increasing the protein content of diets, particularly with red meat, is associated with increased formation of toxic bacterial metabolites resulting in an increased risk of cancer development( Reference Gill and Rowland 69 ). The impact of different protein, carbohydrate and fibre (as non-starch polysaccharide) intakes is shown by a study in which seventeen obese men were provided with an initial maintenance diet (85 g protein, 360 g carbohydrate, 21·9 g non-starch polysaccharide, 116 g fat per d) followed by a high-protein, moderate-carbohydrate (139 g protein, 181 g carbohydrate, 12·8 g non-starch polysaccharide, 82 g fat per d) diet and a high-protein, low-carbohydrate (137 g protein, 22 g carbohydrate, 8·8 g non-starch polysaccharide, 143 g fat per d) diet, in a cross-over design, for periods of 4 weeks each. At the ‘normal’ intake levels represented by the maintenance diet, the major phenolic metabolites were ferulic acid and its metabolic conversion products that arise from microbial activity( Reference Russell, Gratz and Duncan 67 ). When volunteers were consuming the low-carbohydrate, high-protein diet, however, their faecal samples showed very low concentrations of the fibre-derived ferulic acid and its metabolites and the phenolic acid showing the highest concentration was phenylacetic acid, derived from amino acid fermentation( Reference Russell, Hoyles and Flint 66 , Reference Russell, Gratz and Duncan 67 ). Furthermore, there were increased concentrations of N-nitrosamines and branched chain fatty acids and decreased levels of beneficial products such as butyrate in the stool samples from the volunteers during the period when they were consuming the high-protein, low-carbohydrate diet( Reference Russell, Gratz and Duncan 67 ). Taken together these data suggest a less favourable gut environment, particularly with respect to colon cancer risk, on the high-protein, low-carbohydrate diet and these changes were only partially reversed by the moderate carbohydrate intake.
Predicting the impact of dietary manipulation on microbiota profiles and metabolites
We have seen that microbiota composition is influenced by diet, and that both the substrate and microbiota composition affects bacterial metabolite formation. It is known that prebiotic supplementation can affect both the composition and activity of the gut microbiota. A prebiotic is defined as a ‘selectively fermented ingredient that results in specific changes in the composition and/or activity of the gastrointestinal microbiota, thus conferring benefit(s) upon host health’( Reference Gibson, Scott and Rastall 70 ). Ideally, prebiotics would be targeted at promoting beneficial bacterial species whose populations are decreased in situations associated with increased disease risk (e.g. metabolic syndrome and ageing). Clearly such specific targeting requires detailed prior knowledge on the substrate utilisation capabilities, and competitive abilities, of the bacterium to be targeted. Substrate utilisation profiles can be established for cultured strains, providing important information on the types of substrate that can be used by a bacterium under ideal growth conditions. Analysis of the supernatant can also provide some indication of the major metabolites produced by a specific bacterium. However, bacteria in the human gut exist within a complex community where there is competition for growth substrates, and cross-feeding of metabolites; thus care has to be taken in extrapolating results from pure culture to the in vivo situation. This is illustrated by work with fructan-based prebiotics (fructo-oligosaccharides and inulin)( Reference Macfarlane, Steed and Macfarlane 71 ). Many studies have focused on establishing an increase in bifidobacterial populations, and sometimes reduced numbers of potentially pathogenic Clostridial species (such as C. perfringens), but only a few have investigated the effect on the gut microbiota as a community. Pure culture work has shown however that a number of butyrate-producing Firmicutes are able to use fructo-oligosaccharides directly for growth( Reference Scott, Martin and Duncan 72 ), and genes for prebiotic degradation were identified in a range of abundant commensal bacteria by functional metagenomic screening( Reference Cecchini, Laville and Laguerre 73 ). Of course, bacteria that are able to utilise prebiotics as substrates for growth in pure culture may not necessarily compete well for the substrate within the mixed community. Nevertheless, a human study investigating the effect of consuming 10 g mixed chain length inulin per day detected increased numbers of both bifidobacteria and F. prausnitzii, a butyrate-producing bacterium in faecal samples( Reference Ramirez-Farias, Slezak and Fuller 74 ).
