


Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous disorder with an estimated incidence of 1:6000 live births.Reference Osborne, Fryer and Webb 1 Two causative genes have been identified. TSC1 is located on chromosome 9q34 and encodes the protein hamartin,Reference van Slegtenhorst, de Hoogt and Hermans 2 whereas TSC2 is found on chromosome 16p13 and encodes tuberin. 3 Hamartin and tuberin form a complex that controls cell growth and proliferation through mechanisms that include inhibition of the mammalian target of rapamycin (mTOR) pathway.Reference Au, Williams, Gambello and Northrup 4 Understanding the pathophysiology of TSC has led to the development of medications directed at inhibiting the mTOR pathway that have been approved to treat various manifestations of TSC.Reference Franz, Belousova and Sparagana 5 - Reference Kotulska, Chmielewski and Borkowska 7
Clinically, TSC is characterized by hamartomatous growths that can occur in virtually any organ, including the skin, brain, heart, and kidneys and rarely the liver.Reference Northrup, Koenig and Au 8 Diagnostic criteria for TSC have recently been updated and outline 11 major and six minor features that allow for a clinical diagnosis of either definite or possible TSC.Reference Northrup and Krueger 9 Central nervous system manifestations include cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas (SEGAs). Epilepsy is the most common neurologic symptom in childhood, reported in 80% to 90% of patients, and is often resistant to medical therapy.Reference Holmes and Stafstrom 10 Onset of epilepsy is typically within the first 2 years of life, and more than one-third of patients may develop infantile spasms.Reference Chu-Shore, Major, Camposano, Muzykewicz and Thiele 11 Tuberous sclerosis-associated neuropsychiatric disorders occur in most individuals with TSC, are often underdiagnosed, and cause significant morbidity.Reference Curatolo, Moavero and de Vries 12 Autism spectrum disorder is the most common psychiatric diagnosis, reported in 17% to 61% of patients,Reference Curatolo, Porfirio, Manzi and Seri 13 whereas approximately half of patients have intellectual disability, often in association with autism.Reference Joinson, O’Callaghan, Osborne, Martyn, Harris and Bolton 14
Recommendations have recently been developed that outline a framework to optimize and standardize the surveillance and care for patients with TSC.Reference Krueger and Northrup 15 These underscore the importance of a multidisciplinary team familiar with the management of TSC. The objective of this study was to assess the manifestations of TSC as seen through the neurology department at a large Canadian provincial children’s hospital. We hope that this will assist in identifying the resources to provide in a multidisciplinary TSC clinic and to optimize ongoing patient care.
METHODS
This retrospective study was approved by the University of British Columbia Clinical Research Ethics Board. All patients were assessed through the Pediatric Neurology clinic at British Columbia Children’s Hospital between 1990 and 2014. Patients were identified through prospectively maintained epilepsy and clinical neurophysiology databases as well as referrals to the TSC clinic, which was established in 2012. Data were obtained by systematic chart review.
Patients were included if they met the diagnostic criteria for definite TSC, as outlined by the 2012 International Tuberous Sclerosis Consensus Conference.Reference Northrup and Krueger 9 Epilepsy was diagnosed by a documented history of two or more unprovoked seizures. Seizure types were classified according to the International League Against Epilepsy Commission on Classification and Terminology.Reference Berg, Berkovic and Brodie 16 Patients were considered seizure-free if there was a minimum of 1 year following the last documented seizure at the time of most recent follow-up. Developmental delay and psychiatric conditions were reported as present when those diagnoses were documented by a pediatrician, psychiatrist, or neurologist. The end point of follow-up was considered as the time of the last documented clinical neurology visit.
Clinical data were analyzed descriptively. The proportions of patients manifesting various clinical features are reported and, where applicable, median values are reported along with the range of values obtained. Because of the population size and retrospective nature, the aim of this study was primarily descriptive, and so comparative statistical analysis was not performed.
RESULTS
Study Population
A total of 81 patients were identified who were born between 1987 and 2014. Genetic testing for TSC is not routinely funded in the province of British Columbia, but six patients (7%) had genetic confirmation of diagnosis (two TSC1, four TSC2). Forty-one (51%) were male. The median age at most recent clinical follow-up was 10.0 years (range, 0.2-23.2)(Figure 1). Eleven children (14%) were diagnosed with TSC antenatally, based on the presence of cardiac rhabdomyomata on prenatal ultrasound. Six patients (7%) had a documented family history of TSC in a first-degree relative.
