Sterol regulatory element binding proteins (SREBP) are a family of membrane-bound transcription factors, which play a critical role in the maintenance of lipid homoeostasis by activating genes encoding enzymes required for the biosynthesis of sterols and fatty acids(Reference Horton, Goldstein and Brown1). To date, there are three kinds of SREBP, including SREBP-1a and -1c derived from utilisation of alternate promoters of one gene that differ in the first exon(Reference Yokoyama, Wang and Briggs2–Reference Shimano, Horton and Shimomura4), and SREBP-2 encoded by a separate gene(Reference Hua, Yokoyama and Wu5). Specifically, SREBP-1a and -1c preferentially activate transcription of genes required for fatty acids and TAG biosynthesis, while SREBP-2 mainly controls transcription of genes required for sterol biosynthesis(Reference Pai, Guryev and Brown6–Reference Horton, Shah and Warrington8). SREBP typically contain a N-terminal transactivated domain possessing a basic helix-loop-helix-leucine zipper (bHLH-Zip) motif and a C-terminal regulatory domain, which are joined together by two membrane-binding regions interrupted by about thirty amino acids (aa)(Reference Horton, Goldstein and Brown1,Reference Shimano9) . They are initially synthesised as inactive precursors that bind to membranes of the endoplasmic reticulum. To influence transcription, the SREBP must be proteolytically cleaved to release the N-terminal transactivated domain so that they can enter the nucleus. Those segments are designed as nuclear (mature) SREBP, which further activate transcription of target genes by binging to the sterol regulatory element (SRE) or classic palindromic E-box within their promoters using bHLH-Zip motif(Reference Shimano9).
Long-chain PUFA (LC-PUFA), especially ARA (20 : 4n-6), EPA (20 : 5n-3) and DHA (22 : 6n-3), are critical in a number of physiological processes, such as cell membrane formation, neurological development, immune response and CVD treatments(Reference McMurchie10–Reference Russo13). The endogenous biosynthesis of LC-PUFA from C18 precursors (18 : 2n-6 and 18 : 3n-3) requires consecutive desaturation and elongation steps catalysed by fatty acyl desaturase (Fad) and elongase of very long-chain fatty acid (Elovl), respectively(Reference Castro, Tocher and Monroig14). Among which, Δ6 Fad not only catalyses the first desaturation step but also catalyses the rate-limiting step in DHA biosynthesis by conversion of 24 : 5n-3 to produce 24 : 6n-3, which is then partially β-oxidised to DHA via the Sprecher pathway(Reference Voss, Reinhart and Sankar15). Therefore, Δ6 Fad has been considered as the key and rate-limiting enzyme in LC-PUFA biosynthesis. Meanwhile, SREBP as important transcriptional regulators of Δ6 Fad have been well demonstrated or indicated by finding SRE within Δ6 Fad promoters in vertebrates, that is, mammals(Reference Matsuzaka, Shimano and Yahagi16,Reference Nara, He and Tang17) and teleosts(Reference Zheng, Leaver and Tocher18–Reference Dong, Zhao and Chen23), but still not in invertebrates including marine molluscs.
The razor clam Sinonovacula constricta is an economically and nutritionally important bivalve species that is widely distributed in the estuarine and intertidal zones along the coasts of the west Pacific Ocean, with a total production of over 823 000 tons and a value of US$ 1·3 billion in 2016(24,Reference Ran, Li and Zhang25) . Particularly, it possesses high levels of EPA and DHA, each accounting for about 10 % of the total fatty acids(Reference Ran, Chen and Ran26). Therefore, S. constricta is an excellent LC-PUFA resource for human nutritional requirement. Importantly, S. constricta is demonstrated as the first marine mollusc to possess all Fad and Elovl activities required for LC-PUFA biosynthesis via the Sprecher pathway(Reference Ran, Xu and Liao27,Reference Ran, Xu and Liao28) . In addition, S. constricta Δ6 Fad is also the first report of a Δ6 Fad in a marine mollusc(Reference Ran, Xu and Liao27). Therefore, S. constricta provides a favourable organism to investigate the regulatory mechanisms of LC-PUFA biosynthesis in marine molluscs.
