


1. Introduction
According to the classical view of biology, RNA has three roles, as a messenger (mRNA) that shuttles information between DNA and proteins, as an adaptor (tRNA) that translates the information stored in mRNA into protein sequence, and as a structural molecule (rRNA) that is part of the ribosome (Fig. 1). Research over the last 25 years has revealed that RNA carries out many other essential functions in the cell. RNA regulates gene expression at the transcriptional and translational levels, and this regulation often arises from the structures adopted by various RNA classes, including ribozymes, riboswitches, and RNA–protein complexes (Doudna & Cech, Reference Doudna and Cech2002; Serganov & Nudler, Reference Serganov and Nudler2013). Since RNA is single stranded it can fold back on itself forming a plethora of secondary and tertiary interactions, as well as complex folding motifs, binding pockets, and active site clefts (Fig. 1). Misfolding and mutations of RNA are characteristics of many cancers and diseases; for example, triplet repeat expansion diseases are associated with Huntington's disease, myotonic dystrophy, and Fragile X syndrome (Osborne & Thornton, Reference Osborne and Thornton2006). Single nucleotide polymorphisms (SNPs) that alter the structural ensemble of RNA sequences also have been associated with genetic diseases (Halvorsen et al. Reference Halvorsen, Martin, Broadaway and Laederach2010). Accordingly, understanding RNA structures and their dynamic regulation is an integral aspect of understanding RNA function.

Fig. 1. The Classical View (top) and the Modern View (bottom) of RNA's role in biology. In the classical view of biology, RNA (top) serves as a messenger molecule between DNA and proteins and proteins have all the main functions in cells. Messenger RNA serves to translate information from DNA to proteins. The modern view of biology (bottom) has emerged in the last 25 years as the field learns more about the many functions of RNA. Non-coding RNA (ncRNA) has vast regulatory functions, some of which include immune responses (‘ppp’ = 5′-triphosphate, which activates PKR) (Nallagatla et al. Reference Nallagatla, Hwang, Toroney, Zheng, Cameron and Bevilacqua2007), thermosensors, ribozymes, riboswitches, and genome editing. In the modern view of biology, proteins still have most cellular functions, but RNA plays essential roles in the cell beyond its classical functions.
The negatively charged phosphate backbone and diverse folds of RNA lead it to interact with cellular components, including metal ions, ligands, and proteins. Binding interactions with these species can change the fold of the RNA (Fig. 2). Monovalent and divalent metal ions are essential for the catalysis of small self-cleaving and large ribozymes both for folding and for active site catalysis (Serganov & Patel, Reference Serganov and Patel2007; Swisher et al. Reference Swisher, Su, Brenowitz, Anderson and Pyle2002). Small molecule-binding refolds riboswitches to regulate gene expression in a positive or negative mode (see Fig. 2) (Garst et al. Reference Garst, Edwards and Batey2011; Serganov & Patel, Reference Serganov and Patel2007).

Fig. 2. RNA interactions with RNA-binding proteins (RBP, left), metal ions (central), and ligands (star, right) can result in structure changes. Unlike typical in vitro conditions, there are other molecules and complex solution conditions in vivo that can interact with RNA and change its structure. These structure changes can result in an RNA with less structure (top left) more structure (top right), or an alternate conformation than the structure that is prevalent in vitro (bottom). Also shown (bottom) are the bacterial expression platforms of riboswitches that switch between two mutually exclusive structures that turn a gene ON (left) or OFF (right) by exposing or sequestering the Shine–Dalgarno sequence (blue).
Functional RNAs, such as tRNA, ribozymes, and riboswitches, are often found in ribonucleoprotein (RNP) complexes, which can help fold metastable RNA structures or induce a conformational change. The Protein Data Bank (PDB) has over 2000 annotated RNA-binding proteins, which include RNA chaperones, helicases, dsRNA-binding proteins, tRNA synthetases, ribonucleases (RNases) and RNA recognition motifs (RRMs) (Gerstberger et al. Reference Gerstberger, Hafner and Tuschl2014). Highly studied RNPs include the ribosome, non-plant RNase P, and the splicesome, which are responsible for the synthesis of proteins, maturation of the 5′-end of tRNAs, and splicing of pre-mRNAs, respectively. Remarkably it is the RNA component that is responsible for catalysis in these three RNPs, while the protein component provides scaffolding (Guerrier-Takada et al. Reference Guerrier-Takada, Gardiner, Marsh, Pace and Altman1983; Nissen et al. Reference Nissen, Hansen, Ban, Moore and Steitz2000).
Crowding plays critical but poorly understood roles in RNA folding. The cellular environment is very complex with up to 40% of the cytosol taken up by macromolecules (Minton, Reference Minton2001; Zimmerman & Trach, Reference Zimmerman and Trach1991). In addition, small molecule metabolites, polyamines and other species occupy volume and interact with RNAs. Macromolecular crowding can drive the compaction of RNA and proteins, while small molecules can either stabilize or destabilize RNAs through interactions with the RNA molecule (Minton, Reference Minton2001).
Nearly all of the biological components that influence RNA structure and function in vivo – biological ion compositions, ligands, proteins, and crowding – are missing during typical in vitro experiments (expanded upon in Table 1). A major goal of current research is to add back these components in order to more closely mimic in vivo conditions. We group studies of RNA folding into three approaches: (1) in vitro studies in dilute solutions; (2) in vivo studies in living cells; and (3) in vivo-like studies that mimic in vivo conditions. We also discuss how in silico methods facilitate each of these approaches. There are advantages and limitations to working in each of these conditions, and experiments in each can yield unique insights into the biological functions of RNAs. Structures and folding pathways of RNA have been studied mostly in dilute in vitro conditions, resulting in fundamental insights into RNA structure and function. However, there is a deep desire to understand how Nature works, and the in vivo environment is very different from typical in vitro solution conditions (Table 1 and Fig. 3). In particular, the majority of thermodynamic experiments studying the energetics of folding and associated pathways (Freier et al., Reference Freier, Kierzek, Jaeger, Sugimoto, Caruthers, Neilson and Turner1986b; Schroeder & Turner, Reference Schroeder and Turner2009) have been conducted in non-biological salt concentrations (London, Reference London1991; Lusk et al. Reference Lusk, Williams and Kennedy1968; Minton, Reference Minton2001; Romani, Reference Romani2007; Truong et al. Reference Truong, Sidote, Russell and Lambowitz2013). There are also myriad RNA–protein interactions in vivo, many of which profoundly affect RNA folding and function.

Fig. 3. Artist's rendition of in vitro conditions (left), in vivo conditions (right) and in vivo-like conditions (center). Typical in vitro solutions are dilute with high monovalent ion concentrations that are very different from cellular conditions. The cellular environment is complex with monovalent and divalent salts, macromolecules, cosolutes, and organelles. In vivo-like conditions (center) bridge in vitro and in vivo conditions and are more complex than in vitro conditions with added synthetic crowding agents and proteins and physiological ion concentrations. However, in vivo-like conditions are still much less complex than those prevailing in vivo.
Table 1. Comparison of in vitro and in vivo solution conditions

Conditions in vitro are the conditions historically used to study RNA. Typical values in the literature are listed in the table, although actual values differ across various studies. In vivo-like conditions, not provided in this table, typically emulate at least one of the conditions missing during in vitro experiments.
a Typically Na+ is used in vitro although K+ is found. Freier et al. (Reference Freier, Kierzek, Caruthers, Neilson and Turner1986a), Xia et al. (Reference Xia, Santalucia, Burkard, Kierzek, Schroeder, Jiao, Cox and Turner1998).
b Feig & Uhlenbeck (Reference Feig, Uhlenbeck, Gesteland, Cech and Atkins1999).
c Typically Mg2+ is used. Herschlag & Cech (Reference Herschlag and Cech1990), Tanner & Cech (Reference Tanner and Cech1996).
d Alberts et al. (Reference Alberts, Bray, Lewis, Roberts and Watson1994), London (Reference London1991), Romani (Reference Romani2007).
e Lusk et al. (Reference Lusk, Williams and Kennedy1968), Truong et al. (Reference Truong, Sidote, Russell and Lambowitz2013).
Over the last few years, in vivo experiments probing RNA structure in living cells have revealed significant differences in many RNA structures as compared to in vitro (Kwok et al. Reference Kwok, Ding, Tang, Assmann and Bevilacqua2013; Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014; Tyrrell et al. Reference Tyrrell, Mcginnis, Weeks and Pielak2013). In vivo studies, while desirable because of their biological relevance, are at the same time limited in that they typically elucidate only the ensemble structure of each RNA transcript, do not deconvolute RNA–protein interactions versus RNA self-structure, and cannot easily perturb or control solution conditions. In particular, biophysical studies that can be readily conducted under highly controlled in vitro conditions are often simply not feasible in vivo. In an effort to gain more insight into the structure and function of RNA in the cellular environment, recent studies have focused on the folding pathway, structure, and function of RNAs under in vivo-like conditions, which mimic conditions in the cell (Desai et al. Reference Desai, Kilburn, Lee and Woodson2014; Dupuis et al. Reference Dupuis, Holmstrom and Nesbitt2014; Kilburn et al. Reference Kilburn, Roh, Behrouzi, Briber and Woodson2013; Nakano et al. Reference Nakano, Miyoshi and Sugimoto2014; Strulson et al. Reference Strulson, Molden, Keating and Bevilacqua2012, Reference Strulson, Yennawar, Rambo and Bevilacqua2013).
In silico prediction and modeling of RNA structure is an important tool used in all three of the above approaches to provide additional insight into RNA structure and function (Dawson & Bujnicki, Reference Dawson and Bujnicki2016; Seetin & Mathews, Reference Seetin and Mathews2012a). Prediction of canonical base pairs, for example, provides testable hypotheses for RNA structure and also provides frameworks for interpreting experimental results. Likewise, experimental data aid in improving in silico structure prediction.
