



The increasing global prevalence of obesity is associated with poor nutritional habits, with increased consumption of high-fat diet (HFD) and high-sugar diet, and sedentarism(Reference Popkin1). This inadequate nutrition during the perinatal period, including maternal nutritional status, is also an important contributing factor for the alarming worldwide obesity rates. In humans and experimental models, early life nutritional insults, such as under- or overnutrition during the perinatal period, increase the risk of obesity and its co-morbidities in adult offspring(Reference Nicholas, Morrison and Rattanatray2). This phenomenon is known as metabolic programming and highlights the intense ontogenetic plasticity in early life, reprogramming gene expression pattern of the offspring to ensure short-term adaptation to the milieu but with detrimental metabolic outcomes throughout life(Reference Hanson, Godfrey and Lillycrop3,Reference Gluckman, Hanson and Buklijas4) . This adaptive mechanism involved in early life events and its impact on later risk of chronic diseases is the basis of the developmental origins of health and disease, a new field of study(Reference Hanson and Gluckman5).
Maternal obesity during the perinatal period increases offspring adiposity and adipocyte hypertrophy, reflecting in high serum leptin, an important hormone involved in energy metabolism homeostasis(Reference Kirk, Samuelsson and Argenton6). Previously, we showed that maternal consumption of isoenergetic HFD during the perinatal period alters breast milk composition and increases body weight, food intake, adiposity, circulating leptin levels associated with impaired leptin signalling at the hypothalamus. Besides that, it alters lipid metabolism in male offspring at weaning(Reference Franco, Fernandes and Rocha7), adolescence(Reference Oliveira, Souza and Souza8) and adult life(Reference Franco, Dias-Rocha and Fernandes9). This metabolic profile increases the susceptibility to develop insulin resistance, dyslipidaemia and non-alcoholic fatty liver disease in the offspring of obese dams, as others have demonstrated(Reference Latouche, Heywood and Henry10–Reference Oben, Mouralidarane and Samuelsson12).
Obesity management has been a major challenge in the last decades, and there are very few pharmacological approaches approved by the international organisations(Reference Krentz, Fujioka and Hompesch13). In addition, the increasing rates of obesity among children and adolescents(Reference Ng, Fleming and Robinson14) require further investigation of possible therapies that could be beneficial and safe for this critical window of development. Furthermore, obese children and adolescents are frequently inserted into obesogenic environments with poor familiar habits that could have programmed their metabolism since perinatal life(Reference Bay, Morton and Vickers15, Reference Birch16). Therefore, it is necessary to investigate potential interventions to prevent or improve metabolic outcomes induced by perinatal maternal dietary choices.
It has been shown that the supplementation of maternal HFD with n-3 PUFA attenuates metabolic injury (such as dyslipidaemia, oxidative stress and insulin resistance) in the offspring in adulthood, compared with maternal HFD rich in SFA(Reference Ramaiyan, Bettadahalli and Talahalli17, Reference Heerwagen, Stewart and de la Houssaye18). Also, the chronic consumption of n-3 PUFA throughout offspring’s life reduced metabolic deleterious outcomes (such as hypertension, hyperleptinaemia and glucose intolerance) in experimental programming models(Reference Wyrwoll, Mark and Mori19, Reference Hou, Ji and Wang20). These metabolic benefits observed with n-3 PUFA or fish oil (FO) supplementation are well characterised in adult animal experimental models and humans. FO consumption reduces adiposity and serum lipids in adult rodents(Reference Souza, Cordeiro and Oliveira21, Reference Rossi, Lombardo and Lacorte22) and humans(Reference Couet, Delarue and Ritz23). n-3 PUFA present a hypolipidaemic effect(Reference Ukropec, Reseland and Gasperikova24, Reference Jump25) by controlling molecular mechanisms involved in the suppression of hepatic lipogenesis and up-regulation of lipid oxidation(Reference Jump25).
We have recently shown that FO intervention in adolescent rats reduced serum lipids only in control offspring but not in those programmed by maternal HFD. This resistance to the hypolipidaemic effect of FO was associated with reduced levels of n-3 PUFA in serum of the adolescent rats programmed by maternal HFD(Reference Oliveira, Souza and Souza8). Nevertheless, FO intake induced molecular changes in hepatic TAG metabolism suggestive of decreased de novo lipogenesis, in both control- and HFD-adolescent offspring. This raises the question of whether FO intervention during adolescence could impact the offspring metabolism in adulthood. Here we tested the hypothesis that FO intervention during adolescence may prevent or attenuate the higher adiposity, hyperleptinaemia, liver steatosis and dyslipidaemia of adult male offspring programmed by perinatal maternal HFD.
Material and methods
Ethical statement
All procedures were approved by the Ethics Committee on Animal Care of the Health Sciences Center, Federal University of Rio de Janeiro (protocol no. IBCCF 140/13), which follows the principles adopted in the UK and Brazil according to Brazilian Law no. 11.794/2008(Reference Drummond26, Reference Marques, Morales and Petroianu27).
Experimental animals and procedures
Wistar rats were obtained from the Center of Reproduction Biology of the Federal University of Rio de Janeiro, Rio de Janeiro, Brazil. They were acclimated and maintained in individual cages with free access to water and food in a room with controlled temperature (23 ± 2°C) and 12 h light–12 h darkness cycles (lights on from 07.00 to 19.00 hours). Wistar rats are experimental models frequently used in evaluation of the consequences of metabolic programming(Reference Franco, Fernandes and Rocha7,Reference Oliveira, Souza and Souza8,Reference Ramaiyan, Bettadahalli and Talahalli17,Reference Wyrwoll, Mark and Mori19,Reference Hou, Ji and Wang20,Reference Zheng, Xiao and Zhang28) , because rodents have short gestation and lactation periods, allowing the rapid evaluation of transgenerational transmission(Reference Armitage, Khan and Taylor29). Besides that, genetic and environmental influences can be carefully controlled in these experimental models. The relevance of our study is that this experimental model simulates the increased consumption of HFD and obesity rates among women in reproductive age(Reference Vahratian30), and its correlation with metabolic obesity among adolescent offspring reported by epidemiological observations(Reference Oken, Rifas-Shiman and Field31). Then, our model allows to investigate the perinatal insult impact on phenotype and molecular changes of offspring(Reference Armitage, Khan and Taylor29), inferring the molecular mechanism involved in the human observations.