In the present study, the characterisation of SREBP and its transcriptional regulation mechanism on Δ6 Fad were investigated in S. constricta. In brief, the S. constricta SREBP was first cloned and its sequence was analysed. Second, the structure of S. constricta Δ6 Fad promoter was characterised, including cloning of the promoter sequence, prediction of SRE by bioinformatic software and determination of the core promoter region by dual luciferase assay. Third, considering the SREBP activate transcription independently by their N-terminal segments (nuclear SREBP) harbouring bHLH-Zip motif in cultured cells and intact animals(Reference Hua, Wu and Goldstein3–Reference Hua, Yokoyama and Wu5,Reference Horton, Shimomura and Brown7,Reference Shimano, Horton and Hammer29) , the partial segment containing bHLH-Zip motif of S. constricta SREBP was subjected to a prokaryotic expression system. Followed by, the purified protein was used to interact with the potential SRE in S. constricta Δ6 Fad promoter by electrophoretic mobility shift assay (EMSA). The results greatly increased our understanding on the regulatory mechanism of LC-PUFA biosynthesis in marine molluscs, which would facilitate optimising the LC-PUFA biosynthetic pathway of bivalves in further studies.
Materials and methods
Cloning and sequence analyses of Sinonovacula constricta sterol regulatory element binding proteins
S. constricta RNA was extracted from fresh-mixed tissues of foot muscle, gill and gonad using a MiniBEST Universal RNA Extraction Kit (TaKaRa). The quality and concentration of the extracted RNA were determined on a 1 % agarose gel and a NanoDrop® ND-1000 (NanoDrop), respectively. A quantity of 1 µg of total RNA was reverse-transcribed into template complementary DNA (cDNA) using a PrimeScript™ RT-PCR Kit (TaKaRa).
The first fragment of S. constricta SREBP cDNA was obtained by searching against its transcriptome data, and only one gene with high homology to vertebrate SREBP was detected. To verify the target sequence, gene-specific primers (SREBP-V-F, V-R in Table 1) were designed using Primer 5 software. The PCR was carried out using LA Taq® Hot Star Version (TaKaRa). The resulting PCR products were purified, cloned into pMD™ 18-T Vector (TaKaRa) and transformed into E. coil DH5α competent cells. The recombinant single colonies successfully grown in Luria-Bertain (LB) plates containing ampicillin (50 µg/ml) were selected, incubated and sequenced (BGI). Based on the verified fragment, gene-specific rapid amplification of cDNA ends (RACE) primers (GSP primers in Table 1) was designed to obtain the full length of S. constricta SREBP by two-round PCR using a SMARTer RACE 5′/3′ kit (Clontech). The obtained PCR products were processed as described above. Finally, the full-length cDNA of S. constricta SREBP was obtained by aligning the first fragment with 5′ and 3′ RACE-PCR fragments.
Table 1. Primers used for cloning of Sinonovacula constricta sterol regulatory element binding proteins (SREBP) and Δ6 fatty acyl desaturase (Fad) promoter, construction of reporter vector and express vector, prokaryotic expression of SREBP-D and electrophoretic mobility shift assay (EMSA) assay*

* Restriction sites of KpnI, XhoI and EcoRI are underlined. The bolded letters in italics indicate the mutation sites.
Using the deduced aa of SREBP from S. constricta and representative mammals, fish and marine molluscs, multiple sequence alignment and phylogenetic tree were conducted using ClustalW 2.1(Reference Larkin, Blackshields and Brown30) and MEGA 7(Reference Kumar, Stecher and Tamura31), respectively. The phylogenetic analysis was performed with the maximum-likelihood approach, and the confidence in the resulting phylogenetic tree branch topology was measured by bootstrapping through 1000 iterations.
Cloning and potential regulatory sites of Sinonovacula constricta Δ6 fatty acyl desaturase promoter
S. constricta genomic DNA was extracted from fresh foot muscle using a Marine Animal DNA Kit (CWBIO) and used as a template for Δ6 Fad promoter cloning. The 2000 bp upstream of Δ6 Fad translation initiation codon (ATG) was obtained from the genomic sequencing data of S. constricta (Reference Ran, Li and Yan32) by querying Δ6 Fad cDNA (GenBank accession number MH220406). To further verify this promoter sequence, PCR was carried out by Mighty AmpTM DNA Polymerase version 3 (TaKaRa) using specific primers (Δ6 Fad promoter-V-F and -R in Table 1). Where necessary, the PCR products were subjected to the same processes as described above.