In this review, we discuss major achievements in describing and understanding RNA folding and structure through in vitro, in vivo, and in silico efforts. The next section introduces the reader to in vitro studies of RNA folding, which set the stage for in vivo and in vivo-like studies of RNA folding. We focus on recent efforts to understand how RNA folds in the cell by bridging the gap between knowledge of RNA structure and folding in vitro and in vivo, which has led to an emerging field that studies RNA under in vivo-like conditions. We also discuss ways in which the accuracy of in silico modeling could be improved with experimentally derived in vivo structure probing data. We conclude by discussing advances needed under cellular-like conditions to better understand how RNA folds in the cell.
2. Setting the stage
2.1 In vitro studies of RNA folding
Most of what we currently know about RNA structure and folding comes from studies completed in vitro, under experimental conditions that favor a folded state. Such studies are typically conducted in dilute solutions with high concentrations (~1 M) of monovalent ions (Freier et al. Reference Freier, Kierzek, Jaeger, Sugimoto, Caruthers, Neilson and Turner1986b) and/or (~10 mM) divalent ions (Herschlag & Cech, Reference Herschlag and Cech1990), especially Mg2+, or under conditions that facilitate population of a desired folding intermediate, for example, by renaturing the RNA at an unusual temperature or salt concentration (Baird et al. Reference Baird, Westhof, Qin, Pan and Sosnick2005). These solution conditions are advantageous for studying folding because they can be chosen such that the RNA folds in an apparent two-state manner or the RNA populates just a single intermediate, but have the drawback that they differ profoundly from in vivo conditions, which have predominantly ~140 mM K+ and 0·5–3 mM Mg2+ (expanded upon in Table 1).
An advantage of using high concentrations of monovalent salts is that they compete with trace polyvalent metal ions and hydroxide ions for the phosphate backbone thereby reducing RNA degradation. In addition, high monovalent salt conditions minimize end fraying of RNA hairpins, favoring two-state folding (Freier et al. Reference Freier, Kierzek, Jaeger, Sugimoto, Caruthers, Neilson and Turner1986b). As we describe below, the thermodynamics and kinetics of systems, ranging from simple RNAs, such as hairpins and bulges, to complex RNAs and RNPs, such as ribozymes and the ribosome, have been well characterized. Many aspects of the RNA-folding process can be understood by the application of techniques and the systematic manipulation of conditions only possible under in vitro or under in vivo-like environments.
2.1.1 Major advances: elucidating RNA folding pathways in vitro
With the invention of various enzymological methods, such as PCR, cloning, T7 transcription and chemical synthesis, RNA preparation has advanced to the point where RNA of almost any sequence and length can be studied (Hoseini & Sauer, Reference Hoseini and Sauer2015; Li et al. Reference Li, Wen, Shen, Lu, Huang and Chang2011; Milligan et al. Reference Milligan, Groebe, Witherell and Uhlenbeck1987; Mullis, Reference Mullis1990). A wide variety of techniques have been applied to the study of RNA in vitro (Table 2). The earliest studies on RNA were conducted on homoribopolymers, such as polyU and polyA, which revealed that stacking – the non-bonded interactions between the surfaces of the bases – contributes to RNA stability (Richards et al. Reference Richards, Flessel and Fresco1963; Suurkuusk et al. Reference Suurkuusk, Alvarez, Freire and Biltonen1977). These studies also provided the first indications that individual RNAs adopt structure. An early breakthrough was from studies of tRNA, which could be isolated from living systems owing to its high cellular abundance, which led to insights into RNA tertiary structure. The cloverleaf base pairing of tRNA had been first predicted from sequence alignments of sequence variants (Levitt, Reference Levitt1969). Solving the crystal structure of tRNA confirmed its cloverleaf secondary structure and revealed novel tertiary interactions (Kim et al. Reference Kim, Quigley, Suddath, Mcpherson, Sneden, Kim, Weinzierl and Rich1973; Robertus et al. Reference Robertus, Ladner, Finch, Rhodes, Brown, Clark and Klug1974). The crystal structure of tRNA provided the first direct evidence that RNAs can form complex structures, akin to those of proteins, and that stacking, base pairing, and tertiary contacts all contribute to the adoption of complex three-dimensional (3D) structures (Sussman et al. Reference Sussman, Holbrook, Warrant, Church and Kim1978). With the advent of chemical synthesis techniques, ~100–200 mer of DNA and eventually ~50 mer RNA of any sequence could be made (Matteucci & Caruthers, Reference Matteucci and Caruthers1981; Scaringe et al. Reference Scaringe, Wincott and Caruthers1998; Sierzchala et al. Reference Sierzchala, Dellinger, Betley, Wyrzykiewicz, Yamada and Caruthers2003), with a plethora of atomic modifications. Semi-synthetic approaches were then developed that combine enzymological and chemical synthesis to facilitate the introduction of mutations both at the nucleotide and functional group levels in RNAs of any size (Moore & Sharp, Reference Moore and Sharp1992).
Table 2. Common experimental techniques used to study RNA structure and folding

Thermodynamic and kinetic studies under in vitro conditions provide insight into the complex folding pathways of many functional RNAs. Ribozymes and riboswitches are ideal for the study of RNA folding because their function serves as a readout for the occupancy of the native state (Banerjee et al. Reference Banerjee, Jaeger and Turner1993; Crothers et al. Reference Crothers, Cole, Hilbers and Shulman1974; Mitchell & Russell, Reference Mitchell and Russell2014; Mitchell et al. Reference Mitchell, Jarmoskaite, Seval, Seifert and Russell2013; Rook et al. Reference Rook, Treiber and Williamson1998). Major themes are that large RNAs fold on a rugged pathway through populated intermediates, largely in a hierarchical manner, where secondary structures form before tertiary contacts, as demanded by the topologies of these complex RNAs (Fig. 4) (Brion & Westhof, Reference Brion and Westhof1997; Mitchell & Russell, Reference Mitchell and Russell2014; Solomatin et al. Reference Solomatin, Greenfeld, Chu and Herschlag2010; Tinoco & Bustamante, Reference Tinoco and Bustamante1999; Wan et al. Reference Wan, Suh, Russell and Herschlag2010). It is informative to consider these principles on several specific RNAs. Using temperature-dependent nuclear magnetic resonance (NMR) and relaxation kinetics, the mechanism of tRNA unfolding was elucidated (Crothers et al. Reference Crothers, Cole, Hilbers and Shulman1974; Hilbers et al. Reference Hilbers, Robillard, Shulman, Blake, Webb, Fresco and Riesner1976; Stein & Crothers, Reference Stein and Crothers1976). Five distinct transitions were mapped to the four arms and the tertiary contacts (Crothers et al. Reference Crothers, Cole, Hilbers and Shulman1974). Secondary structures form on a fast timescale (μs to ms) followed by folding of the tertiary structure on a slower timescale (ms to s). In the presence of monovalent metal ions, multiple thermal unfolding transitions are observed for these processes (Stein & Crothers, Reference Stein and Crothers1976). These transitions merge into one as Mg2+ concentrations are increased, revealing that Mg2+ induces an apparent two-state folding. Larger functional RNAs, ribozymes, and riboswitches also fold in a hierarchical manner in vitro (Fig. 4a ).
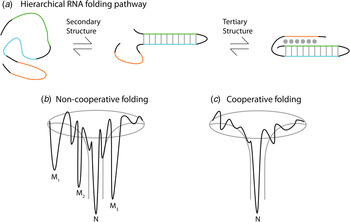
Fig. 4. Depiction of the hierarchical RNA folding pathway and folding funnels for non-cooperative and cooperative folding. (a) RNA folds in a hierarchical manner in which secondary structures form followed by tertiary structure. Hierarchical folding can be (b) rugged and non-cooperative in which the pathway intermediates are populated and the RNA can form misfolds (Mi) before populating the native state (N), or folding can occur in a (c) cooperative manner in which the intermediates do not populate and the RNA folds in a single transition.
The Azoarcus group I ribozyme was used to determine the influence of tertiary interactions on RNA folding (Fig. 5). This ribozyme has been shown to fold quickly, with ~80% of the ribozyme folded into the native state in under 50 ms in 15 mM Mg2+ (Rangan et al. Reference Rangan, Masuida, Westhof and Woodson2003). To determine the roles of tertiary interactions in ribozyme folding, the tertiary contact between the P9 GAAA tetraloop and its J5/5a receptor were perturbed (Chauhan & Woodson, Reference Chauhan and Woodson2008). While the WT ribozyme folded in a cooperative manner to the native state, the tetraloop mutant occupied many previously hidden intermediates on the folding pathway, even at 50 mM Mg2+. This study indicated that tertiary contacts promote cooperative RNA folding.
More recently, methods have been developed to study RNA folding on the nucleotide level and at the millisecond timescale (Merino et al. Reference Merino, Wilkinson, Coughlan and Weeks2005; Scalvi et al. Reference Scalvi, Woodson, Sullivan, Chance and Brenowitz1997; Zhuang et al. Reference Zhuang, Bartley, Babcock, Russell, Ha, Herschlag and Chu2000). Experiments using hydroxyl radical mapping yielded insight into the pathway of tertiary structure formation and folding kinetics in the Tetrahymena Group I Intron (Sclavi et al. Reference Sclavi, Sullivan, Change, Brenowitz and Woodson1998). Combined with time resolved small-angle X-ray scattering (SAXS) (Roh et al. Reference Roh, Guo, Kilburn, Briber, Irving and Woodson2010), hydroxyl radical footprinting on the Tetrahymena ribozyme folding pathway uncovered an initial collapse of structure on the millisecond timescale during the dead time of the instrument. During the subsequent time course, tertiary contacts and several intermediates were elucidated (Sclavi et al. Reference Sclavi, Sullivan, Change, Brenowitz and Woodson1998).
