


Introduction
Dravet syndrome (previously known as “severe myoclonic epilepsy of infancy”) was first described by Dr. Charlotte Dravet in 1978.Reference Dravet 1 It is an early-onset treatment-resistant epilepsy syndrome that typically presents during the first year of life. The incidence of Dravet syndrome is estimated at 1 per 22,000-40,000 based on studies in the United Kingdom and Denmark,Reference Brunklaus, Ellis, Reavey, Forbes and Zuberi 2 , Reference Bayat, Hjalgrim and Moller 3 and it affects males twice as often as females. It typically causes an epileptic encephalopathy. In 2001, Claes and colleaguesReference Claes, Del-Favero, Ceulemanns, Lagae, Van Broeckhoven and De Jonghe 4 discovered mutations in the sodium channel alpha1 subunit gene SCN1A in seven individuals with Dravet syndrome. SCN1A mutations, usually de novo, are found in 70 to 80% of patients with Dravet syndrome. It is now appreciated that the electroclinical features of Dravet syndrome are broader than the original description and that the myoclonic seizures or generalized spike and wave are not present in all patients. Focal seizures that may evolve to generalized convulsive seizures are the dominant seizure type in some individuals and are associated with multifocal EEG abnormalities. With the wider application of new genomic technologies in the diagnosis of epilepsy of unknown cause, it is recognized that other genetic abnormalities (PCDH19, GABRG2, SCN1B and SCN2A) may cause a similar phenotype as Dravet syndrome.
Clinical Presentation and Evolution of Epilepsy in Dravet Syndrome
Seizure onset is typically in the first year of life, with prolonged, febrile and afebrile hemiclonic or generalized clonic seizures in previously healthy children. They may be associated with vaccinations or hyperthermia, including a warm bath. Over the subsequent months, affected individuals experience recurrent febrile and afebrile seizures that often affect alternate sides of the body. Between one and four years of age, other seizure types develop, including myoclonic and atypical absences, focal seizures and generalized tonic-clonic seizures. Focal seizures, with or without impairment of awareness, may be associated with such prominent autonomic features as pallor, cyanosis and drooling, and may evolve into a focal motor or bilateral convulsive seizure. Tonic seizures are reported to be uncommon in Dravet syndrome. In some individuals, myoclonic seizures do not develop and other seizure types, particularly focal or multifocal seizures, are the predominant seizure types.
In a retrospective surveyReference Xu, Zhang, Sun, Liu, Yang and Xiong 5 of 138 children with Dravet syndrome with SCN1A mutations from China, seizure onset was before the age of 7 months in 77%. 72% of the children in that study had febrile seizures with a duration longer than 15 minutes, and 67% had two or more febrile seizures within a 24-hour period. Seizures were hemiclonic with fever in 80%.Reference Xu, Zhang, Sun, Liu, Yang and Xiong 5 In a study of 96 children who had febrile seizures prior to one year of age, 46 of whom had Dravet syndrome and 50 of whom did not, the factors that correlated significantly with Dravet syndrome were onset of febrile seizures under the age of 7 months, 5 or more seizures, duration of seizures longer than 10 minutes, hemiconvulsions, focal seizures, myoclonic seizures and hot-water-induced seizures.Reference Hattori, Ouchida, Ono, Miyake, Maniwa and Mimaki 6 Reflex seizures are frequent, and the most common trigger is hyperthermia (fever, immersion in hot water, intense physical exercise or high ambient temperature).
Status epilepticus occurs commonly in Dravet syndrome, both the convulsive and non-convulsive types. Non-convulsive status epilepticus is also described in the literature as “obtundation status epilepticus.” Obtundation status epilepticus is characterized by altered awareness, autonomic symptoms and subtle myoclonus that commonly involves the fingers and orobuccal muscles, and it may last for many hours or days.Reference Bureau and Dalla Bernardina 7 Status epilepticus may be life threatening, and the symptoms and signs of non-convulsive status epilepticus may be subtle and difficult to appreciate. Thus, it is of utmost importance to educate parents and caregivers about the early symptoms and signs of non-convulsive status epilepticus. It is also very important to put an individualized emergency (rescue) management plan in place for each individual so as to facilitate early intervention to prevent prolonged status epilepticus and its associated complications.