Online software JASPAR2020 (http://jaspar.genereg.net/) and LASAGNA-Search 2.0 (https://biogrid-lasagna.engr.uconn.edu/lasagna_search/) were used to predict the potential transcription factor binding sites of SREBP on S. constricta Δ6 Fad promoter. In addition, multiple-sequence alignment was performed with Δ6 Fad promoters of S. constricta and some representative marine teleosts, including Dicentrarchus labrax (FP671139.1), Epinephelus coioides (Reference Xie, Fu and Wang22), Salmo salar (AY736067.2), Gadus morhua (FJ859898.1) and Siganus canaliculatus (Reference Dong, Zhao and Chen23).
Construction of report vector and expression vector
To determine the core region of Δ6 Fad promoter that interacts with SREBP, different forward primers harbouring restriction site KpnI (underlined in Table 1) and a common reverse primer harbouring restriction site XhoI (underlined in Table 1) were designed using Primer 5 and used to obtain the full-length promoter fragment (Δ6 Fad-2000) and truncated fragments (Δ6 Fad-1389, –936, –463), respectively. The PCR was carried out using Mighty AmpTM DNA Polymerase version 3 (TaKaRa). The resulting PCR products were purified, digested with the corresponding restriction endonucleases (New England BioLabs) and inserted into similarly digested firefly luciferase plasmids pGL3 basic (Promega). Subsequently, the recombinant plasmids pGL3-Δ6 Fad-2000, −1389, −936 and −463 were transformed into E. coil DH5α competent cells, respectively. The recombinant single colonies successfully grown in LB plates containing ampicillin (50 µg/ml) were selected, incubated and sequenced (BGI). The E. coil DH5α-containing recombinant plasmids with correct sequence were further used to isolate the corresponding recombinant plasmids using Endo-free Plasmid Mini Kit I (Omega).
To obtain the expression vector, PCR fragments corresponding to the open reading frame of S. constricta SREBP were amplified from cDNA template by Mighty AmpTM DNA Polymerase version 3 (TaKaRa) using specific primers harbouring restriction sites of EcoRI and XhoI (SREBP-ORF-F and -R in Table 1). The following operations were the same with the construction of the above reporter vector except that the vector was replaced with the expression vector of pCS2+ (Promega). Finally, the recombinant plasmids of pCS-SREBP were obtained.
Dual luciferase assay
To confirm the core region of Δ6 Fad promoter sufficient to initiate transcription by SREBP, dual luciferase assay was performed to detect the influence of the above reporter vectors on transcription activity. In brief, the human embryonic kidney cells (HEK 293 T) were seeded in ninety-six-well cell culture plates in 100 μl high glucose Dulbecco’s modified Eagle’s medium (Transgen Biotech) with 10 % fetal bovine serum (Transgen Biotech) per well at 37°C in a humidified incubator (ESCO). After growing 24 h to 90 % confluent, the cultural medium was replaced with an Opti-MEM I Reduced Serum Medium (Gibco). Immediately, 50 ng of each reporter plasmids, 150 ng of expression plasmids and 5 ng of Renilla luciferase reporter plasmid PRL-CMV (Promega) in 10 µl Opti-MEM I Reduced Serum Medium were co-transfected into cells using lipofectamine® 2000 Reagent (Invitrogen). Each trial was triplicated. At 6 h after transfection, the culture medium was replaced with fresh Dulbecco’s modified Eagle’s medium. At 24 h after transfection, the Firefly and Renilla luciferase activities were measured using Dual-Glo® Luciferase Assay System E2920 (Promega) by the varioskan flash 3001 (Thermo). Finally, the relative luciferase activity was calculated using the ratio of firefly luciferase:Renilla luciferase and normalised by the control reporter.