The folding pathways of large functional RNAs have proven to be quite complex with intermediates that can be trapped for minutes to hours (Banerjee & Turner, Reference Banerjee and Turner1995; Chadalavada et al. Reference Chadalavada, Senchak and Bevilacqua2002; Zarrinkar et al. Reference Zarrinkar, Wang and Williamson1996). For example, 90% of the Tetrahymena ribozyme is found in a misfolded state that transitions to the native state with hour timescale kinetics (Banerjee & Turner, Reference Banerjee and Turner1995), and the hepatitis delta virus (HDV) ribozyme folds through numerous intermediates, some long-lived (Chadalavada et al. Reference Chadalavada, Senchak and Bevilacqua2002). Long-lived misfolded intermediates are often very similar in structure to the native RNA and typically arise from a secondary structure mispairing or an incorrect 3D topology (Mitchell et al. Reference Mitchell, Jarmoskaite, Seval, Seifert and Russell2013; Treiber et al. Reference Treiber, Rook, Zarrinkar and Williamson1998; Wan et al. Reference Wan, Suh, Russell and Herschlag2010). For instance, a long-lived intermediate occurs in the Tetrahymena ribozyme where P3 is docked correctly but the topology of the ribozyme is incorrect (Mitchell & Russell, Reference Mitchell and Russell2014; Mitchell et al. Reference Mitchell, Jarmoskaite, Seval, Seifert and Russell2013). To fold into the native state, this misfold needs to undergo a global unwinding of structure. Importantly, the extent to which these pathways and intermediates are populated in vivo is unknown. Indeed, some of these folding intermediates are affected by the method by which the RNA is purified. For example, the wild-type HDV ribozyme has the optimal rate of catalysis when the ribozyme is folded co-transcriptionally, as opposed to being renatured prior to assay (Chadalavada et al. Reference Chadalavada, Cerrone-Szakal and Bevilacqua2007). In addition, choice of flanking sequences can profoundly affect the activity of small and large ribozymes (Cao & Woodson, Reference Cao and Woodson1998; Chadalavada et al. Reference Chadalavada, Knudsen, Nakano and Bevilacqua2000).
2.1.2 Major advances: applying biophysical techniques to study RNA folding in vitro
Using optical melting, a set of thermodynamic parameters have been established to estimate folding free energies from sequence and structure alone (Andronescu et al. Reference Andronescu, Condon, Turner and Mathews2014; Lu et al. Reference Lu, Turner and Mathews2006; Turner & Mathews, Reference Turner and Mathews2010; Xia et al. Reference Xia, Santalucia, Burkard, Kierzek, Schroeder, Jiao, Cox and Turner1998). The nearest-neighbor model predicts the free energy and stability of an RNA from each base pair's nearest neighbor, along with initiation, symmetry, and terminal-AU base pair terms. Nearest-neighbor terms for certain loops, those regions without canonical base pairs, have also been determined (Mathews et al. Reference Mathews, Disney, Childs, Schroeder, Zuker and Turner2004). As noted below, these experimental parameters have been incorporated in RNA structure prediction programs that find the lowest free energy structures for an input RNA sequence (Mathews, Reference Mathews2006; Reeder et al. Reference Reeder, Hochsmann, Rehmsmeier, Voss and Giegerich2006; Seetin & Mathews, Reference Seetin and Mathews2012a). Parameters to account for complicated tertiary interactions and loops are still being revised (Liu et al. Reference Liu, Shankar and Turner2010b, Reference Liu, Diamond, Mathews and Turner2011; Lu et al. Reference Lu, Turner and Mathews2006). The nearest-neighbor parameters currently available were measured under highly folding in vitro conditions of 1 M NaCl.

Fig. 5. Different RNA structures can be populated under in vitro, in vivo, and in vivo-like conditions. RNA structures induced by the cellular environment, including proteins and crowding, are shown in the two outermost structures. The conditions in vitro favor the population of a structure that may not always be the functional RNA structure (center two structures). Depending on the in vivo-like conditions chosen, specific RNA structures will be populated.
Low-resolution methods provide information about the structure of RNA on both the global and nucleotide length scales. Although these techniques do not give atomic resolution, they have significantly faster throughput than crystallography or NMR structures while still providing insight into the fold and function of RNA. SAXS and Förster Resonance Energy Transfer (FRET) provide low-resolution information on the overall fold of an RNA. RNA is particularly amenable to SAXS because the phosphate backbone is electron-rich and scatters X-rays well. Different solution conditions can be prepared and examined quickly by SAXS to elucidate RNA structural changes. The structures of several functional RNAs and RNA–protein complexes have been explored using SAXS, including ribozymes, riboswitches bound and unbound to ligand, and the spliceosome (Pollack, Reference Pollack2011). FRET studies, in which acceptor and donor fluorophores are attached to the RNA at key locations, have helped elucidate folding intermediates (Walter, Reference Walter2001). Using single-molecule FRET, or smFRET, the Tetrahymena ribozyme was found to fold into multiple conformations, nearly all of which were active, indicating that the ribozyme populates multiple native states (Solomatin et al. Reference Solomatin, Greenfeld, Chu and Herschlag2010). Upon exposure to denaturant, the ribozyme re-populated the native conformations, indicating the results are independent of original conformation. Both SAXS and smFRET have been applied to RNA folding under in vivo-like conditions, as discussed below (Paudel & Rueda, Reference Paudel and Rueda2014; Strulson et al. Reference Strulson, Yennawar, Rambo and Bevilacqua2013).
Structure probing methods serve essential roles in elucidating the structures of functional RNAs at the nucleotide level. Several chemical probes have been employed to attack and modify the RNA bases, sugar, and backbone, in order to reveal the base pairing status of the nucleotides. Commonly used chemical probes include dimethyl sulfate (DMS), carbodiimide tosylate (CMCT), and SHAPE reagents, which allow selective 2′-hydroxyl acylation – each of which is analyzed by primer extension via reverse transcription. Commonly used enzymatic probes are RNases T1, V1, and S1. Targets of these probes and methods of readout are provided in Fig. 6. Structure probing of RNAs in vitro has revealed very complex structures, as well as binding sites of ligands, metal ions, and proteins. As discussed below, structure probing with chemical probes can be used in vivo as well.

Fig. 6. RNA modifications by DMS, SHAPE reagents (selective 2′-hydroxyl acylation analyzed by primer extension), and CMCT (1-cyclohexyl-(2-morpholinoehyl)carbodiimide metho-p-toluene). SHAPE reagents modify the 2′-hydroxyl on the sugar of all four ribonucleobases. SHAPE reagents include 1M7 (1-methyl-7-nitroisatoic anhydride), NMIA (N-methylisotoic anhydride), and NAI (2-methylnicotinic acid imidazolide). DMS modifies the N1 of A and the N3 of C as well as the N7 of G. CMCT modifies N3 of U and N1 of G. The chemical modifications (except N7 of G) can be detected immediately by RT followed by gel electrophoresis or high-throughput sequencing, and the enzymatic cleavages, which cleave single- and double-stranded RNA, can be read out through gel electrophoresis or high-throughput sequencing.
Very recently, RNA structure in vitro has been probed genome-wide at the nucleotide level, utilizing the power of next-generation sequencing. Several methods have been developed to map entire transcriptomes. Parallel Analysis of RNA Structure (PARS) cleaves double-stranded regions with RNase V1 and single-stranded regions with RNase S1, and FragSeq cleaves single-stranded regions with nuclease P1 (Kertesz et al. Reference Kertesz, Wan, Mazor, Rinn, Nutter, Chang and Segal2010; Underwood et al. Reference Underwood, Uzilov, Katzman, Onodera, Mainzer, Mathews, Lowe, Salama and Haussler2010). In PARS, RNA is extracted from cells and aliquots are separately exposed to each nuclease, the digested RNA is converted to cDNA through reverse transcription, and then deep sequenced to map the reverse transcriptase stops to the genome. A PARS score is determined from the log ratio of V1/S1 sequencing reads, where a high PARS score indicates more RNA structure (Kertesz et al. Reference Kertesz, Wan, Mazor, Rinn, Nutter, Chang and Segal2010). In FragSeq, RNA is extracted from cells, and one aliquot is treated with P1 nuclease and a second aliquot is untreated (Underwood et al. Reference Underwood, Uzilov, Katzman, Onodera, Mainzer, Mathews, Lowe, Salama and Haussler2010). RNA-seq is then performed on each aliquot, and a cutting score is determined for each mapped nucleotide that indicates the propensity to be cut by P1 nuclease. The cutting score is then used to annotate RNA secondary structures and/or to restrain RNA secondary structure prediction. Genome-wide studies in several organisms, both in vitro and in vivo, have found that there is significantly more structure in the coding regions than the untranslated regions of RNAs (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014; Kertesz et al. Reference Kertesz, Wan, Mazor, Rinn, Nutter, Chang and Segal2010; Li et al. Reference Li, Zheng, Vandivier, Willmann, Chen and Gregory2012; Wan et al. Reference Wan, Qu, Zhang, Flynn, Manor, Ouyang, Zhang, Spitale, Snyder, Segal and Chang2014; Zheng et al. Reference Zheng, Ryvkin, Li, Dragomir, Valladares, Yang, Cao, Wang and Gregory2010). There is also less structure in the start and stop codons than in the rest of a transcript, which presumably facilitates read-through by the ribosome.