With respect to the evolution of epilepsy over time, seizures tend to become less frequent and severe in adolescence and adulthood. Fever sensitivity persists but has less impact. Myoclonic, atypical absence and focal seizures with altered awareness are less common in adulthood.Reference Genton, Velizarova and Dravet 8 The most common seizure type in adulthood is generalized tonic-clonic, which may be focal in onset and occurs mainly during sleep. Patients may also experience bilateral or asymmetrical posturing, which may be followed by tonic vibratory or clonic movements. However, there are few studies that describe in detail the electroclinical features of the seizure types in Dravet syndrome during adolescence or adulthood. Ictal recordings of five adults revealed subclinical focal seizures in three, focal seizures that evolved into bilateral convulsive seizures (secondarily generalized tonic-clonic seizures) in three, obtundation status in one and tonic events in another patient.Reference Genton, Velizarova and Dravet 8 In a long-term studyReference Caraballo and Fejerman 9 of 53 children aged 4 to 14 years who were followed for from 3 to 14 years, seizure frequency was weekly in 74%, monthly in 13% and daily in 13%. The most common treatments employed in this study were valproic acid, clobazam and a ketogenic diet.
Takayama and colleaguesReference Takayama, Fujiwara, Shigematsu, Imai, Takahashi and Yamakawa 10 described 64 patients followed at one epilepsy center in Japan. The median age at last follow-up was 30 years (range=19-45), and only 1 of 44 individuals with typical Dravet syndrome was in remission, in contrast with 4 of 20 individuals with atypical Dravet syndrome. In this study, atypical Dravet syndrome was characterized by absence of myoclonic and atypical absence seizures.
EEG Evolution in Dravet Syndrome
The EEG is typically normal during the first year of life. Subsequently, the EEG background may be normal or slow. Sleep architecture is typically preserved. Generalized spike and wave, polyspike wave and multifocal spikes are common.
Few studies have carefully documented the evolution of EEG findings over time in Dravet syndrome. Speechio and colleaguesReference Specchio, Balestri, Trivisano, Japaridze, Striano and Carotenuto 11 described EEG findings in 22 children with Dravet syndrome over the first five years following diagnosis. In all these children, the EEG background was normal, but background slowing was seen in 27% after six months. Epileptiform discharges were present in 27% at seizure onset and in 64% by 5 years follow-up. Multifocal epileptiform discharges were observed in 57%, focal epileptiform discharges in 29%, and 14% had generalized epileptiform discharges. A photoparoxysmal response was seen in 9% at seizure onset and in 41% by the end of the study. In a study of 16 children, Korff et al.Reference Korff, Laux, Kelley, Goldstein, Koh and Nordli 12 reported that the EEG was abnormal in 81%. However, at the time of seizure onset, the EEG was abnormal in only 25% of the children. The most common epileptiform pattern consisted of generalized spike wave discharges.Reference Korff, Laux, Kelley, Goldstein, Koh and Nordli 12
In a study of 23 patients in whom seizures were recorded during video-EEG monitoring, Kim and colleaguesReference Kim, Nordli, Berg, Koh and Laux 13 divided patients into three groups: 0-5 years (n=7), 6-10 years (n=11) and 11 years and older (n=5). Slowing of the EEG background was more common in older patients and a photoparoxysmal response in younger children. Epileptiform discharges were found in most patients, and the most common pattern was multifocal epileptiform discharges.
Nabbout et al.Reference Nabbout, Desguerre, Sabbagh, Depienne, Plouin and Dulac 14 also described five adolescents with Dravet syndrome who had bifrontal spike and slow wave and generalized fast polyspikes in sleep, features similar to Lennox–Gastaut syndrome. In another study of EEG evolution in 24 patients with Dravet syndrome, Genton et al.Reference Genton, Velizarova and Dravet 8 described normal EEG background in 8 and slow and disorganized background in 11. Epileptiform activity was multifocal in 11, focal in 7 and generalized in 6. Photosensitivity and pattern sensitivity tended to disappear by 20 years of age.