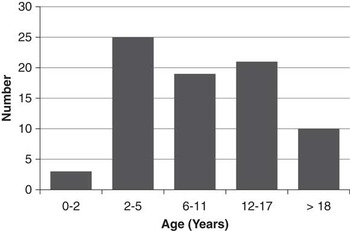
Figure 1 Number of Patients by Age at most recent follow-up.
Epilepsy
Seventy-four patients (91%) had a diagnosis of epilepsy. The median age at seizure onset was 12 months (range, 0-168)(Table 1). The initial seizure type was most commonly focal (66%), followed by epileptic spasms (26%) and generalized (5%). Fifteen patients (20%) had a documented history of status epilepticus, occurring at a median age of 15 months (range, 2-180). At the most recent follow-up, 45 (61%) were seizure-free for a median 4.5 years (range, 1-15), including eight (11%) who were seizure-free off antiseizure medications. Patients were on a median of one antiseizure medication (range, 0-5) and had failed a median of two medications (range, 0-10).
Table 1 Epilepsy Details
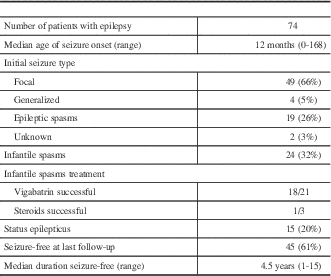
Overall, 32% of patients developed infantile spasms at a median age of 5 months (range, 0.25-30). Vigabatrin successfully treated spasms in 18 of 21 patients (86%) in whom it was tried, whereas corticosteroids were successful in one of three patients (33%).
Those younger than age 12 months at first seizure most often had epileptic spasms as their initial seizure type (54%), whereas those 12 months of age and older at seizure onset most often had focal seizures as their initial seizure type (84%)(Figure 2). A total of 71% of children who had seizure onset after 12 months of age were seizure-free at most recent follow-up, compared with 49% seizure-free in those who had seizure onset before 12 months of age.
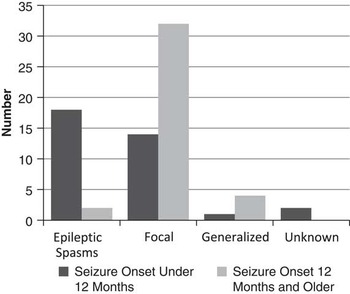
Figure 2 Seizure type and age at onset.
Epilepsy Surgery
Nineteen patients (26% of those with epilepsy) underwent epilepsy surgery on at least one occasion (Table 2). Surgery was performed following video-electroencephalogram (EEG) monitoring, high-resolution magnetic resonance imaging (MRI) and neuropsychological testing. In some cases, functional imaging using subtraction ictal SPECT coregistered with MRI. Rarely, positron emission tomography using alpha-methyl-tryptophan was performed. In those who underwent surgery, the median age at seizure onset was 5 months (range, 0-22). Their initial seizure type was focal in 12 (63%), epileptic spasms in six (32%), and generalized in one (5%). Twelve (63%) eventually developed multiple seizure types and all had focal seizures. These patients had failed a median of four antiseizure medications (range, 2-10). The median age at first surgery was 4.8 years (range, 1.1-15.6), whereas the last follow-up occurred a median 2.4 years (range, 0.2-16.2) following the initial surgery.
Table 2 Epilepsy Surgery Population

The initial surgery in 17 children was tuberectomy with resection of epileptogenic cortex guided by electrocorticography, whereas total corpus callosotomy was performed in two children. Surgery was most commonly in the frontal lobe (12 patients). Surgical complications were reported in four patients (21%), all of whom had a postoperative hemiparesis. This resolved completely in two patients, whereas two children had persistent mild hemiparesis that improved with time, but did not recover fully. The motor deficits were predicted based on the resected tuber involving or being adjacent to the motor cortex. One patient also developed chemical meningitis on postoperative day 3, which resolved completely with steroids.
Four patients (21%) underwent a second epilepsy surgery because of ongoing seizures, at a median of 23 months (range, 7-32) following the first surgery. One patient initially underwent a complete corpus callosotomy for disabling drop attacks, after which his seizures became focal in nature and he became seizure-free following a subsequent right frontal resection. A second patient had extension of a previous right frontal resection, which resulted in a seizure-free outcome. A third patient underwent a multilobar resection following a previous frontal resection, but did not have a reduction in seizure frequency. Finally, the fourth patient developed drop attacks following initial frontal tuber and cortical resection, and then underwent a total corpus callosotomy. This patient experienced a 50% to 90% reduction in seizure frequency.