Using a method similar to PARS but differing in that the RNA structure is probed at several temperatures, PARTE (Parallel Analysis of RNA Structures with Temperature Elevation) was used to obtain the folding free energies for yeast transcripts genome-wide in vitro (Wan et al. Reference Wan, Qu, Ouyang, Kertesz, Li, Tibshirani, Makino, Nutter, Segal and Chang2012). RNA from yeast was folded between 30 and 75 °C and exposed to RNase V1 followed by deep sequencing. By examining the melting temperatures (T m) of RNAs, non-coding and coding RNAs could be distinguished and RNAs with distinct cellular functions could be identified. Functional non-coding RNAs (ncRNAs) were found to have a higher T m on average than mRNAs.
Three methods that utilize DMS chemistry to determine transcriptome-wide RNA structure were recently published: Structure-seq, DMS-sequencing (DMS-seq), and modification sequencing (Mod-seq) (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014; Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014; Talkish et al. Reference Talkish, May, Lin, Woolford and Mcmanus2014). To date, only DMS-seq has been applied in vitro and all of the methods have been applied in vivo. These methods are described in more detail below.
2.1.3 Benefits and limitations of in vitro studies
Many of the foundational experiments on RNA folding and structure have come from in vitro experiments, and numerous underlying mechanisms of RNA folding and function have been discovered in vitro. Studies in vitro have revealed the folding pathways and structures of RNAs. More recently, methods have been developed to probe the structure of RNAs genome-wide. Major advances include elucidating fast formation of secondary structure and slow formation of the tertiary contacts, understanding of RNA folding energetics, establishment of nearest-neighbor parameters, and determination of structures of functional RNA motifs. The complex structures that RNA adopts enable diverse functions. Experimental techniques, ranging from structure probing to kinetic methods, have been applied to RNA across diverse pH, salt, and temperature conditions.
The major limitation of in vitro experiments is that the solution conditions are very different from the cellular environment and unavoidably lack many of the components present in cells, which can influence RNA folding and function. These limitations necessitate the development of experiments and techniques under in vivo and in vivo-like conditions to determine how RNAs fold and respond to cellular environmental conditions.
2.2 In vivo studies of RNA folding
In the previous section, we provided an overview of RNA folding in vitro. In this section we discuss recent advances made in vivo to understand RNA folding. We note that RNA structure has also been explored to a lesser extent in cellular extracts. Experiments in extracts contain more proteins bound to RNA than in vitro experiments but less than in vivo studies, as supported by recent comparisons of low DMS reactivity assignments amongst in vitro, extract, and in vivo studies (Ding et al. Reference Ding, Kwok, Tang, Bevilacqua and Assmann2015). Studies in extracts for RNAs with high positive predictive value (PPV) between reactivities in vitro and in silico, such as the ribosome, have been shown to be biologically relevant (Ding et al. Reference Ding, Kwok, Tang, Bevilacqua and Assmann2015; Moazed et al. Reference Moazed, Stern and Noller1986a). Likewise, for RNAs with low PPV between reactivities in vitro and in silico, studies in extracts might not provide the full complement of interactions. While experiments in cell extracts share many similarities with in vivo conditions, thermodynamic assays cannot be easily performed in extracts due to the denaturation and signal of other biomolecules.
An ultimate goal of RNA-folding studies is to understand how RNA behaves in the cell. The majority of the methods developed to study RNA in vivo are structure probing, where several chemicals known to penetrate the cell membrane are applied to modify RNA. Structure probing has been used to study the structures of RNAs in vivo on both the single gene and genome-wide levels, and has resulted in a breadth of information regarding structures that RNA forms inside living cells. These studies have revealed novel in vivo RNA folds, RNA–protein interactions, and novel regulatory roles.
2.2.1 Major advances: transcript-specific RNA structure mapping in vivo
Structure probing of RNA in vivo uses small chemicals such as SHAPE reagents, DMS, and CMCT, which penetrate cells and modify solvent-accessible regions of the RNA (Bloomfield et al. Reference Bloomfield, Crothers and Tinoco2000; Ehresmann et al. Reference Ehresmann, Baudin, Mougel, Romby, Ebel and Ehresmann1987). Structure probing methods using chemicals have revealed that for some transcripts there are significant differences between RNA structures formed in vivo and in vitro. We first describe in vivo structure probing experiments on single transcripts, followed by experiments across a genome.
The first in vivo nucleic acid structure probing study was from the Gilbert laboratory, where binding of multiple proteins to their cognate sites was observed using DMS modification (Nick & Gilbert, Reference Nick and Gilbert1985). Structure probing is outlined in Fig. 6. Briefly, DMS methylates adenine and cytosine on the Watson–Crick face and guanine on the Hoogsteen face. The modification on A and C is read out directly by stops in reverse transcription (RT) one position before the methylated base, while the methylated G is treated with aniline to create an abasic site followed by RT read out, which again stops one position before the modified base (Bloomfield et al. Reference Bloomfield, Crothers and Tinoco2000; Ehresmann et al. Reference Ehresmann, Baudin, Mougel, Romby, Ebel and Ehresmann1987). The RT can be read out in a gene-specific fashion by polyacrylamide gel electrophoresis (PAGE) or capillary electrophoresis (CE), and in a library fashion with next-generation sequencing (see the next section) (Kwok et al. Reference Kwok, Ding, Tang, Assmann and Bevilacqua2013).
The first report of RNA structure comparisons in vivo and in vitro came from the Cech laboratory (Zaug & Cech, Reference Zaug and Cech1995). Structure probing with DMS was used to map the structures of two known protein-bound RNAs, telomerase RNA and U2 snRNA, as well as the Tetrahymena ribozyme. Protections from reactivity in vivo compared with in vitro indicate either protein protection or gain of base pairing, while enhancements of reactivity indicate refolding to expose RNA bases. Telomerase RNA and U2 snRNA showed different reactivity patterns in vivo versus in vitro, consistent with the influence of protein binding on DMS reactivity. As expected, the group I ribozyme had very similar nucleotide reactivity in vivo and in vitro, demonstrating that the ribozyme is not protein-bound and self-splices without protein assistance in vivo.
Our group investigated structures of high and low abundance RNAs, also on a gene-specific basis, and compared DMS and SHAPE reactivities in vivo and in vitro. For low abundance RNAs we developed a gene-specific ligation-mediated PCR (LM-PCR) approach (Kwok et al. Reference Kwok, Ding, Tang, Assmann and Bevilacqua2013). These studies, which were in the model plant species Arabidopsis thaliana, revealed in vivo footprinting on high abundance 25S rRNA and 5·8S rRNA, as well as on the low abundance U12 snRNA. We showed that different bases in 5·8S rRNA are methylated in vivo and in vitro, which provided evidence for 5·8S rRNA refolding in vivo. These studies also provided critical control reactions that strongly supported DMS modification of RNA occurring in vivo and DMS being completely quenched prior to workup of the in vivo reaction. These controls apply equally to the genome-wide studies in the next section.
2.2.2 Major advances: genome-wide RNA structure mapping in vivo
Recently, several groups including ours have developed high-throughput methods to probe RNA structure in living cells transcriptome-wide. These studies revealed significant differences in RNA structure in vivo compared to in vitro and in silico predicted (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014; Kwok et al. Reference Kwok, Tang, Assmann and Bevilacqua2015; Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014; Talkish et al. Reference Talkish, May, Lin, Woolford and Mcmanus2014). Three separate methods using DMS to probe RNA structure in vivo were published in 2014: Structure-seq (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014), DMS-seq (Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014), and Mod-seq (Talkish et al. Reference Talkish, May, Lin, Woolford and Mcmanus2014), each of which utilizes the next-generation sequencing to probe RNA structure transcriptome-wide.
Each of these studies revealed novel information on RNA structure and possible regulatory functions of those structures. In Structure-seq, the PPV describes the fraction of base pairs in the in vivo DMS-restrained predicted structure that are also predicted in the unrestrained in silico predicted structure (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014). Of the greater than 10 000 mRNAs evaluated in this fashion, most had a PPV value far from unity, with a maximum PPV of the distribution slightly <0·4. This observation indicates that the in vivo structures of many RNAs cannot be predicted well purely in silico, using only sequence information and thermodynamic parameters originally derived in vitro. We also observed that the mRNAs with the lowest PPV distribution (bottom 5%) were enriched in annotations of biological function of stress and stimulus response, while the mRNAs with the highest PPV distribution (top 5%) were enriched in housekeeping functions (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014).
One possibility is that housekeeping RNAs have well-defined folds, while stress-related RNAs have ill-defined folds or adopt many folds. DMS-seq in yeast found that certain mRNAs are less structured in vivo than naked, protein-free RNA in vitro, and under in vivo ATP depletion the mRNAs on a whole become more structured, with the implication that ATP-dependent processes contribute to RNA unfolding. It is likely that a range of factors in vivo contribute to RNA structure (Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014). Mod-seq was used to reveal the binding location of the L26 protein by deletion in yeast; upon L26 deletion, 58 nucleotides became more reactive to DMS in vivo and most of these nucleotides were located in the 5·8S–25S rRNA interface where L26 is known to bind (Talkish et al. Reference Talkish, May, Lin, Woolford and Mcmanus2014).
Individual copies of a given RNA sequence can adopt different conformations owing to the single-stranded nature of RNA. Indeed, this may be the origin of the low PPV value in the stress-related genes (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014) in that structure probing methods reveal the average of all populated structures at some instant in time. There is experimental evidence that some transcripts appreciably populate multiple structures in vitro. Using the PARS method, ~4% of mRNAs had both high RNase V1 and RNase S1 activity, which cleave paired and unpaired RNA, respectively, under in vitro conditions (Wan et al. Reference Wan, Qu, Zhang, Flynn, Manor, Ouyang, Zhang, Spitale, Snyder, Segal and Chang2014). The high extents of cleavage by both nucleases suggest that populations of those mRNAs adopt multiple conformations simultaneously in vitro, and potentially in vivo.