Twenty-six female Wistar rats, 60 d old and weighing 190–220 g, were randomly distributed in two experimental groups: standard diet (STD) or HFD (thirteen per group). The STD group received an STD for rodents (9 % of the energy as fat), and HFD group received an HFD (28⋅6 % of the energy as fat – lard was used as the major fat source) for 8 weeks before mating and during pregnancy and lactation(Reference Franco, Fernandes and Rocha7). During mating, two female rats of the same group were housed with one male rat (150 d old). After this, female rats were housed in individual cages during pregnancy and lactation to avoid stressful environment. The maternal comportment was evaluated, and the maternal stress was discarded by absence of killed puppies by dams. At birth, litters were adjusted to six males for each dam aiming to improve lactation performance(Reference Fischbeck and Rasmussen32). STD and HFD offspring received STD (the same perinatal STD) during the postnatal period, from weaning (21 d old) until adulthood (150 d old).
Our HFD is moderately high-fat compared with most experimental models that generally use diets with 50 % of the energy as fat(Reference Zheng, Xiao and Zhang28, Reference Yokomizo, Inoguchi and Sonoda33). The HFD was manually produced in our laboratory using the ingredients described at Table 1, including the commercial STD chow. Diet composition followed the American Institute of Nutrition recommendations (AIN-93) for micronutrients(Reference Reeves, Nielsen and Fahey34) and was described before(Reference Franco, Fernandes and Rocha7–Reference Franco, Dias-Rocha and Fernandes9). Both diets contained approximately 16⋅3 kJ/g of energy (isoenergetic) and their macronutrients compositions are described at Table 2. The HFD was composed by a moderate increased lipid content with protein and carbohydrate contents within the range recommended for rodents (AIN-93), which avoided the negative impact of low-protein or low-carbohydrate diets(Reference de Oliveira, Gomes and Miranda35, Reference Koski and Fergusson36). The differences between macronutrient compositions of diets were not tested statistically, because we estimated the diet composition by ingredients composition described at ingredient label. The fatty-acid compositions of diets were analysed by gas chromatography. The HFD showed higher SFA content than STD (P < 0⋅01), and its detailed fatty-acid composition is described at Table 3.
Table 1. Diet composition (g/kg) on the basis of the American Institute of Nutrition (AIN)-93G diet formulation

HFD, high-fat diet; BHT, butylated hydroxytoluene.
Table 2. Diet macronutrient composition
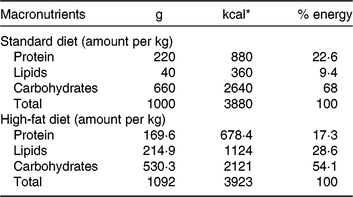
* To convert kcal to kJ, multiply by 4·184.
Table 3. Fatty acid composition of the experimental diets (µg/mg of chow)
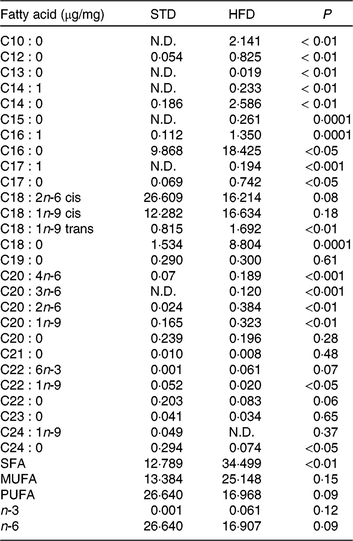
STD, standard diet; HFD, high-fat diet; N.D., not detected.
* Fatty acids were quantified by GC/MS determining peak-area ratios with the 9 : 0 and 19 : 0 internal standards. We used three samples per group.
In the adolescence period(Reference Schneider37), two male rats of each litter were randomly allocated at soyabean oil (SO) or FO group in individual cages. The remaining pups were used in another experimental design(Reference Oliveira, Souza and Souza8). Then, the experimental design had four experimental groups: STD or HFD offspring that received SO (source of n-6 PUFA; Liza®, Cargill) (n 12 and 9, respectively) and STD or HFD offspring that received FO (source of n-3 PUFA; Fagron) (n 12 and 9, respectively). Oil administration was given daily by gavage (4 ml/kg body weight) from 25 to 45 d old. Each capsule of FO had 95⋅5 mg/ml of EPA and 61 mg/ml of DHA, resulting in a dose of 0⋅244 g/kg of DHA and 0⋅380 g/kg per d. This DHA dose adjusted to human(Reference Reagan-Shaw, Nihal and Ahmad38) is in a dose range used in human experimental design with FO supplementation during the adolescence period(Reference McNamara, Strimpfel and Jandacek39–Reference de Ferranti, Milliren and Denhoff41). SO was used as control to exclude lipid load effect, ensuring that the results are derived from the different types of fatty acids used in the treatment. Furthermore, SO is a lipid source recommended for rodents according to the AIN-93(Reference Reeves, Nielsen and Fahey34), and represents the main fatty acid present in the Western diet (n-6 PUFA)(Reference Spector42).
We performed an oral glucose tolerance test with the animals at 147 d of age. After 12-h fasting, capillary blood glucose was measured from a tail incision, followed by oral glucose (d- (+) -glucose; VETEC) administration (2 mg/g of body weight). Glycaemia was also measured at 15, 30, 60 and 90 min after glucose administration (One Touch Ultra Mini device; Johnson and Johnson).
During the experiment, we did not observe adverse events or features of stressed animals, such as aggressive behaviour, hair loss or body weight loss. At 150 d of age, rats were killed by decapitation. Serum was obtained from trunk blood (after centrifugation at 1000 g, 4°C, 20 min) and kept frozen at −20°C for measurements of hormone levels and biochemical parameters. Representative depots of visceral white adipose tissue (retroperitoneal and epididymal) were dissected, weighed and returned to eviscerated carcass, to evaluate the adiposity. Liver was harvested, snap frozen in liquid nitrogen and stored at −70°C prior to extraction of total lipid, protein and RNA.