Figure 3 illustrates seizure outcome following surgery based on the most recent follow-up. Nine patients (47%) were seizure-free, with a median seizure-free duration of 2.5 years (range, 1-9). Only two patients (11%) had less than a 50% reduction in seizure frequency following epilepsy surgery. At most recent follow-up, these patients were on a median of two antiseizure medications (range, 0-5), including one patient who was seizure-free off medication.
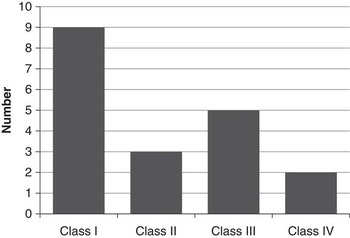
Figure 3 Seizures outcome following epilepsy surgery (Engel Classification).
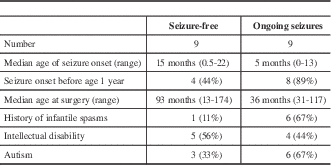
In patients who underwent resective surgery, compared with those with ongoing seizures, patients who were seizure-free tended to have an older age at seizure onset, less frequent history of infantile spasms, and a lower rate of autism (Table 3).
Table 3 Comparison of seizure-free patients and those with ongoing seizures following surgery
Structural Central Nervous System Abnormalities
All patients had neuroimaging available for review: 78 had at least one brain MRI and 3 had at least one head computed tomography, but no MRI. Cortical tubers were identified in 80 patients (99%). Seventy-nine patients (98%) had subependymal nodules on neuroimaging. Fifteen patients (19%) had evidence of a SEGA on neuroimaging, first identified at a median age of 9.5 years (range, 1-17.5)(Figure 4A). Of these, four underwent surgical resection of the SEGA.
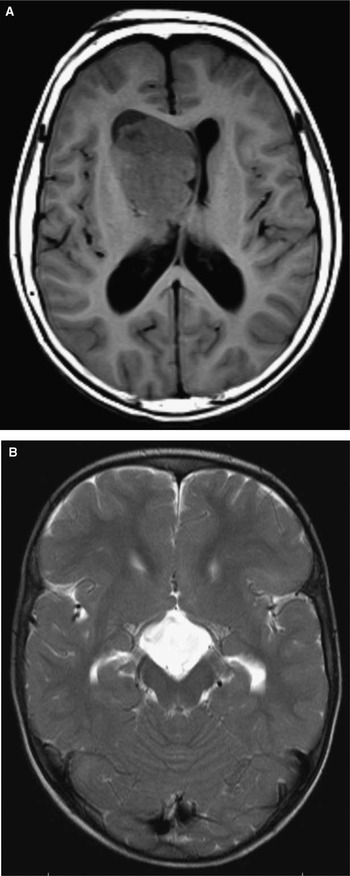
Figure 4 (A) Large right intraventricular SEGA (T1 weighted axial image); (B) Suprasellar cyst (T2 weighted axial image).
Other imaging findings included a pituitary prolactinoma that was identified in one patient who developed secondary central hypothyroidism. Another patient developed a suprasellar arachnoid cyst with secondary growth hormone deficiency (Figure 4B), requiring endoscopic cyst fenestration. A third developed a meningioma, which was surgically resected. One patient had a cerebellar tuber.
Cardiac
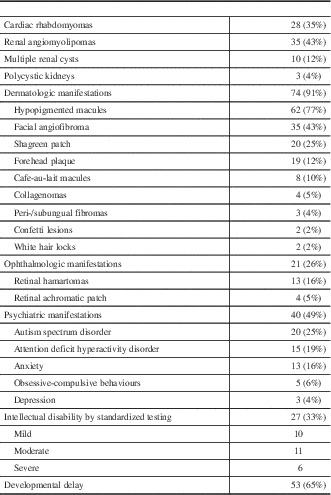
Cardiac rhabdomyomata were documented in 28 patients (35%)(Table 4). However, only three patients (11%) had clinical symptoms, which were all related to arrhythmia. This included one patient who developed supraventricular tachycardia and secondary congestive heart failure at the age of 17 months that was treated with amiodarone, which was later discontinued. A second patient developed fetal supraventricular tachycardia at 30 weeks’ gestation, requiring treatment with flecainide, which was discontinued at age 1 year. A third patient had inducible ventricular tachycardia, requiring an implantable defibrillator. No other patients required cardiac surgery and no cardiac abnormalities were reported in patients without a history of rhabdomyomata.