Genome-wide studies revealed a triplet periodicity in mRNA nucleotide reactivity in yeast, mouse, and humans in vitro (Incarnato et al. Reference Incarnato, Neri, Anselmi and Oliviero2014; Wan et al. Reference Wan, Qu, Zhang, Flynn, Manor, Ouyang, Zhang, Spitale, Snyder, Segal and Chang2014), as well as in Arabidopsis in vivo (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014; Kertesz et al. Reference Kertesz, Wan, Mazor, Rinn, Nutter, Chang and Segal2010). The triplet repeat in reactivity is observed in the coding sequence but not in the untranslated regions. At present the mechanism behind the periodicity is not understood. Observation of the repeat in vitro suggests that occupancy of ribosomes is not necessary. Additional studies under in vitro, in vivo, and in vivo-like conditions will be necessary to attain a molecular-level understanding of the triplet periodicity in mRNA.
High-throughput sequencing has been coupled with CLIP (crosslinking and immunoprecipitation) to probe RNA-binding protein sites transcriptome wide in HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation) and PAR-CLIP (photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation) (Hafner et al. Reference Hafner, Landthaler, Burger, Khorshid, Hausser, Berninger, Rothballer, Ascano, Jungkamp, Munschauer, Ulrich, Wardle, Dewell, Zavolan and Tuschl2010; Licatalosi et al. Reference Licatalosi, Mele, Fak, Ule, Kayikci, Chi, Clark, Schweitzer, Blume, Wang, Darnell and Darnell2008; Weyn-Vanhentenryck et al. Reference Weyn-Vanhentenryck, Mele, Yan, Sun, Farny, Zhang, Xue, Herre, Silver, Zhang, Krainer, Darnell and Zhang2014). Studies using both of these methods on specific proteins have revealed novel sites of protein binding to RNA as well as possible protein regulatory functions (Hafner et al. Reference Hafner, Landthaler, Burger, Khorshid, Hausser, Berninger, Rothballer, Ascano, Jungkamp, Munschauer, Ulrich, Wardle, Dewell, Zavolan and Tuschl2010; Licatalosi et al. Reference Licatalosi, Mele, Fak, Ule, Kayikci, Chi, Clark, Schweitzer, Blume, Wang, Darnell and Darnell2008). Briefly, in HITS-CLIP, RNA is crosslinked to proteins, the protein of interest is isolated through IP, the RNA is reverse transcribed and amplified through PCR, then high-throughput sequencing is performed and reads are mapped to the genome (Licatalosi et al. Reference Licatalosi, Mele, Fak, Ule, Kayikci, Chi, Clark, Schweitzer, Blume, Wang, Darnell and Darnell2008). In PAR-CLIP, cells are grown with a photoactivatable nucleoside (4-thiouridine or 5-bromouridine) in the media to facilitate crosslinking with proteins upon exposure to 365 nm radiation (Hafner et al. Reference Hafner, Landthaler, Burger, Khorshid, Hausser, Berninger, Rothballer, Ascano, Jungkamp, Munschauer, Ulrich, Wardle, Dewell, Zavolan and Tuschl2010).
Genome-wide structure data have recently been used to identify certain sites of RNA–protein interactions. The method icSHAPE was used to probe RNA structure in mouse embryonic stem cells in vivo and in vitro (Spitale et al. Reference Spitale, Flynn, Zhang, Crisalli, Lee, Jung, Kuchelmeister, Batista, Torre, Kool and Chang2015). The difference in nucleotide reactivity in vitro and in vivo matched binding sites of the protein Rbfox2, previously identified with iCLIP experiments. This methodology was tested again and successfully identified RNA-binding sites of another RNA-binding protein, HuR. Using this type of analysis, certain RNA–protein interactions and associated RNA structural rearrangements can be distinguished using bioinformatics with experimental genome-wide mapping data.
2.2.3 Major advances: quantification of cellular factors in vivo
In vivo quantification of all the cellular factors known to affect RNA folding would both allow more accurate interpretation of in vivo RNA structure datasets and allow design of in vivo-like experiments that would more faithfully mimic in vivo conditions. Although such a comprehensive view of the inner workings of living cells has yet to be achieved, tremendous strides have been made in technique development for in vivo monitoring of cellular parameters relevant to RNA structure, including divalent ion concentrations, pH, reactive oxygen species (ROS), certain cosolutes, and RNA molecules themselves. Almost all of these techniques in vivo rely on a fluorescent readout, and thus advances in probe technology have gone hand-in-hand with advances in microscopy, although only the former topic is discussed here.
Fluorescent reporters are of three types: synthetic dyes, genetically encoded reporters, and reporters that incorporate both synthetic dyes and genetically encoded elements. Genetically encoded reporters typically rely on the cellular factor interacting with and altering the readout from a naturally fluorescent protein from jellyfish, green fluorescent protein (GFP), or its engineered variants (Tsien, Reference Tsien2010), the gene for which can be transformed into the system of interest. The ideal sensor will be minimally invasive and will have high specificity, brightness, and signal-to-noise ratios, a dynamic range that can accurately report the range of concentrations observed in vivo, and response kinetics that are as fast as the natural changes in the probed constituent. The best sensors are also ratiometric, which allows signal normalization to take into account such factors as photobleaching and heterogenous dye distribution. It is important to note that the cellular environment differs among various cellular compartments and organelles. For example, the microenvironments of mitochondria (De Michele et al. Reference De Michele, Carimi and Frommer2014) and chloroplasts (Stael et al. Reference Stael, Wurzinger, Mair, Mehlmer, Vothknecht and Teige2011) (both of which have their own genomes and thus local RNA transcription) are quite different from the microenvironment of the nucleus, and both differ from the cytosolic environment. Ideally, a sensor would also have the capacity to be specifically targeted to an organelle or subcellular location where RNA-folding events of interest occur; for example, sensors that are genetically encoded can be fused to sequences that confer organelle-specific targeting (Choi et al. Reference Choi, Swanson and Gilroy2012).
Cations of particular relevance to RNA structure are heavy metals, which tend to destabilize and degrade RNA, Mg2+, which tends to promote RNA folding, and H+ (pH), which affects RNA catalysis. In addition, K+ and Na+ promote formation of the special RNA structure, the G-quadruplex. In vivo concentrations of Mg2+ (London, Reference London1991; Lusk et al. Reference Lusk, Williams and Kennedy1968; Romani, Reference Romani2007; Truong et al. Reference Truong, Sidote, Russell and Lambowitz2013) and K+ as well as pH changes are all within the concentration ranges that can affect RNA structure. Among these cations, sensors based on GFP and its variants are available for Mg2+ (Lindenburg et al. Reference Lindenburg, Vinkenborg, Oortwijn, Aper and Merkx2013), Pb2+ (Nadarajan et al. Reference Nadarajan, Ravikumar, Deepankumar, Lee and Yun2014), Hg2+ (Hu et al. Reference Hu, Hu, Chen and Wang2013), and H+ (Tantama et al. Reference Tantama, Hung and Yellen2011). A number of synthetic pH sensors are also available (Yang et al. Reference Yang, Cao, He, Yang, Kim, Peng and Kim2014). Both genetically encoded and synthetic sensors of ROS are also available (Pouvreau, Reference Pouvreau2014; Swanson et al. Reference Swanson, Choi, Chanoca and Gilroy2011), which could be applied to study how ROS are associated with genetic diseases (Fimognari, Reference Fimognari2015) or environmental conditions (Jaspers & Kangasjärvi, Reference Jaspers and Kangasjärvi2010) that affect RNA structure in vivo.
As discussed in Section 3.2.2, synthetic and biological cosolutes typically destabilize RNA structure. In one early report, sucrose, which is the circulating ‘energy currency’ in plants, was reported to destabilize RNAs in vitro (Gao et al. Reference Gao, Gnutt, Orban, Appel, Righetti, Winter, Narberhaus, Müller and Ebbinghaus2016; Lambert & Draper, Reference Lambert and Draper2007). While the in vitro effects occurred at significantly higher concentrations than prevail in the cytosol proper, in microdomains close to the sites of sugar transporters, sucrose, and other sugars could perhaps be present at significantly higher concentrations and consequently affect RNA structure locally; moreover, weakly folded RNAs, such as certain mRNAs, may be more susceptible to such cosolutes. In a possibly analogous situation, while resting Ca2+ levels in the cell cytosol are 100–200 nM, Ca2+ concentrations as high as 100 mM have been reported at the mouths of Ca2+ channels (Tang et al. Reference Tang, Reddish, Zhuo and Yang2015a). Lipid anchoring of recently developed sucrose and glucose sensors (Fehr et al. Reference Fehr, Lalonde, Lager, Wolff and Frommer2003; Lager et al. Reference Lager, Looger, Hilpert, Lalonde and Frommer2006) to probe the near membrane microenvironment of sugar transporters could allow evaluation of this hypothesis.
The physical microenvironment and the localization of RNA, both of which can impact RNA structure, vary across cellular regions and organelles. Accordingly, methods that allow visualization of the spatial location of any specific RNA of interest are also highly desirable (Buxbaum et al. Reference Buxbaum, Haimovich and Singer2015). One of the first technologies developed for RNA visualization was molecular beacons (Santangelo et al. Reference Santangelo, Nitin and Bao2006), which are oligonucleotides tagged with a synthetic fluorophore at one end and a synthetic quencher on the other end. Molecular beacons take on a non-fluorescent stem-loop structure in the absence of a complementary RNA due to the close proximity of the quencher and fluorophore, but exhibit fluorescence upon unfolding and hybridization to the target RNA. Various strategies (Santangelo et al. Reference Santangelo, Nitin and Bao2006) can be employed to introduce molecular beacons into mammalian cells, but they are not genetically encoded. A more widely used strategy for visualization of specific RNAs employs a genetic approach in which an RNA sequence that binds the bacteriophage MS2 protein is inserted into the UTR of the transcript of interest and the organism is engineered to express GFP-tagged MS2, which then binds to the transcript of interest, marking its location (Buxbaum et al. Reference Buxbaum, Haimovich and Singer2015).