Fatty-acid composition of the experimental diets
The GC/MS assay was used to investigate the fatty-acid profile in experimental diets (STD and HFD). The lipid sample was dissolved in a toluene and 1 % sulfuric acid in methanol solution. GC/MS analysis was carried out on a Shimadzu GCMS-QP2010 Plus system, using an HP Ultra 2 (5 % phenyl-methylpolysiloxane), Agilent (25 m ×0⋅20 mm × 0⋅33 μm). Injector was set at 250°C. Column temperature was programmed from 40 to 160°C at 30°C/min, 160 to 233°C at 1°C/min, 233 to 300°C at 30°C/m and held at 300°C for 10 min. Electro ionisation (EI-70 eV) and a quadrupole mass analyser operated in scans from 40 to 440 atomic mass units. Interface was set at 240°C and the ion source at 240°C. The components were identified by comparing their mass spectra with those of the library NIST05 contained in the computer’s mass spectrometer. Retention indices were also used to confirm the identity of the peaks in the chromatogram by Supelco 37 Component FAME Mix (Sigma-Aldrich). Fatty acids were quantified by determining peak-area ratios with the 9 : 0 and 19 : 0 internal standards. We used three samples per group.
Body composition analysis
Total body lipid quantification was performed as previously described(Reference Stansbie, Brownsey and Crettaz43, Reference Souza, Nunes and Paula44). Briefly, eviscerated carcasses were weighed, autoclaved and homogenised in distilled water (1:1). Homogenate (3 g) was hydrolysed in a shaking water bath at 70°C for 2 h with 30 % KOH and ethanol, followed by three successive washes with petroleum ether (Vetec) to extract lipids. The petroleum ether phases containing the lipids were dried at room temperature until constant weight was obtained.
Serum lipid profile
Serum total cholesterol (no. 12505), HDL-cholesterol (no. 12557) and TAG (no. 12528) quantification was performed using commercial kits from Biosystems and an automated A15 spectrophotometer (Biosystems S.A.). The serum VLDL-cholesterol and LDL-cholesterol were estimated according to the Friedewald assumption(Reference Friedewald, Levy and Fredrickson45): VLDL = TAG/5 and LDL = total cholesterol − HDL − TAG/5.
Hormone assays
Serum leptin was measured using specific commercial radioimmunoassay (RIA) kit from Linco Research (no. XL-85K). Total serum thyroxine and total serum triiodothyronine were determined using specific commercial RIA kits from MP Biomedicals (no. 06B-254011 and no. 06B-254215). Serum thyroid-stimulating hormone (TSH) was measured by a specific rat TSH RIA using reagents acquired from the National Hormone and Pituitary Program, as previously detailed(Reference Ortiga-Carvalho and Curty46). Serum adiponectin was measured by a specific rat Adiponectin ELISA Kit from Merck Millipore (no. EZRADP-62K). All samples were evaluated in duplicate within the same assay according to the manufacturer’s instructions.
Real-time PCR analysis
Real-time PCR assay was used to evaluate the relative mRNA expression of genes encoding PPARα (Ppara), hepatic lipase (Lipc), carnitine palmitoyl transferase-1a (CPT-1a: Cpt1a), leptin (Lep), PPARγ (Pparg), IL-1β (Il1b), IL-6 (Il6), IL-10 (Il10) and TNF-α (Tnf).
Total RNA was isolated from retroperitoneal adipose tissue samples using a commercial RNeasy lipid tissue mini kit (Qiagen), and from liver samples using a commercial SV Total RNA Isolation System (Promega). Total RNA was reverse transcribed using 1 μg of total RNA and a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Products were amplified on Eppendorf MasterCycler RealPlex (Eppendorf) using Maxima SYBR Green qPCR Master Mix (Thermo Scientific). Cycle parameters were 50°C for 2 min and 95°C for 10 min, followed by forty cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 45 s. The intron spanning primers sequence used in the present study are described in Table 4. Primers were synthesised and tested by Integrated DNA Technologies. Samples and negative controls were evaluated in duplicate in the same assay. Efficiency of each PCR reaction was calculated using a serial cDNA dilution standard curve, and varied from 96 to 100 %. Product purity was confirmed by a single peak in the melting curve analysis. Relative mRNA levels were calculated using standard curve method and normalised by mRNA levels of the reference genes Polr2a (liver) or Rplp0 mRNA (adipose tissue). Results are expressed relative to values of control group (STD offspring that received SO during the adolescence period).
Table 4. Primer sequences for gene expression evaluation

Western blotting analysis
We used the Western blotting assay to evaluate the hepatic content of fatty acid synthase (FAS), CPT-1a, and signal transducer and activator of transcription 3 (STAT3 and pSTAT3). The content of cyclophilin (Cyclo) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. Liver samples were homogenised in lyses buffer pH 6⋅4 (50-mm HEPES, 1-mm MgCl2, 10-nm EDTA, and 1 % Triton X) with protease inhibitor cocktail complete (Roche Diagnostics). After centrifugation of the homogenates, the total protein content of the supernatant was quantified using the PierceTM BCA Protein assay kit (Thermo Scientific). Total protein was separated by SDS–PAGE and transferred onto a polyvinylidene difluoride membrane (Hybond-P 0⋅45 µm PVDF; Amersham Biosciences). Membrane was blocked with 5 % non-fat dry milk (Molico, Nestlé) or 5 % bovine serum albumin (INLAB) according to the protein of interest for 1 h at room temperature. Then, membrane was incubated overnight at 4°C with primary antibodies: anti-FAS (Cell Signaling; 1:1000 dilution), anti-CPT-1a (Abcam; 1:3000 dilution), anti-STAT3 (Santa Cruz; 1:400), anti-pSTAT (Santa Cruz; 1:1600) and loading control anti-Cyclo (Thermo Scientific; 1:40 000 dilution) or anti-GAPDH (Cell Signaling; 1:3000 dilution). Membranes were washed and incubated with peroxidase-labelled anti-rabbit IgG antibody (Amersham Biosciences, BKM, England; 1:10 000 dilution) or anti-mouse IgG antibody (Cell Signaling; 1:10 000) for 3 h at room temperature. All blots were then washed and incubated with a luminogen detection reagent (Amersham ECL Prime Western Blotting Detection reagent; Amersham Biosciences or SuperSignal® West Femto Maximum Sensitivity Substrate; Thermo Scientific). Chemiluminescent signal was detect by ImageQuant LAS 4000 equipment followed by densitometric analyses (GE Healthcare Life Sciences).