Table 4 Systemic manifestations of TSC
Renal
Thirty-five patients (43%) had at least one renal angiomyolipoma (AML) noted on ultrasound, including 15 with multiple AMLs. One patient with multiple AMLs underwent a partial nephrectomy because of concern of a renal cell carcinoma raised on renal MRI. Pathology of the excised lesion demonstrated a fat-poor AML. No other patients have undergone any treatment for their AMLs to date and no renal malignancies were identified in this population. An additional 15 patients (19%) had multiple renal cysts on ultrasound, whereas two children (2%) had polycystic kidney disease. Only one patient had impaired renal function, with renal ultrasound demonstrating a single AML and multiple renal cysts, but has not required treatment. An additional patient with multiple AMLs and renal cysts developed hypertension, requiring treatment with an ACE inhibitor, with no other cause for the hypertension found.
Dermatologic
A total of 74 patients (91%) had at least one dermatologic manifestation of TSC. The most common findings were hypopigmented macules in 62 (77%), facial angiofibromas in 35 (43%), and Shagreen patch in 20 (25%). Only one patient younger than 6 years old was noted to have facial angiofibromas. Treatment was documented in nine patients with facial angiofibromas and included laser therapy, cryotherapy, and use of a topical mTOR inhibitor. The frequency of less common skin findings is outlined in Figure 8.
Ophthalmologic
Twenty-one patients (26%) had at least one eye finding. Retinal hamartomata were seen in 13 patients (16%). This included one patient with markedly decreased vision, to the level of counting fingers, related to a hamartoma involving the optic nerve. Retinal achromatic patch was seen in 5%, whereas strabismus was noted in 4%. One patient had a unilateral Horner’s syndrome without an identifiable cause.
Psychiatric
Forty patients (49%) had at least one documented psychiatric diagnosis or behavioral abnormality. The most common diagnosis was autism spectrum disorder, reported in 20 patients (25%), all of whom had epilepsy. This was followed by attention deficit hyperactivity order (ADHD) in 15 (19%) and anxiety in 13 (16%). Common behavioral concerns included sleep difficulty (12%), aggression (11%), and impulsivity (9%). Thirty-two patients (40%) were assessed as having intellectual disability, either on standardized testing (27) or clinically (5). For individuals with intellectual disability diagnosed on formal testing, it was classified as mild in 10, moderate in 11, and severe in six. Fifty-three patients (65%) had a history of developmental delay. When classified by age of seizure-onset, rates of intellectual disability were similar in those with seizure onset before 12 months (43%) compared with after 12 months (37%).
When only those 52 patients who were school-aged (6 years and older) at last follow-up are considered, 32 (62%) had at least one psychiatric diagnosis. Autism was present in 17 school-aged children (33%), all except one of whom had intellectual disability. ADHD was present in 15 (29%) and anxiety in 11 (21%). Twenty-nine (56%) had intellectual disability, 27 based on standardized testing. Of all school-aged patients, 12 (23%) were functioning at grade level, seven (13%) were below grade level, 18 (35%) were in a modified or life skills program, and the school level was unknown in the remainder.
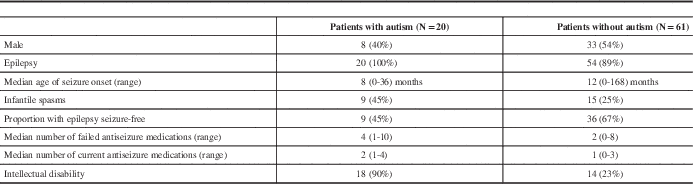
All patients diagnosed with autism spectrum disorder had a history of epilepsy. Patients with autism tended to have an earlier age of seizure onset, a higher rate of infantile spasms, more treatment-resistant epilepsy, and had higher rates of intellectual disability (Table 5).
Table 5 Comparison of patients with and without Autism Spectrum Disorder
Miscellaneous
One patient developed a lumbar hamartoma requiring surgical resection. Pathological examination demonstrated a benign nodular fibrous hamartoma. One patient was born with bilateral cleft lip and palate and hemivertebrae at T1 and T5, whereas another had congenital lymphedema of the left arm.