A different type of RNA marker has been developed recently based on the GFP fluorophore. GFP is fluorescent because the folded protein immobilizes the 4-hydroxy-benzylidene-imidazolinone (HBI) fluorophore encoded by a cyclized and subsequently oxidized Ser–Tyr–Gly tripeptide. RNA aptamers have been identified that analogously immobilize and thus induce fluorescence of a related synthetic fluorophore, DFHBI [(Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one]. The sequence of the RNA aptamer is genetically incorporated into the gene of interest and upon RNA expression and administration of the membrane-permeant fluorophore and its immobilization by the RNA aptamer, fluorescence is observed that marks the location of the target RNA (Paige et al. Reference Paige, Wu and Jaffrey2011). The RNA aptamer, dubbed Spinach, as well as the second generation aptamer Spinach2, both require addition of exogenous Mg2+ to fold properly; such addition could obviously also affect native RNA structures. The third generation Spinach reporter, Broccoli, eliminates this requirement (You & Jaffrey, Reference You and Jaffrey2015).
Spinach aptamers can be further modified to read out concentrations of cellular metabolites by fusion of the Spinach aptamer with other aptamer sequences (identified by artificial selection) that selectively bind small molecules (Paige et al. Reference Paige, Duc, Song and Jaffrey2012), or by incorporation of the Spinach aptamer into prokaryotic riboswitches (You et al. Reference You, Litke and Jaffrey2015). Riboswitch-based reporters have the advantage of having undergone natural selection that confers high affinity and specificity for the metabolite of interest, but are not currently ratiometric. Ratiometric sensors based on FRET between CFP and YFP, variants of GFP, have been engineered for several metabolites, including those with relevance to RNA structure. For example, FRET-based sensing of ATP concentration (Imamura et al. Reference Imamura, Huynh Nhat, Togawa, Saito, Iino, Kato-Yamada, Nagai and Noji2009) could be relevant to RNA structure because of the ATP requirement for the activity of RNA helicases (Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014). In summary, the future is bright for in vivo quantification of a plethora of the metabolites and physical properties that affect RNA structure. Quantification of cellular factors in vivo will play an important role in designing artificial cytoplasms to conduct in vivo-like studies of RNA folding.
2.2.4 Benefits and limitations of in vivo studies
Studies in vivo have shown that RNA can adopt different structures in vivo and in vitro, and have led to fresh insights on how the cellular environment affects RNA folding across a genome. Novel RNA structure motifs and RNA–protein interactions have been demonstrated through genome-wide in vivo experiments. In addition, novel RNA regulatory pathways have been identified by such studies.
Since some RNAs have been shown to fold and function differently under cellular conditions, the question arises, “Why not study RNA solely in living cells instead of in dilute solution conditions?” The reality is that methods for directly studying RNA folding in vivo are limited, and most current in vivo approaches rely on structure probing methods that do not probe RNA thermodynamics or folding pathways. Experiments done in vivo provide information only on the average RNA structure in a cell or organism and lack information on RNA dynamics, the folding process, and the presence of multiple populated structures of the same transcript. These limitations motivate in vivo-like studies to understand the influence of cellular conditions on RNA folding. Before moving to the in vivo-like section, we consider the important role that in silico studies play in both in vitro and in vivo studies.
2.3 In silico studies of RNA folding
Studies in vitro and in vivo described above yielded insights into RNA folding and structure that were informed by in silico structure prediction tools. Structure probing experiments, for example, typically use in silico prediction tools to model structure that is guided by the experimental data. In the subsections below, we describe advances in predicting RNA structure from one sequence, from multiple sequences, and with experimental data. Limitations of each approach are provided as well.
2.3.1 Major advances: RNA structure prediction from one sequence in silico
The most popular approaches to predict RNA structure use dynamic programming algorithms to efficiently search the set of possible structures (Eddy, Reference Eddy2004) and folding free energy nearest-neighbor rules to estimate folding stability (Turner & Mathews, Reference Turner and Mathews2010). The dynamic programming algorithms guarantee that every structure allowed by the set of folding rules is considered, except for those containing pseudoknots (see below). This means, for example, that the lowest free energy conformation will be found for programs that find lowest free energy structures, i.e. the most probable structure at equilibrium.
The accuracy of RNA structure prediction from sequence alone, in terms of fraction of known pairs correctly predicted, is stubbornly limited to ~70% (Hajiaghayi et al. Reference Hajiaghayi, Condon and Hoos2012; Lu et al. Reference Lu, Gloor and Mathews2009), and accuracy is lower for long sequences (>1000 nucleobases) such as small and large ribosomal RNAs and mRNAs (Doshi et al. Reference Doshi, Cannone, Cobaugh and Gutell2004) or for sequences that fold to more than one conformation at equilibrium. In silico predictions of base pairs presently rely on a parameterization of stabilities determined in vitro rather than in vivo, and these parameters are based on relatively few experiments, as compared to all possible folded sequences.
In response to this moderate success rate, a number of in silico methods have been developed to predict alternative structures, as reviewed previously (Mathews, Reference Mathews2006). Programs generate sets of alternative hypotheses for the structure (suboptimal structures) (Wuchty et al. Reference Wuchty, Fontana, Hofacker and Schuster1999; Zuker, Reference Zuker1989), feasible structures in equilibrium with each other (stochastic samples) (Ding & Lawrence, Reference Ding and Lawrence2003), or estimates for base pairing probabilities (partition function calculations) (McCaskill, Reference Mccaskill1990). Each of these three methods is described in turn. Suboptimal structures are those with similar free energy to the lowest free energy structure. Certain suboptimal structures can sometimes be more representative of the biological structure than the in silico-estimated lowest free energy structure, and can be viewed as alternative models or alternative hypotheses for the in vivo structure. Stochastic samples are rigorous samples from the equilibrium (Boltzmann) ensemble. They are useful for estimating ensemble statistics for the secondary structure of an RNA. Partition function calculations provide pairing probability estimates; more probable pairs in predicted structures are more likely to occur in the accepted structure (Mathews, Reference Mathews2004).
2.3.2 Major advances: RNA structure prediction from multiple sequences in silico
The accuracy of in silico folding can be dramatically improved by using additional information to guide the folding. In this section, we discuss using homologous sequences to guide the folding, while in the next section we discuss applying experimental data. Multiple homologous sequences, commonly called an RNA family, can be used to estimate the common secondary structure (Seetin & Mathews, Reference Seetin and Mathews2012a) because structure is generally conserved to a greater extent than sequence for RNAs. Due to sequence variation, the number of base pairs conserved across a family is smaller than the number of base pairs adopted by each sequence. With enough sequences, conserved pairs stand out as positions of covariation, where compensating base pair changes are observed. Covariation is a change in sequences where one biological species, for example, will have an AU base pair, but another species will have a GC pair at the homologous position. During evolution, two separate changes occurred in sequence (a compensating change) that conserved the base pair.
Three approaches are used to estimate the biologically conserved structure from a set of homologous sequences (Reeder et al. Reference Reeder, Hochsmann, Rehmsmeier, Voss and Giegerich2006; Seetin & Mathews, Reference Seetin and Mathews2012a). In the first approach, the available sequences are aligned, and then used to restrain the in silico prediction. This approach is typically the fastest, but generally works best when the pairwise sequence identity of all the homologs is high (75% or higher). These programs are exemplified by RNAalifold (Bernhart et al. Reference Bernhart, Hofacker, Will, Gruber and Stadler2008) and TurboFold (Harmanci et al. Reference Harmanci, Sharma and Mathews2011). Programs in the second set predict the structures for each sequence first and then compare the predicted structures to find those common to all sequences. This approach works well when the structure is highly conserved and is exemplified by RNAcast (Reeder & Giegerich, Reference Reeder and Giegerich2005). The third approach is to simultaneously align and fold sequences to find the common structure and sequence alignment. This is the best approach to use when the sequences are diverse (pairwise sequence identity for some sequence pairs below 75%) because low pairwise identity makes sequence alignment challenging. Programs in this class include Dynalign/Multilign (Fu et al. Reference Fu, Sharma and Mathews2014; Xu & Mathews, Reference Xu and Mathews2011), Foldalign (Torarinsson et al. Reference Torarinsson, Havgaard and Gorodkin2007), LocARNA (Will et al. Reference Will, Reiche, Hofacker, Stadler and Backofen2007), PARTS (Harmanci et al. Reference Harmanci, Sharma and Mathews2008), and RAF (Do et al. Reference Do, Foo and Batzoglou2008).
The accuracy of in silico prediction of conserved structures from a set of homologous sequences can be much higher, than for predictions from single sequences. For example, often an additional 20% or more of the known base pairs can be correctly predicted using multiple homologs as compared to predictions using a single sequence (Xu & Mathews, Reference Xu and Mathews2011). For a given set of sequences, however, it is not always obvious which approach or program to use, and, therefore, it is probably best to try more than one program to develop hypotheses about the in vivo structure. To date, no program can completely automate comparative sequence analysis. Manual comparison is still required for the most accurate RNA secondary structure determination.