TLC
Liver total lipids were extracted by the method of Bligh &Dyer(Reference Bligh and Dyer47) with modifications. After incubation in chloroform–methanol–water solution (2:1:0⋅4, by vol.), samples were centrifuged (1500 g for 20 min at 4°C). Then, the chloroform was added to the supernatant. After centrifugation (1500 g for 20 min) the organic phase was removed and dried in nitrogen. The extracted lipids were analysed by TLC for neutral lipids, using a DC Silicagel 60 plate (Merck Millipore) and standards lipids. The plates were submerged for 10 s in Charring solution (3 % CuSO4 and 8 % H3PO4 (v/v)), then they were dried and heated to 110°C for 5–10 min. TLC plates were analysed by densitometry (GE Healthcare Life Sciences), defining hepatic free cholesterol, NEFA and TAG content.
Statistical analysis
The sample size was calculated by using the G*Power 3.1.9.2 program(Reference Faul, Erdfelder and Lang48), based in our previous observations of maternal HFD effect on plasma and molecular parameters of the adult offspring(Reference Franco, Dias-Rocha and Fernandes9) and our previous observations of FO intake effect on plasma and molecular parameters in adolescents rats(Reference Oliveira, Souza and Souza8). Regarding plasma parameters, a sample size of nine animals per group would provide the appropriate power (1 − β = 0·8) to identify significant differences (α = 0·05) in the variables analysed, using a two-tailed test. For molecular parameters, a sample size of six animals per group would provide the appropriate power (1 − β = 0·8) to identify significant differences (α = 0·05) in the variables analysed, using a two-tailed test. The statistical comparisons were performed using the software GraphPad Prism (GraphPad Software Inc.) showing results as means with their standard errors. Two-way ANOVA were used considering two variables (maternal diet and oil intervention) and significant differences at P < 0⋅05. Tukey’s post-test was used to evaluate differences between columns in only each maternal condition. In each analysis of post-test, we corrected the result for multiple comparisons using statistical hypothesis testing and reported multiplicity adjusted P value for each comparison, using a confidence level of 0⋅05. To data correlation analysis, normality was assessed by the Kolmogorov–Smirnov test, followed by non-parametric Spearman’s correlation test.
Results
Maternal HFD increased body weight (1⋅09-fold; P < 0⋅0001; Fig. 1(A)), food intake (1⋅08-fold; P < 0⋅0001; Fig. 1(B)), retroperitoneal adiposity (1⋅33-fold; P < 0⋅01; Fig. 1(C)) and total body fat (1⋅21-fold; P < 0⋅01; Fig. 1(D)) of adult male offspring. Regardless of maternal diet, FO intake in the adolescence period did not change body weight or food intake in adulthood (Fig. 1(A) and 1(B)), but it decreased retroperitoneal adipose tissue mass (approximately 19 %, P < 0⋅05; Fig. 1(C)) and showed a tendency to reduce total body fat in adult offspring (P = 0⋅06; Fig. 1(D)).
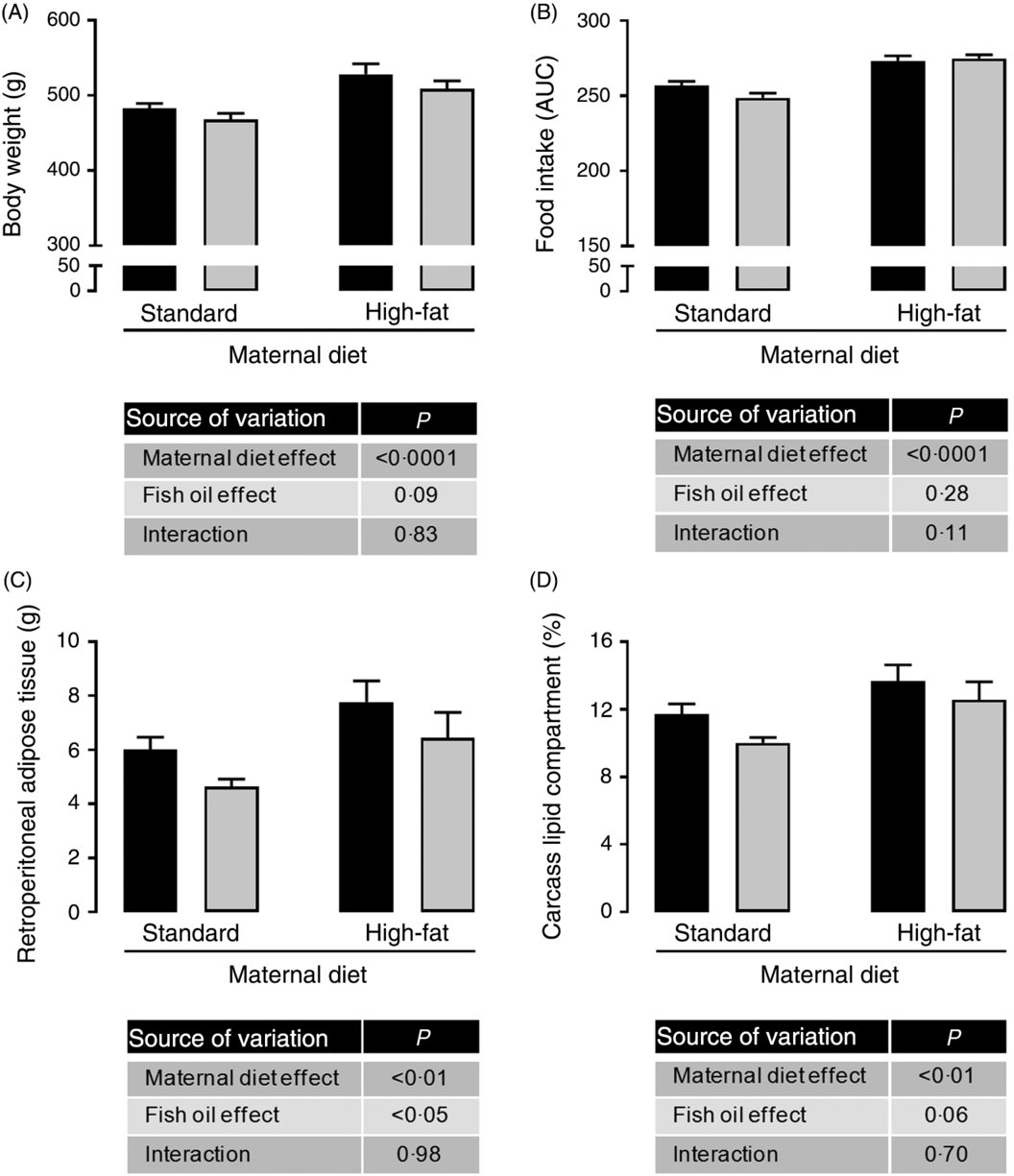
Fig. 1. Effects of fish oil consumption during the adolescence period on adult male offspring from dams that received a standard or high-fat diet. Offspring received daily oral intervention with soyabean oil (![]() ) or fish oil (
) or fish oil (![]() ) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Body weight at 150 d old. (B) Food intake after oil withdraw (AUC). (C) Retroperitoneal adipose tissue mass. (D) Carcass lipid compartment. Data are means, with their standard errors represented by vertical bars (n 9–12). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05.
) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Body weight at 150 d old. (B) Food intake after oil withdraw (AUC). (C) Retroperitoneal adipose tissue mass. (D) Carcass lipid compartment. Data are means, with their standard errors represented by vertical bars (n 9–12). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05.
Because we observed maternal diet and FO effects on adiposity of adult offspring, we evaluated adipose tissue gene expression. There was no effect of maternal HFD neither of FO on PPAR-γ, IL-1b, IL-6, IL-10 and TNF-α mRNA expression (data not shown), but maternal HFD promoted 1⋅6-fold increase in leptin mRNA expression in retroperitoneal adipose tissue (P < 0⋅05; Fig. 2(A)). This increased leptin mRNA expression was accompanied by approximately 1⋅75-fold increase in serum leptin of the maternal HFD offspring (P < 0⋅05; Fig. 2(B)). Interestingly, the FO intake during adolescence could attenuate the mRNA expression and serum levels of leptin in both STD and HFD adult offspring (P < 0⋅05; Fig. 2(A) and 2(B)). Concomitantly, in the liver, the pSTAT3/STAT3 protein content ratio, which is an important intracellular mediator of leptin action, was significantly reduced in maternal HFD adult offspring (P < 0⋅01), but it was not affected by FO intake (Fig. 2(C)).
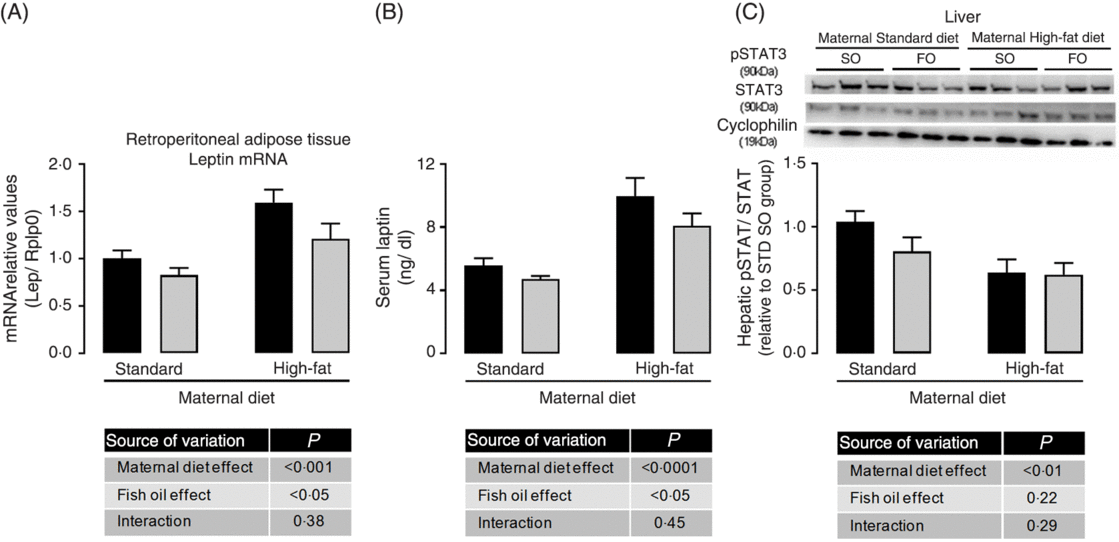
Fig. 2. Effects of fish oil consumption during the adolescence period on leptin levels and hepatic signalling in adult male offspring from dams that received a standard (STD) or high-fat diet. Offspring received daily oral intervention with soyabean oil (SO; ![]() ) or fish oil (FO;
) or fish oil (FO; ![]() ) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) mRNA expression of leptin (Lep) in retroperitoneal adipose tissue. (B) Serum leptin levels. (C) Hepatic pSTAT/STAT ratio by Western blotting analysis. Data are means, with their standard errors represented by vertical bars (n 8–10). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. To convert leptin in ng/dl to ng/l, multiply by 10.
) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) mRNA expression of leptin (Lep) in retroperitoneal adipose tissue. (B) Serum leptin levels. (C) Hepatic pSTAT/STAT ratio by Western blotting analysis. Data are means, with their standard errors represented by vertical bars (n 8–10). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. To convert leptin in ng/dl to ng/l, multiply by 10.
Maternal HFD did not change serum adiponectin, serum TSH and serum thyroid hormones triiodothyronine and thyroxine, but the offspring that received FO during adolescence showed approximately 18 % reduced serum triiodothyronine in adult life, regardless of the maternal diet (Table 5).
Table 5. Effects of fish oil (FO) consumption during the adolescence period on serum hormones and on metabolic parameters of adult male offspring from dams that received a standard or high-fat dietFootnote *
(Mean values with their standard errors; n 9–12)

SO, soyabean oil; OGTT, oral glucose tolerance test.
* Statistical analysis was performed using two-way ANOVA.
† To convert triiodothyronine in ng/dl to ng/l, multiply by 10. To convert thyroxine in μg/dl to μg/l, multiply by 10. To convert cholesterol in mg/dl to mmol/l, multiply by 0·0259.
Maternal HFD did not change oral glucose tolerance (Table 5), but promoted dyslipidaemia in adult offspring. We observed increased serum TAG (1⋅24-fold; P < 0⋅05; Fig. 3(A)), serum total cholesterol (1⋅12-fold; P < 0⋅01; Fig. 3(B)), HDL and VLDL, without changes in serum LDL (Table 5) in adult offspring from HFD dams. However, these metabolic parameters were not changed by FO intake during adolescence of these animals.