DISCUSSION
This study outlines the spectrum of TSC manifestations observed at a provincial pediatric neurology referral centre. Epilepsy was the most common symptom presenting in childhood, occurring in 91% of our patients, with most patients having seizure onset in the first 2 years of life. This is similar to the rate of 75% to 93% reported in other studies.Reference Chu-Shore, Major, Camposano, Muzykewicz and Thiele 11 , Reference Devlin, Shepherd, Crawford and Morrison 17 , Reference Au, Williams and Roach 18 Although epilepsy was typically treatment resistant in these patients, with most failing multiple antiseizure medications, 61% were at least 1 year seizure-free at the time of their last follow-up. This is better than recent reports in the literature, where only 32% to 49% achieved a period of 1-year seizure-freedom.Reference Overwater, Bindels-de Heus and Rietman 19 , Reference Vignoli, La Briola and Turner 20 The correlation between later age of seizure onset and improved seizure outcome has previously been reported in these studies. Approximately one-third of TSC patients have been reported to develop infantile spasms, most often in the first year of life,Reference Saxena and Sampson 21 , Reference O’Callaghan, Harris and Joinson 22 which is similar to our experience. We found that 86% of TSC patients with infantile spasms who were treated with vigabatrin had cessation of their spasms, comparable to the response rate of up to 95% previously reported.Reference Hancock and Osborne 23 Given the ascertainment of patients through a neurology clinic, our study may overestimate the frequency and severity of epilepsy in the overall population with tuberous sclerosis complex.
The role of surgical treatment for treatment-resistant focal epilepsy is increasing, with evidence that outcomes following surgery are superior to medical therapy.Reference Engel, McDermott and Wiebe 24 TSC is different from most focal epilepsies in that there is an underlying genetic abnormality and there are often multiple potentially epileptogenic lesions located outside of the area of resection. Despite this, outcomes from epilepsy surgery in TSC are generally positive, with up to 55% to 60% of patients reported to become seizure-free and only 10% having a reduction in seizures of less than 50%.Reference Jansen, van Huffelen, Algra and van Nieuwnhuizen 25 - Reference Krsek, Jahodova and Kyncl 27 Outcomes in our study population were very similar, with 47% being seizure-free and 10% having less than a 50% improvement following surgery.
The results of studies looking at outcome predictors for seizure-freedom following epilepsy surgery in TSC have been inconsistent. Clinical variables reported to be associated with a good postoperative outcome include the absence of tonic or generalized seizures, a milder degree of intellectual impairment, seizure onset after the first year of life, preoperative hemiparesis, unifocal EEG abnormalities, and concordance between EEG and neuroimaging studies.Reference Jansen, van Huffelen, Algra and van Nieuwnhuizen 25 , Reference Krsek, Jahodova and Kyncl 27 - Reference Zhang, Hu, Zhang, Meng, Chen and Zhang 29 Our study was not designed to examine predictors of outcome following surgery, although those with recurrent seizures after surgery tended to be younger at seizure onset and had a higher rate of infantile spasms. Some of the surgical patients in our cohort were analyzed as part of a recent multicenter, international, retrospective cohort study, which found that larger surgical resections (greater than a tuberectomy) were associated with a longer duration of seizure freedom postoperatively.Reference Fallah, Rodgers and Weil 30 The surgical complication rate in our cohort was low, similar to that reported in other studies,Reference Weiner, Carlson and Ridgway 31 with transient or mild hemiparesis being the most common complication. This was a predictable risk associated with frontal resection of tubers in close proximity to the motor area. Thus, our experience demonstrates that epilepsy surgery is a safe and effective treatment option in carefully selected children with TSC and treatment-resistant epilepsy. Further work is needed to determine the best methods by which to select patients for surgery and whether surgery may improve neurodevelopmental outcomes.Reference Liu, An and Yang 32 , Reference Zaroff, Morrison, Ferraris, Weiner, Miles and Devinsky 33
The high rate of cortical tubers and subependymal nodules found in our population is similar to that previously described.Reference Rosser, Panigrahy and McClintock 34 The ascertainment of patients through a neurology center is likely biased towards a higher rate of central nervous system findings compared to the general TSC population. 15% of our patients developed SEGAs, including two patients who presented symptomatically with dramatic growth in their SEGAs. Under the current guidelines, which recommend surveillance neuroimaging every 1 to 3 years,Reference Krueger and Northrup 15 these patients may have had their growing SEGAs discovered sooner and been candidates for intervention at an earlier point, with potentially less complicated courses. Other rare neuroimaging findings included meningioma, pituitary adenoma, and arachnoid cyst. There have been rare reports of these associated with TSC in the literature,Reference Castillon-Benavides, Salinas-Lara, Ponce-Guerrero, Leon, Gelista and Tena-Suck 35 - Reference Boronat, Caruso, Auladell, Van Eeghen and Thiele 37 which may represent incidental associations or a common pathogenic mechanism.