2.3.3 Major advances: RNA structure prediction in silico restrained with experimental data
Another type of information used to guide in silico prediction of RNA structure is experimental structure mapping. Such mapping data can come from in vitro or in vivo experiments and are used to restrain structure prediction (Lorenz et al. Reference Lorenz, Wolfinger, Tanzer and Hofacker2016; Sloma & Mathews, Reference Sloma and Mathews2015). The effects of experimental structure restraints have been well studied using in vitro probing data on structured ncRNAs. Over 85% of known pairs can be correctly predicted using in vitro SHAPE, DMS, or enzymatic cleavage data (Cordero et al. Reference Cordero, Kladwang, Vanlang and Das2012; Deigan et al. Reference Deigan, Li, Mathews and Weeks2009; Eddy, Reference Eddy2014; Hajdin et al. Reference Hajdin, Bellaousov, Huggins, Leonard, Mathews and Weeks2013; Ouyang et al. Reference Ouyang, Snyder and Chang2013; Washietl et al. Reference Washietl, Hofacker, Stadler and Kellis2012; Wu et al. Reference Wu, Shi, Ding, Liu, Hu, Yip, Yang, Mathews and Lu2015; Zarringhalam et al. Reference Zarringhalam, Meyer, Dotu, Chuang and Clote2012) when the extent of accessibility is quantified using capillary/gel electrophoresis or deep sequencing counts. This is a dramatic improvement over the above-mentioned 70% limit in the absence of mapping data. Using in vivo mapping data to improve the accuracy of structure prediction has not yet been well studied, although mapping data overlaid on known structures suggests that, for structured ncRNAs such as rRNAs, the existing methods should improve structure prediction accuracy (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014). We recently developed a pipeline called StructureFold to fold RNAs across a genome using restraints from experimental data, which works with Structure-seq data (Tang et al. Reference Tang, Bouvier, Kwok, Ding, Nekrutenko, Bevilacqua and Assmann2015b), and the RNAstructure program and can accommodate other data and folding algorithms.
2.3.4 Challenges with in silico modeling of RNA secondary structure
Despite its widespread use, RNA secondary structure prediction has known limitations. First, the nearest-neighbor parameters are based on a limited number of experiments measured in vitro in 1 M NaCl rather than in vivo-like conditions, and there are probably many sequences that are not predicted well with those parameters (Andronescu et al. Reference Andronescu, Condon, Turner and Mathews2014; Mathews et al. Reference Mathews, Disney, Childs, Schroeder, Zuker and Turner2004). On the one hand, for a limited number of simple RNAs melted in physiological K+ and Mg2+ concentrations, the stability is often similar to that in 1 M NaCl (Diamond et al. Reference Diamond, Turner and Mathews2001; Jaeger et al. Reference Jaeger, Zuker and Turner1990; Jiang et al. Reference Jiang, Kennedy, Moss, Kierzek and Turner2014; Schroeder & Turner, Reference Schroeder and Turner2000). However, for a 5S ribosomal RNA loop E motif, for example, an appreciable difference in stabilities was found between buffers with and without Mg2+ (Serra et al. Reference Serra, Baird, Dale, Fey, Retatagos and Westhof2002). Second, although enthalpy parameters are available for structure prediction between 10 and 60 °C (Lu et al. Reference Lu, Turner and Mathews2006), predictions are generally made at 37 °C, which is relevant to humans, but not the majority of organisms. Third, finding lowest free energy structures assumes that RNAs fold to equilibrium, i.e. kinetics do not control folding. In favor of this assumption, an in vivo study of ribozymes suggested that RNAs fold to equilibrium to a greater extent in yeast cells than in vitro (Mahen et al. Reference Mahen, Harger, Calderon and Fedor2005). Also, in vitro structure mapping studies of annealed ribosomal RNAs were consistent with in vivo structures (Moazed et al. Reference Moazed, Stern and Noller1986b). However, some sequences are kinetically trapped, such as transcriptional riboswitches (Seetin & Mathews, Reference Seetin and Mathews2012a; Wickiser et al. Reference Wickiser, Cheah, Breaker and Crothers2005a; Wickiser et al. Reference Wickiser, Winkler, Breaker and Crothers2005b). Therefore, it is unclear to what extent factors such as non-physiological ionic conditions and cotranscriptional folding play roles in shaping the folding of RNA.
A fourth limitation of the most popular programs for in silico folding is that they cannot predict pseudoknots (Liu et al. Reference Liu, Mathews and Turner2010a). A pseudoknot occurs when there are base pairs between nucleotides in two different loops. Formally, a pseudoknot is composed of two or more base pairs, defined by indices i base paired to j and i′ base paired to j′, where the order of the nucleotides is i < i′ < j < j′. Pseudoknotted pairs are a small fraction of total base pairs in known structures but often occur in highly structured and functional RNAs. For programs that predict pseudoknots, the accuracy is shockingly low (<5%) (Bellaousov & Mathews, Reference Bellaousov and Mathews2010), although the use of multiple homologous sequences to identify conserved pseudoknots improves the accuracy (Seetin & Mathews, Reference Seetin and Mathews2012b). Recently, it was also shown that in vitro SHAPE mapping data can guide in silico structure prediction, including pseudoknots, and achieve over 90% accuracy at predicting known base pairs (Hajdin et al. Reference Hajdin, Bellaousov, Huggins, Leonard, Mathews and Weeks2013). The program that implements this, ShapeKnots, is limited, however, to sequences of 600 nucleotides or fewer.
Although structure mapping data and sequence comparison are each used to guide in silico modeling of RNA secondary structure, little has been done until recently to combine the two approaches for additional synergy. The secondary structures of three long ncRNAs were modeled with the aid of structure mapping data: HOTAIR (with in vitro SHAPE, DMS, and terbium) (Somarowthu et al. Reference Somarowthu, Legiewicz, Chillón, Marcia, Liu and Pyle2015), SRA (with in vitro SHAPE, DMS, in-line probing, and RNase V1 digestion) (Novikova et al. Reference Novikova, Hennelly and Sanbonmatsu2012), and XIST (with in vivo DMS mapping) (Fang et al. Reference Fang, Moss, Rutenberg-Schoenberg and Simon2015). For each of these studies, sequence comparison, i.e. the verification that the structures are conserved and the identification of compensating base pair changes, was subsequently used to further support the structure model.
Two software programs were enhanced to combine structure mapping data and sequence comparison to improve structure prediction. Sükösd et al. (Reference Sükösd, Knudsen, Kjems and Pedersen2012) reported PPfold, a program that uses a probabilistic approach to predict structure and can be guided by SHAPE mapping data and/or sequence covariation as estimated from a sequence alignment. Recently, SHAPE data were used to inform sequence alignment and then RNAalifold to predict the conserved structure for the aligned sequences (Lavender et al. Reference Lavender, Lorenz, Zhang, Tamayo, Hofacker and Weeks2015b). The key observation is that homologous nucleotides, i.e. those that align, have similar SHAPE reactivities and thus the differences in SHAPE reactivity can be included as an additional metric in the scoring of alignments. This approach demonstrated an improved accuracy of base pair prediction by RNAalifold as compared to consensus structure prediction or SHAPE guided structure prediction alone. Both of these approaches were used to model HIV RNA structure using mapping data and sequence comparison (Lavender et al. Reference Lavender, Gorelick and Weeks2015a; Sükösd et al. Reference Sükösd, Andersen, Seemann, Jensen, Hansen, Gorodkin and Kjems2015).
3. Bridging the gap between in vitro and in vivo RNA folding using in vivo-like studies
3.1 The gap
The previous sections outlined major contributions of RNA-folding studies in vitro and in vivo to our understanding of how RNA behaves, while considering the important roles that in silico approaches play. In vitro studies provide the fundamentals of RNA thermodynamics and kinetics, RNA structural motifs, and genome-wide RNA structure trends. In vivo structure probing methods reveal RNA structural trends related to biological functions and regulatory roles of RNA genome-wide. We discussed how several research teams have used genome-wide in vivo structural probing to uncover that, in general, RNAs do not adopt the same structures in vivo as in vitro. Since structure generally dictates function, understanding differences between RNA folding in vivo and in vitro can illuminate biological function. Toward accomplishing this goal, RNA folding and function studies have been increasingly conducted under conditions that mimic the cellular environment.
The dilute solution conditions traditionally used to study RNA in vitro are vastly different from the cellular environment. The cellular environment is a complex solution containing biopolymers, metabolites, dilute free salts, and organelles, with 20–40% of the cellular volume occupied by macromolecular crowders (Minton, Reference Minton2001; Zimmerman & Trach, Reference Zimmerman and Trach1991). As such, there is no single cellular environment to which RNA is exposed. As an mRNA passes from the nucleus to the cytosol, solution conditions change; in eukaryotes, the cell is compartmentalized and as the RNA is transported to different regions its fold can change.
It is of interest to consider the differences between RNA structure in eukaryotic and prokaryotic organisms. Functional RNAs have intricate structures with tertiary contacts that assemble secondary structures close in space. Cations, typically Mg2+, neutralize the negative charge of the phosphate backbone and promote tertiary structures. Free Mg2+ concentrations in prokaryotic and eukaryotic cells are different, ~1·5–3·0 and 0·5–1·0 mM, respectively (London, Reference London1991; Lusk et al. Reference Lusk, Williams and Kennedy1968; Romani, Reference Romani2007; Truong et al. Reference Truong, Sidote, Russell and Lambowitz2013). Structured RNAs such as ribozymes, riboswitches, and thermosensors are found frequently in prokaryotes, where free Mg2+ levels are higher. Although a few ribozymes and one riboswitch have been identified in eukaryotes, they appear to be rare, and proteins are typically involved in forming requisite tertiary structures (Kubodera et al. Reference Kubodera, Watanabe, Yoshiuchi, Yamashita, Nishimura, Nakai, Gomi and Hanamoto2003; Roth et al. Reference Roth, Weinberg, Chen, Kim, Ames and Breaker2014; Salehi-Ashtiani et al. Reference Salehi-Ashtiani, Luptak, Litovchick and Szostak2006). Lambowitz and co-workers demonstrated that prokaryotic group II introns fold poorly in eukaryotic cells, although they could select variant RNAs that fold into active conformations at eukaryotic low Mg2+ concentrations (Truong et al. Reference Truong, Sidote, Russell and Lambowitz2013). Studies in our laboratory indicate that the eukaryotic innate immune sensor PKR is activated by prokaryotic RNAs under eukaryotic low Mg2+ conditions, leading to the speculation that riboswitches and ribozymes may be selected against in eukaryotes to aid in discriminating self and non-self at the RNA level (Hull & Bevilacqua, Reference Hull and Bevilacqua2015, Reference Hull and Bevilacqua2016; Hull et al. Reference Hull, Anmangandla and Bevilacqua2016). To date, there are no studies that compare the structures of eukaryotic and prokaryotic RNAs genome-wide, but such information would be valuable.