Fig. 3. Effects of fish oil consumption during the adolescence period on serum and hepatic lipid content in adult male offspring from dams that received a standard (STD) or high-fat diet. Offspring received daily oral intervention with soyabean oil (SO; ![]() ) or fish oil (FO;
) or fish oil (FO; ![]() ) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Serum TAG. (B) Serum total cholesterol. (C) Liver mass. (D) Hepatic free cholesterol content. (E) Hepatic NEFA content. (F) Hepatic TAG content. Data are means, with their standard errors represented by vertical bars (n 9–12). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. Tukey’s post-test was used to evaluate differences between columns in each maternal condition and results are represented as the bars at the top of the graph. The adjusted P value was * P = 0·0116 for liver mass (C) and * P = 0·0068 for hepatic TAG content (D). To convert TAG in mg/dl to mmol/l, multiply by 0·0113. To convert cholesterol in mg/dl to mmol/l, multiply by 0·0259.
) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Serum TAG. (B) Serum total cholesterol. (C) Liver mass. (D) Hepatic free cholesterol content. (E) Hepatic NEFA content. (F) Hepatic TAG content. Data are means, with their standard errors represented by vertical bars (n 9–12). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. Tukey’s post-test was used to evaluate differences between columns in each maternal condition and results are represented as the bars at the top of the graph. The adjusted P value was * P = 0·0116 for liver mass (C) and * P = 0·0068 for hepatic TAG content (D). To convert TAG in mg/dl to mmol/l, multiply by 0·0113. To convert cholesterol in mg/dl to mmol/l, multiply by 0·0259.
Because FO and leptin are known to regulate hepatic lipid metabolism, we investigated lipid metabolism markers in the liver of adult offspring. Maternal HFD promoted higher liver weight (1⋅14-fold; P < 0⋅0001; Fig. 3(C)), and it was reduced in 10 % by FO intake in STD offspring but not that of HFD group (P < 0⋅05; Fig. 3(C)). Both maternal HFD and FO intake during adolescence decreased hepatic content of free cholesterol (Fig. 3(D)) in adult offspring. However, neither maternal HFD nor FO intake during adolescence changed hepatic cholesterol ester content (data not shown). FO intake reduced hepatic NEFA content (P < 0⋅05; Fig. 3(E)), regardless of maternal diet. Interestingly, maternal HFD promoted 1⋅6-fold higher hepatic TAG content in adult offspring, which was restored to STD offspring levels by FO intake during adolescence of these animals (Fig. 3(F)). FO intake during adolescence reduced hepatic TAG content in approximately 29–40 %, independent of maternal diet (Fig. 3(F)).
We investigated the hepatic content of key enzymes of lipid synthesis and oxidation pathways. Although we did not observe statistically significant differences among experimental groups in hepatic FAS and CPT-1a protein abundance (Fig. 4(A) and 4(B)), the maternal HFD offspring showed a strong trend to decreased CPT-1a protein abundance (P = 0⋅07; Fig. 4(B)). We observed a significant negative correlation among hepatic CPT-1a protein abundance and hepatic TAG content (P < 0⋅05; Fig. 4(C)), suggesting that impairment in lipid oxidation pathway could be involved in hepatic TAG accumulation. mRNA levels of hepatic CPT-1, hepatic lipase (Lipc) and PPAR-α showed the same profile of CPT-1a protein abundance, however there were no statistical significant differences among the experimental groups (Fig. 4(D), 4(E) and 4(F)).
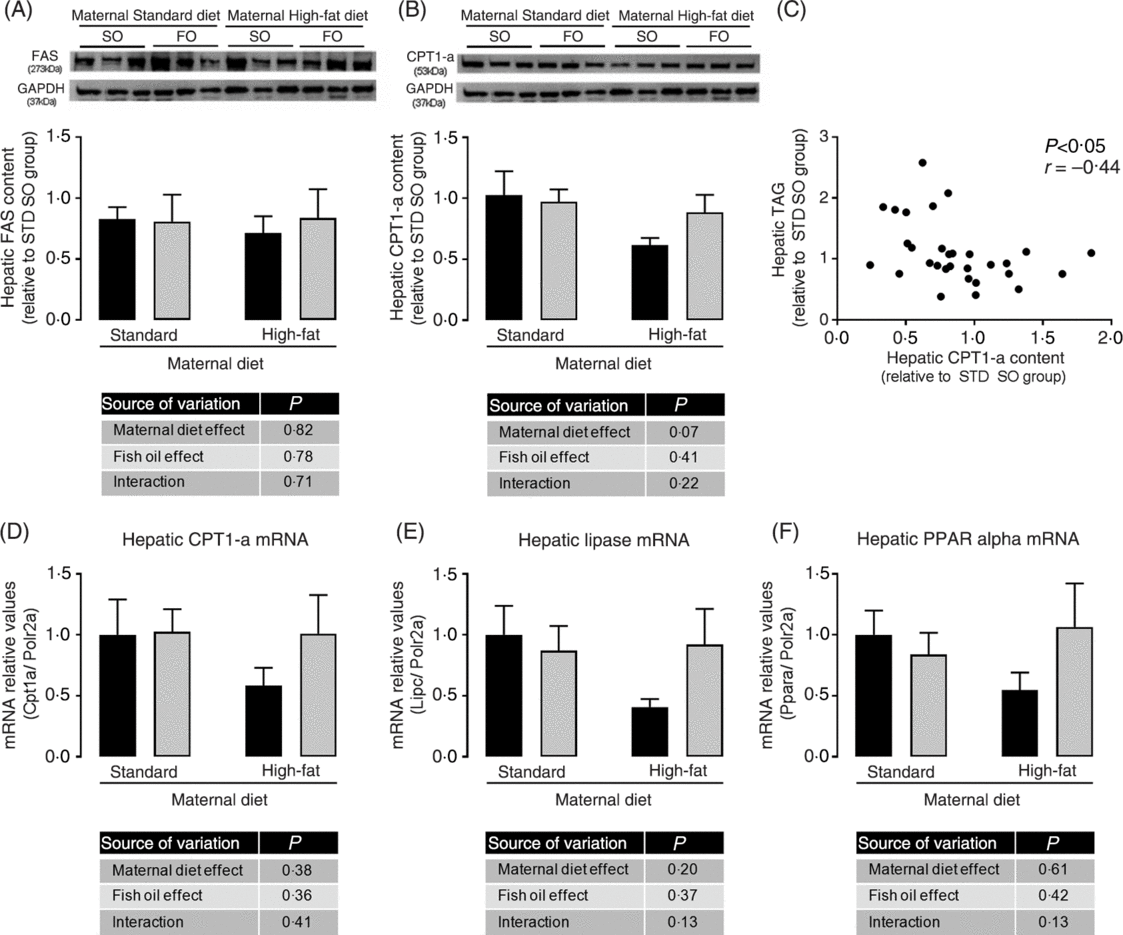
Fig. 4. Effects of fish oil consumption during the adolescence period on hepatic expression of key targets of lipid metabolism in adult male offspring from dams that received a standard (STD) or high-fat diet. Offspring received daily oral intervention with soyabean oil (SO; ![]() ) or fish oil (FO;
) or fish oil (FO; ![]() ) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Hepatic fatty acid synthase (FAS) protein abundance. (B) Hepatic carnitine palmitoyl transferase-1a (CPT-1a) protein abundance. (C) Correlation among hepatic CPT-1a protein abundance and hepatic TAG content. (D) Hepatic carnitine palmitoyl transferase-1a (Cpt1a) mRNA expression. (E) Hepatic lipase (Lipc) mRNA expression. (F) Hepatic PPARα (Ppara) mRNA expression. Data are means, with their standard errors represented by vertical bars (n 6–9). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