Prokaryotic expression and purification of the truncated sterol regulatory element binding proteins
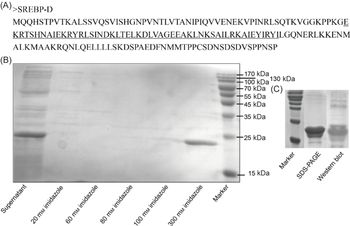
According to the function predication of S. constricta SREBP using online software of ExPASy-PROSITE (https://prosite.expasy.org/), the functional fragment of the SREBP N-terminal containing bHLH-Zip motif was identified (marked as SREBP-D) and used for prokaryotic expression. In brief, the corresponding cDNA sequence of SREBP-D was amplified by Mighty AmpTM DNA Polymerase version 3 (TaKaRa) using specific primers harbouring restriction sites of EcoRI and XhoI (SREBP-D-F and -R in Table 1). The following operations were the same with the construction of the above reporter vector except that the vector was replaced with the expression vector of pET-28a (Novagen). The obtained recombinant plasmids of pET-SREBP-D were transformed into Rosetta (DE3) competent cells. Next, the recombinant cells were cultured in the LB medium containing ampicillin (50 µg/ml) and chloromycetin (34 µg/ml) at 37°C. When the bacteria grew to the mid-logarithmic phase, isopropyl thiogalactoside was added at a final concentration of 0·4 mm and incubation at 16°C for 12 h. Subsequently, the bacteria were collected by centrifugation at 8000 g at 4°C. The obtained cell pellet was sonicated and used for protein extraction using His60 Ni Gravity Columns (Clontech). The protein impurities and the target protein were eluted with different concentrations of imidazole, and the eluted target protein was concentrated with an ultrafiltration centrifugal tube (UFC501096, Solarbio Science & Technology Co. Ltd). The concentrated SREBP-D was subjected to SDS-PAGE and confirmed by Western blot using Anti-6 × His rabbit polyclonal antibody (Sangon Biotech Co. Ltd), and its concentration was determined by the Enhanced BCA Protein Assay Kit (Beyotime Biotech Co. Ltd).
Electrophoretic mobility shift assay
The double-stranded 3′-biotinylated probes (50 nm) of the predicted binding sites (W primers in Table 1) were synthesised using an EMSA Probe Biotin Labeling Kit (Beyotime). Meanwhile, the corresponding unlabelled wild and site-direct mutant probes (10 µm) were synthesised (W and M primers in Table 1). Using Chemiluminescent EMSA Kit (Beyotime), the reaction mixture of above-mentioned SREBP-D (1·5 µg) and corresponding probes was incubated at 25°C for 30 min and then ran on a 6 % non-denaturing polyacrylamide gel in an ice water bath. The target bands were then transferred to a positively charged nylon membrane (Beyotime). After being cross-linked by UV light and conjugated by Streptavidin-HRP, the results were detected by using BeyoECL Moon reagent (Beyotime).
Statistical analyses
Statistical analyses of relative luciferase activity were conducted by one-way ANOVA, together with pairwise multiple comparisons by Newman–Keuls tests (SPSS 22.0). The values are represented as mean values and standard deviations, and P < 0·05 was considered statistically significant.
Results
Sequence and phylogenetics of Sinonovacula constricta sterol regulatory element binding proteins
The full length of mRNA transcript for S. constricta SREBP was 4222 bp, with a 5′ untranslated region of 20 bp, an open reading frame (ORF) of 3438 bp and a 3′ untranslated region of 764 bp. The ORF encoded a protein of 1146 aa, containing a basic bHLH-Zip motif and two membrane binding regions interrupted by 21 aa (Fig. 1). The detailed sequence information was deposited in the GenBank database with accession number of MK584917. The aa sequence of S. constricta SREBP had the highest identity with that of Chlamys nobilis of 49·83 %. Notably, compared with SREBP-2 (33·16–36·24 %) of Homo sapiens, Mus musculus and Danio rerio, S. constricta SREBP protein exhibited relatively higher identities with their SREBP-1 (38·21–41·12 %) (Table 2). The phylogenetic results showed that the vertebrate SREBP-1, SREBP-2 and marine molluscan SREBP were clustered into three separate groups, respectively (Fig. 2).

Fig. 1. Partial peptide sequence alignment of sterol regulatory element binding proteins (SREBP) from Sinonovacula constricta and representative organisms. Identical residues are shaded red, and similar residues are shaded blue. The solid line marked portion indicates the basic helix-loop-helix-leucine zipper structure, and the dotted line marked portion indicates the membrane binding domains. Representative species of SREBP include Homo sapiens SREBP-1 (NP_001005291.1) and SREBP-2 (NP_004590.2), Mus musculus SREBP-1 (NP_035610.1) and SREBP-2 (NP_150087.1), Danio rerio SREBP-1 (NP_001098599.1) and SREBP-2 (NP_001082935.1) and Chlamys nobilis SREBP (AHB60716.1).