There is still an opportunity to add to the literature on the evolution of the electroclinical features of Dravet syndrome with SCN1A mutations over time, and additional ictal EEG data would be very informative.
Family History of Epilepsy/Febrile Seizures
The first report of SCN1A mutations in epilepsy was in the syndrome of genetic epilepsy with febrile seizures plus.Reference Escayg, MacDonald, Meisler, Baulac, Huberfeld and An-Gourfinkel 15 A family history of epilepsy or febrile seizures has been observed in 15-35% of individuals with Dravet syndrome, and the most common phenotype seen in affected relatives is genetic epilepsy with febrile seizures plus.Reference Singh, Andermann, Whitehouse, Harvey, Keene and Seni 16 Dravet syndrome has been reported in monozygotic twins and rarely in zygotic twins. There are also rare reports of two or more affected children in the same family. Somatic or germline mosaic mutations may explain an unaffected or mildly affected parent, and in one studyReference Depienne, Trouillard, Saint-Martin, Gourfinkel-An, Bouteiller and Carpentier 17 mosaicism was found in 7% of families with Dravet syndrome. The observation of a positive family history of febrile seizures and epilepsy in individuals with Dravet syndrome and de novo mutations in SCN1A suggest that the mode of inheritance is polygenic and that other modifier genes such as SCN9A contribute to the phenotype.
Genetic Testing in Dravet Syndrome
The first mutations in the SCN1A gene in seven individuals with Dravet syndrome were discovered by Claes and colleaguesReference Claes, Del-Favero, Ceulemanns, Lagae, Van Broeckhoven and De Jonghe 4 in 2001. It is now recognized that SCN1A mutations are seen in 75-80% of individuals with Dravet syndrome. More than 500 mutations have been reported in individuals with Dravet syndrome. Mutations are truncating in 40-50%, missense in 40% and splice site in the remainder. Whole-gene deletions are observed in 2-3%, and gene duplications are rare. Most gene mutations are de novo, but familial mutations occur in 5-10% and are usually missense.Reference Claes, Del-Favero, Ceulemanns, Lagae, Van Broeckhoven and De Jonghe 4 , Reference Nabbout, Gennaro, Dalla Bernardina, Dulac, Madia and Bertini 18 , Reference Wallace, Hodgson, Grinton, Gardiner, Robinson and Rodriguez-Casero 19
Truncating, nonsense and frameshift mutations and gene deletions correlate with the classical Dravet syndrome phenotype with an earlier age at onset.Reference Marini, Mei, Temudo, Ferrari, Buti and Dravet 20 , Reference Marini, Scheffer, Nabbout, Suls, De Jonghe and Zara 21 Missense mutations that affect the pore-forming region are associated with a more severe phenotype. Individuals who test negative for SCN1A sequence-based mutations may have SCN1A exon deletions or chromosomal rearrangements involving SCN1A and contiguous genes, which may be detected using multiplex ligation-dependent probe amplification (MPLA) and comparative genome hybridization (CGH). The Genetics Commission of the International League Against Epilepsy (ILAE) has recently published guidelines for SCN1A testing in clinical practice.Reference Hirose, Scheffer, Marini, De Jonghe, Andermann and Goldman 22 Their recommendations are summarized in Table 1.