The systemic manifestations of TSC in our population are similar to those reported in the literature. We found a rate of cardiac rhabdomyomata of 35%, compared with the 30% to 50% reported rate.Reference Rosser, Panigrahy and McClintock 34 , Reference Hinton, Prakash, Romp, Krueger and Knilans 38 Only three of our patients became symptomatic from their rhabdomyomata, all related to arrhythmias, including one with congestive heart failure secondary to arrhythmia. Arrhythmia is the most common cardiac complication, reported in up to 25% of patients with rhabdomyomata,Reference Sciacca, Giacchi and Mattia 39 whereas only 2% to 5% of patients develop significant hemodynamic compromise and heart failure. The natural history of cardiac rhabdomyomata is spontaneous regression, so most patients will have improvement in their cardiac status over time without surgical intervention. Similarly, the 43% rate of angiomyolipomas in this study is comparable to the 40% to 80% previously reported.Reference Rosser, Panigrahy and McClintock 34 , Reference Korula, Ekbote, Kumar, Danda, Agarwal and Chaturvedi 40 The rate of renal cysts in our study, 19%, is at the low end of the 17% to 47% rate reported in these studies. Although typically asymptomatic in childhood, these lesions tend to increase in size over time and can lead to hypertension, hemorrhage, and end-stage renal disease.Reference Dixon, Hulbert and Bissler 41 Thus, the low rate of renal dysfunction and need for intervention in our population would be expected to change as patients age. Retinal hamartomas are the most common ophthalmic manifestation of TSC, reported in 44% of a population-based study.Reference Rowley, O’Callaghan and Osborne 42 The low rate in our population, 16%, is likely an underestimate related to the retrospective nature of the study. We describe one patient born with bilateral cleft lip and palate, which has only rarely been described in association with TSC.Reference Scully 43 One patient in our study was born with congenital focal lymphedema, which is an uncommon association with TSC and may reflect a shared pathophysiology.Reference Hoshiai, Oguma, Sato, Konishi and Minami 44
Psychiatric and behavioral comorbidities were very common, affecting half of patients in our study. Autism was the most common problem, diagnosed in 20%. The reported rates of autism in TSC range from 17% to 61%.Reference Curatolo, Porfirio, Manzi and Seri 13 Patients with autism in our study tended to have a younger age at seizure onset, a higher rate of infantile spasms, and more treatment resistant epilepsy—factors that have previously been suggested as risk factors for developing autism in the TSC population.Reference Bolton, Park, Higgins, Griffiths and Pickles 45 , Reference Cusmai, Moavero, Bombardieri, Vigevano and Curatolo 46 The majority of patients with autism had comorbid intellectual disability, with the overall prevalence of intellectual disability in our study population being 40% and most often in the moderate-severe range. Previous studies have reported a rate of intellectual disability around 50% in TSC patients, with a significant portion falling in the profound range.Reference Joinson, O’Callaghan, Osborne, Martyn, Harris and Bolton 14 ADHD was the most prevalent diagnosis after intellectual disability and autism, occurring in 29% of our school-aged patients, while other studies have reported a rate of approximately 30% to 60%.Reference D’Agati, Moavero, Cerminara and Curatolo 47 The slightly lower rate of autism and other neuropsychiatric diagnoses compared with the literature may reflect, in part, the absence of standardized screening or reporting of neuropsychiatric manifestations when these children were assessed. Therefore, these comorbidities may have been missed or not accurately captured in the retrospective review. Early screening for autism spectrum disorders in children with TSC should be recommended, and some research groups are looking for early biomarkers of autism in this population. Additionally, a portion of the cohort was at a young age at last follow-up and many neuropsychiatric problems may not become evident until children are older. Indeed, if only children of school age in this cohort are considered, one-third had a diagnosis of autism and 56% had intellectual disability.