Table 2. Amino acid identity between Sinonovacula constricta sterol regulatory element binding proteins (SREBP) and SREBP (-1/-2) of Homo sapiens, Mus musculus, Danio rerio or Chlamys nobilis (Percentages)


Fig. 2. Phylogenetic tree comparing the deduced amino acid sequences of sterol regulatory element binding proteins (SREBP) from Sinonovacula constricta (bold fonts) and representative organisms. The tree was constructed using the maximum-likelihood approach with MEGA 7. The horizontal branch length is proportional to amino acid substitution rate per site. The numbers represent the frequencies with which the tree topology presented was replicated after 1000 iterations. An asterisk indicates the predicted SREBP genes of Crassostrea gigas (genome ID: 10758), Aplysia californica (genome ID: 443), Crassostrea virginica (genome ID: 398), Octopus bimaculoides (genome ID: 41501) and Mizuhopecten yessoensis (genome ID: 12193) with complete genomes currently available.
Sequence and bioinformatics of Sinonovacula constricta Δ6 fatty acyl desaturase promoter
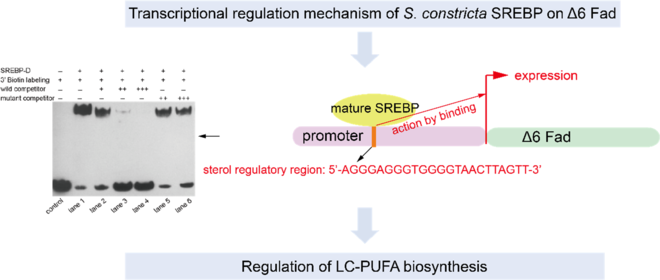
The detailed sequence information of the cloned 2000 bp of S. constricta Δ6 Fad promoter was deposited in the GenBank database with accession number of MK584921. As shown in online Supplementary Fig. S1, three putative binding sites for SREBP on S. constricta Δ6 Fad-2000 bp were detected by in silico analysis. Specifically, one binding site (5′-AGGGAGGGTGGGGTAACTTAGTT-3′) was aligned with the conserved SRE region of teleost Fads2 promoters (Fig. 3), while no feature was found in the distribution of the other two binding sites. In addition, no similar element with the two conserved NF-Y regions of teleost Fads2 promoters was predicted in the S. constricta Δ6 Fad promoter (Fig. 3).

Fig. 3. Alignment of fatty acyl desaturase 2 (Fads2) promoter sequences from Sinonovacula constricta and representative marine teleosts including Dicentrarchus labrax, Epinephelus coioides, Siganus canaliculatus, Gadus morhua and Salmo salar. Identical residues are shaded red, and similar residues are shaded blue. The sequences are all numbered relative to the transcription start site. The conserved NF-Y and SRE regions of the teleost Fads2 promoters are shown in the frames, respectively. The putative SRE of Sinonovacula constricta Δ6 Fad promoter is highlighted by a solid line.
Transactivation activities of 5′-truncated Sinonovacula constricta Δ6 fatty acyl desaturase promoter by sterol regulatory element binding proteins
When co-transfection with a report plasmid containing 5′-truncated S. constricta Δ6 Fad promoter and an expression plasmid containing S. constricta SREBP in HEK 293 T cells, the maximal relative luciferase activity was detected in samples transfected with pGL3-Δ6 Fad-2000 or −1389 (Fig. 4). Transfection with pGL3-Δ6 Fad-936 significantly reduced the luciferase intensity. Transfection with additional deleted report vector of pGL3-Δ6 Fad-463 did not further reduce the transcriptional activity, which was comparable with the luciferase readout of the control transfected with empty report and expression plasmids.