Interestingly, fructan-based prebiotics stimulate both bifidobacteria numbers and butyrate production( Reference Falony, De Vuyst, Charlampopoulos and Rastall 75 ) even though bifidobacteria produce lactate and not butyrate. In part, this is likely to reflect the direct stimulation of butyrate-producing species; in addition, other members of the microbial community are able to convert lactate into butyrate, as was discussed earlier. The conversion of lactate into butyrate via bacterial cross-feeding was demonstrated in an experiment in which E. hallii and B. adolescentis were cultured together and independently on starch( Reference Belenguer, Duncan and Calder 76 ). In pure culture B. adolescentis grows well, producing acetate, lactate and formate, while E. hallii cannot grow on starch. In co-culture, however, acetate declines and lactate completely disappears, being converted into butyrate by the action of E. hallii. Meanwhile a second type of cross-feeding was revealed in experiments with Roseburia spp., which lack the ability to utilise lactate. These species were only able to utilise fructo-oligosaccharides for growth when co-cultured with a Bifidobacterium species because of the release of breakdown products such as fructose, again resulting in butyrate formation( Reference Belenguer, Duncan and Calder 76 , Reference Falony, Vlachou and Verbrugghe 77 ). Bifidobacteria themselves differ in their competitive ability to utilise fructans of varying chain length for growth, indicating that prebiotic supplementation has strain-specific effects( Reference Falony, Lazidou and Verschaeren 78 , Reference De Vuyst and Leroy 79 ), and these effects will also be host-specific, depending on the commensal bifidobacteria population. The strain-specificity of substrate degradation is also apparent from co-culture experiments. R. inulinivorans was able to outcompete a strain of B. longum ( Reference Scott, Martin and Chassard 80 ), whereas a different strain of B. longum out-competed R. inulinivorans during growth on long-chain inulin( Reference De Vuyst and Leroy 79 ). Similarly it has been shown that different strains or species of bifidobacteria also differ in their ability to use another class of prebiotics, arabinoxylan oligosaccharides, for growth( Reference Rivière, Moens and Selak 81 ).
More generally, cross-feeding of breakdown products is a widespread feature in the utilisation of complex carbohydrate polysaccharides by gut microbial communities( Reference Flint, Duncan and Scott 82 , Reference Flint, Scott and Duncan 83 ). Because Gram-negative Bacteroides species are able to sequester soluble polysaccharides and perform substrate degradation largely in the periplasm( Reference Martens, Koropatkin and Smith 7 ), it is unlikely that many short-chain substrates will escape to be used by other bacteria. In Gram-positive Firmicutes that lack a periplasmic space, however, hydrolysis is more likely to happen extracellularly, with the potential for competition from other bacteria for breakdown products. This has been illustrated in co-culture experiments with the starch degrader R. bromii, which is apparently unable to take up glucose and some α(1–6) breakdown products of starch degradation( Reference Ze, Duncan and Louis 13 ).
Conclusion: the need for integrative approaches and modelling
It should be apparent that the complex interactions that occur between gut bacteria make simple predictions of dietary effects extremely difficult. However, the field of theoretical modelling can offer some help and some hope in tackling the behaviour of microbial communities and their metabolites. For example, experimental data from a simple batch co-culture experiment examining lactate cross-feeding have been successfully modelled( Reference Muñoz-Tamayo, Laroche and Walter 84 ). A far more ambitious project is to model the whole community by defining a manageable number of functional groups within the gut microbiota. This has met with some success in modelling the impact of a one unit pH change( Reference Walker, Duncan and McWilliam Leitch 85 ) upon the microbial community in an anaerobic continuous flow fermentor system( Reference Kettle, Louis and Holtrop 86 ). The models referred to so far aim at quantitative predictions of metabolite concentrations, fluxes and bacterial populations within the system and therefore depend on information from representative cultured bacterial isolates on growth rates, cell sizes, substrate preferences and pH tolerances. Another active, but quite distinct, area of modelling starts from bacterial genomes (genome-scale metabolic models) in order to predict the metabolic outputs of interactions between selected bacterial species( Reference Shoaie, Karlsson and Mardinoglu 87 – Reference El-Semman, Karlsson and Shoaie 89 ). Such genome-based modelling is essentially qualitative in nature, but there should be scope for the two approaches to inform each-other in the future( Reference Walker, Duncan and Louis 90 ). In conclusion, modelling of the complex gut microbial ecosystem appears essential not only for gaining a better understanding of the system and its response to dietary change, but ultimately for designing strategies to alleviate disease and maintain gut health.
Financial Support
The Rowett Institute of Nutrition and Health is funded by the Scottish Government (SG-RESAS).
Conflicts of Interest
None.
Authorship
The authors contributed equally to the writing of the manuscript.