Table 1 Predominant seizure types in Dravet syndrome
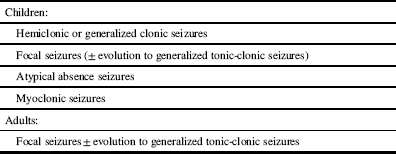
The phenotype of SCN1A mutations may be heterogeneous even within the same family, and it is likely that modifying genes are important in determining the severity of epilepsy and possibly comorbidities.Reference Brunklaus, Ellis, Reavey, Forbes and Zuberi 2 In animal models, SCN8A may restore a normal seizure threshold in the presence of SCN1A mutations.Reference Martin, Tang, Ta and Escayg 23 The compensatory up-regulation of SCN3A may mitigate the effects of Nav1.1 deficiency. Variants in CACNA1A genes may also modify the phenotype in Dravet syndrome. In a studyReference Ohmori, Ouchida, Kobayashi, Jitsumori, Mori and Michiue 24 of 48 patients with Dravet syndrome and SCN1A mutations, CACNA1A variants were observed in 20 subjects. In individuals with CACNA1A variants, absence seizures, earlier age at seizure onset and more frequent prolonged seizures before the age of one year were more common than in Individuals without CACNA1A variants.Reference Ohmori, Ouchida, Kobayashi, Jitsumori, Mori and Michiue 24 SCN9A mutations have also been observed in some individuals with SCN1A mutations and may be associated with a more severe clinical phenotype.Reference Mulley, Hodgson, McMahon, Iona, Bellows and Mullen 25 , Reference Singh, Pappasa, Dahle, Claes, Pruess and De Jonghe 26
In children where SCN1A mutations are not found, several other genes have been reported, including CHD2, GABRA1, GABARG2, STXBP1, SCN1B, SCN2A and PCDH19.Reference Suls, Jaehn, Kecskés, Weber, Weckhuysen and Craiu 27 - Reference Depienne, Bouteiller, Keren, Cheuret, Poirier and Trouillard 29 However, the electroclinical phenotypes associated with these other gene abnormalities are often somewhat atypical for Dravet syndrome. With the wider application of next-generation sequencing in the evaluation of epilepsy of unknown cause, it is likely that the electroclinical features associated with gene mutations other than SCN1A will be better described.
Pathophysiology of Dravet Syndrome
Several pathophysiological mechanisms have been reported in Dravet syndrome, and the most accepted is the “interneuron hypothesis,” where SCN1A mutations result in inhibition of the GABAergic inhibitory interneurons, resulting in excessive excitation. This mechanism is supported by a mouse model.Reference Cheah, Yu, Westenbroek, Kalume, Oakley and Potter 30 Nav1.1 was preferentially expressed in the axon initial segments of the parvoalbumin-positive interneurons, the main type affected in Dravet syndrome.Reference Yamakawa 31 Studies using models involving human-derived induced pluripotent stem cell neurons suggest that both GABAergic inhibitory neurons and glutamatergic excitatory neurons in the forebrain of patients with Dravet syndrome are hyperexcitable with increased sodium current density, overall resulting in network hyperexcitability.Reference Liu, Lopez-Santiago, Yuan, Jones, Zhang and O’Malley 32 SCN1A haploinsufficiency may result in a compensatory increase in the sodium current via expression of other sodium channels.Reference Chopra and Isom 33
Neuroimaging in Dravet Syndrome
Structural brain imaging using MRI is usually normal in Dravet syndrome, but imaging abnormalities are described in some patients. In a reviewReference Barba, Parrini, Coras, Galuppi, Craiu and Kluger 34 of 120 patients in Italy with Dravet syndrome, malformations of cortical development were detected in 4. In another study,Reference Gaily, Anttonen, Valanne, Liukkonen, Träskelin and Polvi 35 of 18 children with Dravet syndrome who had MR imaging after 3 years of age, 7 had hippocampal sclerosis or loss of grey–white definition in the temporal lobe. In a studyReference Pérez, García-Pentón, Canales-Rodríguez, Lerma-Usabiaga, Iturria-Medina and Román 36 comparing nine patients with Dravet syndrome and SCN1A mutations and nine controls without seizures, globally reduced grey and white matter volumes were seen in individuals with Dravet syndrome.