Fig. 4. Transcriptional activity of the segmentally truncated Sinonovacula constricta Δ6 fatty acyl desaturase (Fad) promoter regulated by sterol regulatory element binding proteins (SREBP). pRL-CMV is the Renilla luciferase expression plasmid (internal reference); pGL3-basic, pGL3-Δ6 Fad-2000, -1389, -936 and -463 are the empty firefly luciferase reporter plasmid and the recombinant pGL3-basic plasmid inserted with 2000 bp, 1389 bp, 936 bp and 463 bp upstream of S. constricta Δ6 Fad promoter, respectively. pCS2+ and pCS-SREBP are the empty protein expression vector plasmid and the recombinant pCS2+ plasmid inserted with the open reading frame (ORF) sequence of S. constricta SREBP, respectively. ‘+’ and ‘–’ indicate that the plasmid was transfected into the HEK 293T cells or not, respectively. Relative luciferase activity was expressed as mean values and standard deviations (n 3), which was first calculated by the ratio of firefly luciferase activity:Renilla luciferase activity, and then normalised by the corresponding result of the control 1. Values sharing a common letter above the bar graph were not significantly different (P ≥ 0·05).
Binding activities of the truncated sterol regulatory element binding proteins on putative regulatory sites in Δ6 fatty acyl desaturase promoter
The truncated SREBP protein containing bHLH-Zip motif (Fig. 5(A)) was successfully expressed in Rosetta (DE3) cells and presented in the supernatant after ultrasonication (Fig. 5(B)). The purified protein was further verified by Western blot (Fig. 5(C)) and subjected to EMSA analysis. Though three putative binding sites were predicted by bioinformatic analysis, only one sequence (5′-AGGGAGGGTGGGGTAACTTAGTT-3′) was validated (Fig. 6). In brief, when only 3′ biotin labelling sequence was added to the reaction, no blocked band was produced (Fig. 6 control). When 3′ biotin labelling sequence was incubated with the truncated SREBP-D, an obviously band of DNA-protein complex was formed (Fig. 6 lane 1). The specificity of this binding was verified by adding wild or mutated sequences. Specifically, when incubated with the increasing amount of wild sequence, the intensity of this band was weakened and further disappeared (Fig. 6 lanes 2–4). In contrast, when incubated with excess mutated sequence, the intensity of this band was still very strong (Fig. 6 lanes 5–6) with minor damage, which probably due to excess mutations that hinder the binding of the labelling sequence to the protein.

Fig. 5. Amino acid sequence (A), prokaryotic expression (B) and purified protein (C) of Sinonovacula constricta sterol regulatory element binding protein (SREBP)-D. The underlined portions are the bHLH-Zip motif of S. constricta SREBP.

Fig. 6. Binding activity of Sinonovacula constricta sterol regulatory element binding protein (SREBP)-D to the predicted regulatory site of Δ6 fatty acyl desaturase (Fad) promoter. ‘+’ and ‘–’ indicate that the component was added or not, respectively. ‘+’, ‘++’ and ‘+++’ mean that the amount of unlabelled wild or mutant competitor probe is 100-, 500- and 1000-fold, respectively, of that of the 3′ biotin-labelling probe.
Discussion
The newly cloned S. constricta SREBP contained a diagnostic bHLH-Zip domain and two interrupted membrane binding domains that conserved in typical vertebrate SREBP(Reference Horton, Goldstein and Brown1), indicating that SREBP were highly conserved in functional regions during evolution. The phylogenic tree analysis revealed that vertebrate SREBP-1, SREBP-2 and marine molluscan SREBP were clearly clustered into three separate groups, indicating that they were significantly differentiated during species evolution. Interestingly, consistent with the finding that there was one SREBP in C. nobilis (Reference Liu, Zhang and Zheng33), only one SREBP was detected in S. constricta and other five molluscs (Crassostrea gigas, Aplysia californica, Crassostrea virginica, Octopus bimaculoides and Mizuhopecten yessoensis) by interrogating their genomes (Fig. 2). The result was different with the evidence of three SREBP (-1a, -1c and -2) in mammals(Reference Horton, Goldstein and Brown1) and two SREBP (-1 and -2) in teleosts(Reference Dong, Tan and Cai20,Reference Tay, Kuah and Shu-Chien21,Reference Minghetti, Leaver and Tocher34) . This might be explained by that the invertebrates considered cannot biosynthesise sterol de novo, and the role of SREBP in regulating sterol metabolism might be subsequently acquired by gene duplication (producing SREBP-2) in chordates when diverged from arthropods and nematodes(Reference Rawson35). Consistently, Liu et al.(Reference Liu, Zhang and Zheng33) reported that C. nobilis SREBP shows more similarity with vertebrate SREBP-1 because its expression exhibits a positive correlation with the lipid content changes in ovary. However, in particular, the de novo sterol biosynthesis has been reported in some invertebrates, that is, echinoderms(Reference Carson and Lennarz36). Therefore, whether the single SREBP found in invertebrates play both regulatory roles in sterol and fatty acid biosynthesis or not needs further investigation.