This study is limited by its retrospective nature because there was no standardized protocol for investigation, surveillance, or management in the study population. As a result, the reported rates of TSC manifestations may be an underestimation. Furthermore, recruitment through neurology department records may have missed a proportion of children with TSC without neurologic manifestations. However, this study is likely an accurate reflection of the TSC-associated neurologic manifestations seen at a tertiary referral centre. Overall, this study reinforces the spectrum of TSC manifestations in childhood, placing particular emphasis on the burden of neurologic morbidity. Further prospective studies will be useful in confirming these findings. In particular, formal neuropsychiatric screening will provide useful adjunctive information for future studies in this population. The tuberous sclerosis-associated neuropsychiatric disorders checklist has recently been developed as a guide to assist physicians in identifying neuropsychiatric concerns through discussion with patients and their families.Reference de Vries, Whittemore and Leclezio 48
These results have contributed to the ongoing development of a TSC clinic at British Columbia Children’s Hospital. Given the prominence of epilepsy in this population, the clinic is directed by two pediatric epileptologists (MC, AD) who are also involved in the epilepsy surgery program, supported by neurology nurses familiar with TSC. Thus, patients who are potential candidates for epilepsy surgery may be identified early. For those patients who undergo epilepsy surgery, the neurosurgeon may attend the clinic for coordinated follow-up. A medical geneticist also attends the clinic to provide genetic counseling as required. To address the potential underestimation of the rate of neuropsychiatric comorbidities identified in our study population, standardized screening has been instituted. Patients between the age of 18 months and 3 years are screened for autism at each visit using the Modified Checklist for Autism in Toddlers, Revised with Follow-up.Reference Robins, Casagrande, Barton, Chen, Dumont-Mathieu and Fein 49 Those with a positive screen are then referred to a developmental pediatrician for formal assessment. School-aged children are screened annually for ADHD using the SNAP-IV 26 Rating Scale 50 and additional challenges are sought using the Strengths and Difficulties QuestionnaireReference Goodman and Goodman 51 and the Pediatric Quality of Life Inventory.Reference Varni, Seid, Knight, Uzark and Szer 52 A neuropsychiatrist with expertise in genetic disorders now participates in the TSC clinic. It is hoped that the identification of patient needs through this prospective screening will further facilitate the direct involvement of neuropsychology and clinical psychology in the TSC clinic. Because manifestations outside the nervous system are common, our patients are routinely assessed and followed as required by other disciplines including cardiology, nephrology, dermatology, and ophthalmology. However, because our results demonstrate that these systemic manifestations are most often asymptomatic, these disciplines do not necessarily need to be involved at every visit. Therefore, we envision the TSC clinic as a combination of a core “physical clinic,” focusing on neurologic and neuropsychiatric manifestations, supported by a network of specialists knowledgeable in TSC, but who do not necessarily attend the physical TSC clinic. The development of this clinic is an ongoing process and we plan to regularly reassess its structure and the services it provides though prospective data collection and feedback from our patients, their families, and other health care professionals.
CONCLUSION
TSC is a challenging disease, both for clinicians and for affected patients and their families. As described in this study, they are often faced with treatment resistant epilepsy, complex neuropsychiatric manifestations, and systemic medical complications that evolve with age. In recognition of these issues, we have established a dedicated tuberous sclerosis clinic that continues to evolve in an effort to improve patient care. It is hoped that this clinic will facilitate appropriate monitoring of the disorder and, hopefully, reduce complications for patients. Transition to adult services remains challenging because of the multidisciplinary nature and complexity of this lifelong disease. Increased collaboration between pediatric and adult specialists will help to improve care for patients with TSC across the lifespan.
Disclosures
AD is a site co-investigator and received research support and honoraria from Novartis. MC is a site principal investigator and received research support and honoraria from Novartis. CW, CS, HC, MA, SP, KA, PS, and AS have nothing to disclose.
Statement of Authorship
CW was responsible for developing the data collection database, collecting and analyzing data, and primary manuscript development. CS assisted in analysis of epilepsy data. HC and MA assisted in database development and data collection. SP and KA were involved in primary clinical evaluation of patients. PS and AS were involved in primary clinical evaluation of patients and assisted in analysis of epilepsy surgery data. AD and MC were involved in primary clinical evaluation of patients and assisted in data analysis. MC supervised all aspects of manuscript development. All authors participated in the editing of the final manuscript.