) from 25 to 45 d of age, and evaluations were performed in adult life (150 d old). (A) Hepatic fatty acid synthase (FAS) protein abundance. (B) Hepatic carnitine palmitoyl transferase-1a (CPT-1a) protein abundance. (C) Correlation among hepatic CPT-1a protein abundance and hepatic TAG content. (D) Hepatic carnitine palmitoyl transferase-1a (Cpt1a) mRNA expression. (E) Hepatic lipase (Lipc) mRNA expression. (F) Hepatic PPARα (Ppara) mRNA expression. Data are means, with their standard errors represented by vertical bars (n 6–9). Statistical analysis was performed using two-way ANOVA and is described in the table at the bottom of each graph, considering significant differences at P < 0·05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
In the present study, we originally demonstrated that FO consumption just during the adolescence period was able to reprogramme offspring metabolism in adult life, improving metabolic outcomes induced by perinatal maternal HFD. We showed attenuation of visceral adiposity, serum leptin and hepatic TAG that could contribute to a healthier life of adult offspring. These observations suggest the adolescence as a critical period in which dietary choices may impact adult health, revealing a critical window susceptible to metabolic reprogramming.
In the present study, maternal HFD and STD were isoenergetic, and HFD was moderately high-fat compared with other maternal HFD models(Reference Zheng, Xiao and Zhang28, Reference Yokomizo, Inoguchi and Sonoda33). We have been showing that moderate maternal HFD consumption during perinatal life increases body weight, adiposity and leptin levels in male offspring at weaning(Reference Franco, Fernandes and Rocha7), adolescence(Reference Oliveira, Souza and Souza8) and 180 d old(Reference Franco, Dias-Rocha and Fernandes9), and here the same profile was observed at 150 d old. In experimental models of obesity, n-3 PUFA consumption decreases adiposity and serum lipids(Reference Rossi, Lombardo and Lacorte22, Reference Wang, Storlien and Huang49). However, these beneficial effects are usually observed during or at the end of treatment. In contrast, we previously showed that FO consumption during adolescence did not change body weight, adiposity or serum leptin in STD or HFD offspring right after the intervention(Reference Oliveira, Souza and Souza8). Interestingly, in the present study, we showed that FO intervention during adolescence, independent of maternal diet, promoted long-term effects, reducing visceral adiposity and serum leptin in adult life.
Dyslipidaemia is a common finding of many metabolic programming models(Reference Lima Nda, de Moura and Passos50, Reference Zambrano, Martinez-Samayoa and Rodriguez-Gonzalez51). However, in our maternal HFD model we did not observe dyslipidaemia in adolescent offspring, neither changes in liver TAG content(Reference Oliveira, Souza and Souza8), but they presented a molecular profile suggestive of hepatic lipid metabolism impairment, with increased sterol regulatory-element binding protein 1 and acetyl CoA carboxylase expression, reduced CPT-1a content, and decreased ATP binding cassette subfamily G member 8 expression in liver(Reference Oliveira, Souza and Souza8). We suggest that these early hepatic molecular changes primed for dyslipidaemia progression during adult life, as we observed here in HFD offspring at 150 d of age.
Adult HFD offspring showed increased hepatic TAG content, which may progress to hepatoesteatosis(Reference Kawano and Cohen52). This metabolic profile was improved by FO consumption during adolescence, since it restored the hepatic TAG content to STD offspring levels. The mechanism probably involves multiple FO actions. Here we observed in the liver of adult HFD offspring a trend to reduction of CPT-1a, hepatic lipase and PPAR-α expressions, suggesting impairment of lipid oxidation which seems to be prevented by FO intake during the adolescence. Supporting the hypothesis of increased lipid oxidation as a potential target mechanism for FO in this model, we found a negative correlation between hepatic CPT-1a content and hepatic TAG content. However, the improvement in hepatic lipid metabolism induced by FO was not sufficient to reverse the serum lipid increment found in HFD offspring, suggesting the involvement of other mechanisms or a time-dependent effect.
Hyperleptinaemia and leptin resistance are common outcomes of maternal HFD, as previously observed by our group(Reference Franco, Fernandes and Rocha7–Reference Franco, Dias-Rocha and Fernandes9) and others(Reference Wang, Storlien and Huang49). In the present study, maternal HFD increased leptin mRNA levels in white adipose tissue in parallel to hyperleptinaemia in adulthood, which were attenuated by FO intake during adolescence. In our model, this leptin regulation might be a result of adiposity changes. However, we are not able to reject the possibility of epigenetic mechanisms involvement. Experimental evidences suggests that epigenetic mechanisms may be involved in the maternal HFD(Reference Masuyama and Hiramatsu53) and n-3 PUFA(Reference Shen, Wang and Xia54) impact on leptin regulation in other experimental models.