Neurodevelopment in Dravet Syndrome
Neurodevelopment and formal neurological examination are typically normal at the time of seizure onset. However, there is slowing of the rate of the developmental progress along with variable decline in the developmental quotient over time.Reference Dravet, Bureau, Dalla Bernardina and Guerrini 37 Ataxia and pyramidal signs were present in 59 and 22% of subjects, respectively.Reference Dravet, Bureau, Dalla Bernardina and Guerrini 37 Attention, visual/motor integration, visual perception and executive function are impaired, whereas language may be less involved in some patients.Reference Chieffo, Ricci, Baranello, Martinelli, Veredice and Lettori 38 , Reference Chieffo, Battaglia, Lettori, Del Re, Brogna and Dravet 39
In a studyReference Villeneuve, Laguitton, Viellard, Lépine, Chabrol and Dravet 40 of 21 children aged 6 to 10 years with Dravet syndrome, seen at a single institution and assessed using the WISC and Vineland Adaptive Behaviour scales, no child had a normal IQ after the age of 6 years. Only 5 of the 21 children had a verbal or non-verbal IQ score greater than 60. The children had attention problems, impulsivity, perseverative responses and deficits in planning. Socialization skills assessed using the Vineland scale were significantly better than communication skills. No correlation was observed between age at seizure onset, status epilepticus, number of seizures, myoclonic or absence seizures with IQ. Nabbout and colleaguesReference Nabbout, Chemaly, Chipaux, Barcia, Bouis and Dubouch 41 reported cognitive findings in 67 children with Dravet syndrome, 52 of whom were tested only once and 15 underwent repeat testing. IQ was typically normal before the age of 2 years (mean of 80) but low after age 3 years (mean=48, range=30-69). However, there was no evidence of regression. Attention and hyperactivity were common. No significant correlation was seen between IQ and age at first seizure, number of episodes of status epilepticus, or myoclonic and focal seizures. However, IQ was lower in those with SCN1A mutations. Thus, it was the conclusion of the authors that the encephalopathy was not a pure consequence of epilepsy but that the SCN1A mutation played an additional direct role.
Genton and colleaguesReference Genton, Velizarova and Dravet 8 reported 24 patients with Dravet syndrome followed to 20-50 years of age. Only three individuals lived independently, and the remainder had moderate to severe delay. Ataxia was seen in nine, dysarthria in eight and tremor in seven. In another report of 31 patients, only 1 lived independently, 7 had no language and 9 spoke only a few words.Reference Akiyama, Kobayashi, Yoshinaga and Ohtsuka 42 Catarino et al.Reference Catarino, Liu, Liagkouras, Gibbons, Labrum and Ellis 43 described the long-term follow-up in 22 adults followed to a median of 39 years. Most had intellectual disability, 16 lived in residential care and 6 at home with support. Takayama and colleaguesReference Takayama, Fujiwara, Shigematsu, Imai, Takahashi and Yamakawa 10 reported on long-term outcome in 64 patients followed to a median of 30 years. All had intellectual disability, which was severe in 75%, moderate in 20% and mild in 5%. Only two individuals lived independently.
In Dravet syndrome, there is widespread dysfunction of Nav1.1. Maturation of sodium channels in humans and mice shows that Nav1.1 expression is very low in neonates, and other alpha subunits Nav1.2 and 1.3 compensate. Normally, with increasing age, Nav1.3 declines and Nav1.1 increases. In Dravet syndrome, Nav1.1 fails to increase normally, resulting in widespread dysfunction of Nav1.1. This may explain why seizures do not start until later on, during the first year of life. Furthermore, mouse modelsReference Han, Tai, Westenbroek, Yu, Cheah and Potter 44 of Dravet syndrome demonstrate hyperactivity, stereotypic behaviour and cognitive deficits due to reduced expression of GABAergic interneurons in the forebrain. In a studyReference Bender, Natola, Ndong, Holmes, Scott and Lenck-Santini 45 of mice where SCN1A was selectively reduced but the animals did not develop seizures, cognitive function was affected, providing further evidence that Nav1.1 dysfunction plays a role independent of seizures on cognitive decline.