The transcription factor binding elements are usually distributed in the vicinity of gene promoter; thus, the 2000 bp upstream the translation initiation codon (ATG) of S. constricta Δ6 Fad was cloned. Though very conservative binding sites of NF-Y and SRE exist in teleost Fads2 promoters, this was not the case with S. constricta Δ6 Fad promoter. The result indicated that the DNA sequence of Δ6 Fad promoters from teleosts and marine molluscs were differentiated during evolution, and the transcriptional regulation mechanism of SREBP on Δ6 Fad might be very distinctive in S. constricta.
Overexpression of S. constricta SREBP in HEK 293 T cells induced a significantly higher promoter-driven luciferase activity, indicating that S. constricta SREBP was involved in the transcriptional regulation of Δ6 Fad. Similar results had been found in vertebrates(Reference Nara, He and Tang17,Reference Dong, Tan and Cai20,Reference Tay, Kuah and Shu-Chien21) . By 5′-deletion analysis of S. constricta Δ6 Fad promoter, the region of 936-1389 bp upstream Δ6 Fad promoter was predicted to be responsible for its sufficient transcriptional activation. Based on the in silico analysis of Δ6 Fad promoter, a binding site was validated specifically to interact with SREBP-D protein by EMSA, which was exactly located in the core region identified above. Therefore, it was speculated that this binding site might be the potential element that acted by SREBP. Interestingly, though this site harbouring 5′-GTGGGGTAACT-3′ was aligned in the same location with the conserved SRE (5′-CTCGAATGATC-3′) of teleost Fads2 promoters, the base composition between them was obviously different excepted for the underlined four bases (T, G, T, A). It indicated that the conserved four bases might play a key role in the site recognition and binding on Fads2 promoter by SREBP. In addition, the predicted binding sequence was more identical to the classic SRE sequence (forward sequence 5′-ATCACCCCAC-3′, reverse sequence 5′-GTGGGGTGAT-3′), while the binding site of teleost Fads2 promoters was more similar with the modified SRE sequence of 5′-CTCACACGAG-3′(Reference Shimano9,Reference Amemiya-Kudo, Shimano and Hasty37) . It indicated that the SRE sequence might be differentiated during species evolution.
Honestly, though no direct evidence of SREBP regulating Δ6 Fad expression for LC-PUFA biosynthesis in S. constricta in vivo, some clues have been observed in our previous work. For example, the expressions of SREBP (not published) and Δ6 Fad exhibit similar patterns in S. constricta larvae at early developmental stages and when fed with different microalgae of distinguished LC-PUFA composition(Reference Ran, Kong and Xu38). Besides, an experiment of silencing SREBP in S. constricta has been in progress by our group, which should provide more direct evidence in the near future.
In conclusion, in the present study, the transcriptional activity of S. constricta Δ6 Fad promoter was demonstrated to be activated by SREBP, and the binding site of SREBP on the Δ6 Fad promoter was predicted; thus, the potential molecular mechanism of SREBP regulating Δ6 Fad expression in S. constricta was revealed. This was the first report on the regulatory mechanism of LC-PUFA biosynthesis in marine molluscs, which would facilitate optimising the LC-PUFA biosynthetic pathway of bivalves in further studies.
Acknowledgements
This research was supported by the National Key Research and Development Program of China (2019YFD0900400), Zhejiang Major Science Project, China (2019C02057), Ningbo Science and Technology Research Projects, China (2019B10006), and the Earmarked Fund for Modern Agro-industry Technology Research System, China (CARS-49).
J. X., X. Y. and Z. R. designed the study. Z. R. and F. K. conducted the study and wrote the paper. Z. R. and K. L. analysed the data. All authors have read and approved the final manuscript.
The authors declare that there are no conflicts of interest.
Supplementary material
For supplementary material referred to in this article, please visit https://doi.org/10.1017/S0007114520002068