The JAK2-STAT3 pathway is the major signalling pathway involved in leptin action and its components have been described in the liver(Reference Huynh, Levi and Denroche55, Reference Huynh, Neumann and Wang56). We have previously described in our model of maternal HFD that there are changes in molecular markers of central leptin resistance in male offspring at weaning and adult life (180 d old)(Reference Franco, Fernandes and Rocha7, Reference Franco, Dias-Rocha and Fernandes9). However, the peripheral leptin signalling in programming models is poorly known. In the present study, maternal HFD reduced hepatic pSTAT3/STAT3 ratio, suggesting hepatic leptin resistance in HFD adult offspring. The importance of leptin signalling in liver metabolism was evidenced using a mice model with specific loss of leptin signalling in the liver (deficiency in the leptin-stimulated STAT phosphorylation), which shows hepatic TAG accumulation and serum TAG-rich lipoproteins(Reference Huynh, Levi and Denroche55, Reference Huynh, Neumann and Wang56). Besides that, STAT3 knockout increases the hepatic TAG content(Reference Inoue, Ogawa and Ozaki57). Therefore, the impairment in the activation of the transducer factor STAT3 in the liver of HFD offspring may contribute to hepatic TAG accumulation. Although FO intervention during adolescence attenuated leptin serum levels in adult offspring, it did not change hepatic activation of STAT3. Although n-3 PUFA has reversed the increased leptin expression and leptin receptor expression in HFD mice models(Reference Rossi, Lombardo and Lacorte22, Reference Shen, Wang and Xia54), its effect on leptin signalling pathway remains to be elucidate.
In this work, the effects of FO intake exclusively during adolescence were unable to restore all metabolic outcomes from maternal HFD offspring. However, it reverted the hepatic lipid accumulation and it attenuated the metabolic profile even after FO withdraw. These data highlight the potential of the adolescence period in reprogramming the metabolic profile with effects that persist along life. Adolescence is a period with intense neuronal and hormonal changes, essential to normal development, growth, behaviour and reproductive features(Reference Avendano, Vazquez and Tena-Sempere58). However, these adaptive changes make adolescence a vulnerable period to epigenetic changes, as result of increased plasticity, modifying the transcriptional activity of many genes and highlighting the interplay between genetic, hormonal and environmental changes(Reference Hochberg, Feil and Constancia59). Among neuroendocrine changes at this period, leptin is an important connection among central and peripheral metabolism, modulating energy expenditure and food intake, besides others signals such as the reproductive axis(Reference Avendano, Vazquez and Tena-Sempere58). In our experimental model, the persistent metabolic effects after FO withdraw suggests epigenetic mechanisms involvement in leptin regulation and other genes of energy metabolism control. This hypothesis is reinforced by a previous study that described FO impact on epigenetic mechanisms such as DNA and histone modifications, by methylation(Reference Shen, Wang and Xia54, Reference Lee, Barraza-Villarreal and Biessy60) and acetylation(Reference Song, Na and Baek61), and non-coding RNA(Reference Li, Li and Gao62). n-3 PUFA were able to revert leptin promoter modifications promoted by diet-induced obesity (DIO) in mice(Reference Shen, Wang and Xia54) and to abolish the insulin-induced acetylation on sterol regulatory-element binding protein 1 and cyclo-oxygenase-2 genes in epithelial cells culture(Reference Song, Na and Baek61). These mechanisms were responsible for the down-regulation of leptin expression in DIO mice(Reference Shen, Wang and Xia54) and down-regulation of lipogenesis and inflammation markers in epithelial cells(Reference Song, Na and Baek61), respectively. Then, these observations suggest putative mechanisms by which FO intake may alter specific epigenetic regulatory processes, impacting in energy metabolism in our experimental model. Nonetheless, a detailed analysis of the regulation of epigenetic markers is needed in the future.
Our main limitation in this work was not identifying the exact molecular mechanism involved in this metabolic reprogramming, and the transgenerational transmission of these new features. However, our observations highlight that perinatal metabolic programming did not promote a definitive metabolic outcome. This metabolic profile of adult offspring is reversible, not only during an intervention along life(Reference Wyrwoll, Mark and Mori19, Reference Hou, Ji and Wang20, Reference Mathias, Elmhiri and de Oliveira63), but even with punctual intervention during the adolescence period. This punctual reprogramming during the adolescence period increases the possibilities to prevention strategies for programmed metabolic syndrome(Reference Mathias, Elmhiri and de Oliveira63).
Therefore, in the present study, we showed that the increased susceptibility to obesity and non-alcoholic fatty liver disease determined by early life events, such as maternal HFD, could be attenuated by interventions during the adolescence period. The intervention only during the adolescence period with FO intake did not affect adiposity and leptinaemia at this period of life(Reference Oliveira, Souza and Souza8), but it reduced adiposity, serum leptin and restored normal hepatic TAG content in adult life, demonstrating long-term effects. Then, FO intake during adolescence reduced the susceptibility of STD and HFD offspring to obesity and non-alcoholic fatty liver disease development in adulthood. These data suggest that this susceptibility is a result of many epigenetic modifications along life, including perinatal but also the adolescence period(Reference Bay, Morton and Vickers15, Reference Oosting, Kegler and Boehm64, Reference de Oliveira, Lisboa and de Moura65), evidencing that maternal choices but also adolescent habits have impact on susceptibility to metabolic disorders in adult life.
Conclusion
FO consumption exclusively during adolescence programmed STD offspring and reprogrammed HFD offspring male rats to a healthier metabolic phenotype in adult life. The reduction in visceral adiposity, serum leptin and hepatic TAG were the main beneficial FO effects observed in offspring adulthood. These observations highlight the adolescence period as a critical window to prevent or attenuate the long-term metabolic disturbances in the offspring from obese mothers, possibly by reprogramming gene expression and priming for improvement of metabolic outcomes later in life.
Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa no Estado do Rio de Janeiro (FAPERJ) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The authors are grateful for technical support from Laboratório de Lípideos – LabLip – FCM/UERJ, and for assistance of Luela Dias and Juliana Penna.
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (C. C. P.-M., grant no. 305427/2013-0), Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ) (C. C. P.-M., grant nos. 110.108/2013 and 202.924/2015) (L. L. S., grant no. 201.821/2015) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The funding agencies had no role in the design, analysis or writing of this article.
A. F. P. S., L. S. O., A. C., E. S. and L. L. S. carried out all animal studies and participated in collection and interpretation of data. A. F. P. S. and L. L. S. performed all experimental procedures and analyses. G. E. G. K. and G. C. A. performed biochemical experiments and analyses. L. L. S., C. C. P.-M. and I. H. T. formulated the research question; they delineated the experimental design and supervised the study. L. L. S. and C. C. P.-M. wrote the manuscript.
The authors declare that there are no conflicts of interest.