Comorbidities in Dravet Syndrome
A crouch gait has been described in many reports on Dravet syndrome. In one studyReference Rodda, Scheffer, McMahon, Berkovic and Graham 46 utilizing formal gait analysis in patients with Dravet syndrome who ranged in age from 2 to 34 years, crouch gait was seen in 8 of 9 children aged 13 years and older, in 50% of children aged 6-12 years and in no child younger than 6 years. Fasano and colleagues,Reference Fasano, Borlot, Lang and Andrade 47 in a study of 12 adults with Dravet syndrome, described features of Parkinsonism with bradykinesia, asymmetric rigidity and cogwheeling in 11 (91%). Two patients were treated successfully with levodopa, resulting in significant improvement in their Parkinsonian symptoms. Osteopenia and increased risk of fractures are common in individuals with treatment-resistant epilepsy, particularly when enzyme-inducing drugs are used or there is reduced mobility, but this has not been studied in Dravet syndrome.
Despite sleep concerns being a common complaint in individuals with Dravet syndrome, the literature on this topic is limited. A small, retrospective studyReference Dhamija, Erickson, St Louis, Wirrell and Kotagal 48 of polysomnogram findings in six children with Dravet syndrome referred for sleep assessment found no abnormalities in sleep macroarchitecture despite parental concerns.
SUDEP and Mortality
Mortality is increased significantly in Dravet syndrome, and death may occur at any age but is more frequent during childhood. Death may be due to status epilepticus, SUDEP or accidental death, and it may also be related to seizures associated with drowning or injury. Mortality rates have varied in reported studies from 3.1 to 20.8%, with deaths due to sudden, unexpected death of someone with epilepsy (SUDEP), status epilepticus and accidents (especially drowning) being the most common. In a reportReference Skluzacek, Watts, Parsy, Wical and Camfield 49 based on data from a patient support group, it was estimated that individuals with Dravet syndrome have a risk of SUDEP approximately 15-fold higher than those with other childhood epilepsies.
Studies in Dravet mice modelsReference Kalume, Westenbroek, Cheah, Yu, Oakley and Scheuer 50 suggest that SUDEP is due to increased parasympathetic activity after a generalized tonic-clonic seizure, resulting in lethal bradycardia and electrical ventricular dysfunction. As this finding was seen in mice with the SCN1A mutations selectively targeted to affect the brain, but not if the SCN1A mutations were selectively targeted to affect the heart only, it was proposed that SUDEP is due to a brain effect on the heart after a seizure. Cheah and colleaguesReference Cheah, Westenbroek, Roden, Kalume, Oakley and Jansen 51 proposed that the natural decline in Nav1.3 channel expression with the failure of increase in Nav1.1 channel expression leads to disinhibition of neuronal circuits, treatment-resistant epilepsy and premature death. In a studyReference Auerbach, Jones, Clawson, Offord, Lenk and Ogiwara 52 of mice with an SCN1A mutation, Auerbach observed a twofold increase in sodium current density in ventricular myocytes, resulting in increased excitability, prolongation of action potential duration, which results in QT prolongation, abnormal rhythms and spontaneous deaths in some mice. Thus, it is possible that altered electrical cardiac function may increase the risk of SUDEP, but these animal data have not been confirmed in humans.
Conclusion
Dravet syndrome remains one of the most severe epilepsy syndromes of early childhood and is characterized by very high morbidity and mortality. It impacts physical and cognitive function, mental health, bone health and sleep. Anxiety about the risk of death and SUDEP is very high in parents and caregivers. Early diagnosis of Dravet syndrome is important to avoid anti-seizure medications that exacerbate seizures, but there is a major need for new therapies to improve seizure control and long-term outcomes.
Transition from paediatric to adult services is very challenging, and there is a great need to improve this process.Reference Ceulemans 53 , Reference Kuchenbuch, Chemaly, Chiron, Dulac and Nabbout 54 It is also likely that Dravet syndrome is underdiagnosed in adults with treatment-resistant epilepsy. It is therefore important to educate colleagues who are caring for adults about this epilepsy syndrome.
Disclosures
Mary Connolly has the following disclosures: Novartis, site principal investigator, research support; Novartis, speaker, honorarium; UCB, speaker, honorarium. All funds go to the Epilepsy Research and Development Fund (no personal benefit).
Table 2 ILAE guidelines for SCN1A testingReference Hirose, Scheffer, Marini, De Jonghe, Andermann and Goldman 22