Historically, in vitro experiments lack many of the components of cellular environments, and, moreover, often have high concentrations of salt to fold RNA for thermodynamic and structural studies (Table 1). Thermodynamic studies cannot, however, readily be performed in vivo. The cell prohibits wide variations of temperature, pH, salt, and ligand concentration, all of which are necessary to obtain thermodynamic information. As a result, RNA is being increasingly studied in artificial cytoplasms that mimic aspects of the cellular environment while allowing biophysical studies. Several recent studies focused on mimicking aspects of the in vivo environment in vitro; conditions referred to herein as ‘in vivo-like’ conditions (Fig. 3). Effects of such conditions as cellular concentrations of monovalent and divalent ions and molecular crowding agents on the folding of RNAs have been a theme in a number of recent studies (Desai et al. Reference Desai, Kilburn, Lee and Woodson2014; Dupuis et al. Reference Dupuis, Holmstrom and Nesbitt2014; Nakano et al. Reference Nakano, Kitagawa, Yamashita, Miyoshi and Sugimoto2015; Paudel & Rueda, Reference Paudel and Rueda2014; Strulson et al. Reference Strulson, Boyer, Whitman and Bevilacqua2014; Tyrrell et al. Reference Tyrrell, Weeks and Pielak2015). Experiments under these in vivo-like conditions have the potential to bridge our understanding of observations made in vitro and in vivo.
3.2 Design of artificial cytoplasms and early experiments
In this section, we discuss various methods of mimicking cytoplasmic conditions, including the use of polymers and cosolutes as crowding agents and the use of protocells and synthetic membranes. We also discuss the outcomes of early experiments under these in vivo-like conditions. Finally, directions in which the field needs to move to understand the fold and function of RNA in vivo are suggested.
3.2.1 Polymers
Synthetic crowding agents such as polyethylene glycol, dextran, and ficoll, and small cosolute additives such as methanol, proline, and trimethylamine oxide (TMAO) have been used to mimic the crowded environment of the living cell. Functional RNAs that are well studied in vitro have been used to test the effects crowding agents have on RNA folding. Various methods, including UV melts, SAXS, kinetic techniques, and smFRET, have been used to study RNA under these in vivo-like conditions. Several studies have shown that synthetic crowding agents affect the thermodynamics and function of several RNAs (Dupuis et al. Reference Dupuis, Holmstrom and Nesbitt2014; Kilburn et al. Reference Kilburn, Roh, Guo, Briber and Woodson2010, Reference Kilburn, Roh, Behrouzi, Briber and Woodson2013; Lambert et al. Reference Lambert, Leipply and Draper2010; Strulson et al. Reference Strulson, Boyer, Whitman and Bevilacqua2014). Findings of these studies are that RNAs fold cooperatively, structure becomes compact, and ribozymes cleave faster under in vivo-like conditions (Kilburn et al. Reference Kilburn, Roh, Behrouzi, Briber and Woodson2013; Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009; Strulson et al. Reference Strulson, Yennawar, Rambo and Bevilacqua2013, Reference Strulson, Boyer, Whitman and Bevilacqua2014).
The kinetics of several small and large ribozymes have been probed under in vivo-like conditions and in all reported cases, rates of catalysis have increased in the presence of molecular crowders as compared to dilute solution conditions (Desai et al. Reference Desai, Kilburn, Lee and Woodson2014; Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009; Paudel & Rueda, Reference Paudel and Rueda2014; Strulson et al. Reference Strulson, Molden, Keating and Bevilacqua2012, Reference Strulson, Yennawar, Rambo and Bevilacqua2013). For example, the hammerhead ribozyme has higher catalytic activity, between 3·5 and 6·5 faster than in dilute solutions, in the presence of 10–30% (wt %) PEG200 or PEG8000, suggesting a more populated active state in crowded conditions (Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009). In addition, in vivo-like solution conditions can stabilize ribozymes even in the presence of denaturants. For example, the rate of catalysis of the CPEB3 ribozyme in the presence of 2·5 M of the denaturant urea was recovered by the addition of 30% (w/v) PEG200, PEG8000, or Dextran10, at a rate higher than in buffer alone (Strulson et al. Reference Strulson, Yennawar, Rambo and Bevilacqua2013). SAXS experiments have provided insight into the structural basis for enhanced catalysis, showing that the natively folded state adopts a more compact structure in the presence of molecular crowders under conditions of biological Mg2+ concentrations (Kilburn et al. Reference Kilburn, Roh, Guo, Briber and Woodson2010; Strulson et al. Reference Strulson, Yennawar, Rambo and Bevilacqua2013).
The thermal stability of several functional RNAs has been reported to increase under in vivo-like conditions as compared with in vitro experiments. For instance, in 20% PEG200 or PEG8000 the hammerhead ribozyme retains catalytic activity up to 60 °C, a temperature that thermally denatures the ribozyme in dilute solutions (Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009). Observation of increased hammerhead catalytic activity, up to 270-fold, at high temperatures in crowded conditions indicates a more thermostable RNA under in vivo-like conditions. Interestingly, the individual secondary structure elements of the ribozyme were observed, through optical melting experiments, to be thermally destabilized in molecular crowding agents, suggesting that tertiary structure is stabilized and resulting in more cooperative folding of the ribozyme (Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009). A thermodynamic study from our laboratory using SHAPE structure probing on tRNAphe under in vivo-like conditions showed that tRNA folds in a cooperative manner at biological Mg2+ concentrations in the presence of molecular crowding (Strulson et al. Reference Strulson, Boyer, Whitman and Bevilacqua2014). The observed increase in folding cooperativity with crowding was accompanied by an increase in the temperature of the melting transition for tertiary structure. When the tertiary interactions were removed by mutation of nucleotides in tertiary contacts to uridine, cooperativity was lost and the RNA folded with multiple transitions under all solution conditions, thus indicating that tertiary interactions are vital to cooperative RNA folding under in vivo-like conditions. This effect is similar to that observed under in vitro conditions mentioned above (Chauhan & Woodson, Reference Chauhan and Woodson2008).
The contribution of molecular crowding agents to RNA catalysis and folding has been found to be largest in a background of physiologically low ionic conditions rather than high ionic conditions (Kilburn et al. Reference Kilburn, Roh, Behrouzi, Briber and Woodson2013; Strulson et al. Reference Strulson, Yennawar, Rambo and Bevilacqua2013). In the absence of crowding, physiological concentrations of Mg2+ are not high enough to fold functional RNAs in a two-state manner. This is apparent from the observation of long-lived intermediates and slow folding under these conditions (Banerjee & Turner, Reference Banerjee and Turner1995; Chadalavada et al. Reference Chadalavada, Senchak and Bevilacqua2002; Mitchell et al. Reference Mitchell, Jarmoskaite, Seval, Seifert and Russell2013). However, in the presence of biological crowding conditions and physiological Mg2+, functional RNAs tend to fold in a cooperative manner into compact structures (Desai et al. Reference Desai, Kilburn, Lee and Woodson2014; Dupuis et al. Reference Dupuis, Holmstrom and Nesbitt2014; Strulson et al. Reference Strulson, Boyer, Whitman and Bevilacqua2014; Tyrrell et al. Reference Tyrrell, Weeks and Pielak2015), and ribozymes and riboswitches tend to have higher rates of cleavage and higher ligand-binding affinity (Paudel & Rueda, Reference Paudel and Rueda2014). The addition of more Mg2+ to these conditions does not result in a further increase in the rate of activity or more cooperative RNA folding, indicating that together physiological crowding and Mg2+ conditions fold RNA optimally (Fig. 7).

Fig. 7. Under both in vitro (right, grey) and in vivo-like conditions with molecular crowding (left, pink) RNA fold into their native state that is functional, indicated in this figure by catalysis. High concentrations of Mg2+ (10 mM or higher) are needed to achieve the folded state in vitro compared with in vivo-like crowded conditions where low physiological Mg2+ (0·5 mM) folds the RNA. Reprinted with permission from Strulson et al. (Reference Strulson, Yennawar, Rambo and Bevilacqua2013). Copyright 2016 American Chemical Society.
A recent study explored the structural effects of the molecular crowding agent PEG (ranging in size from the monomer to 35 000 kDa) on the adenine riboswitch (Tyrrell et al. Reference Tyrrell, Weeks and Pielak2015). Using SHAPE chemistry, the reactivity of the riboswitch under in vitro, in vivo, and in vivo-like conditions was explored. The authors found that in low molecular weight PEG (<3350 kDa) the riboswitch had low correlation between reactivity in vivo and in vivo-like conditions, whereas in higher molecular weight PEG (12 000–35 000 kDa) the RNA had a similar reactivity under in vivo and in vivo-like conditions. While this study was limited to a single molecular crowding agent, it is significant because it showed that certain in vivo-like conditions are not accurate cellular mimics.
Recently, the folding of a model RNA was examined in vivo. The Salmonella fourU RNA thermometer hairpin containing a FRET pair was injected into live mammalian cells and reported to have similar melting temperatures and unfolding free energy in vivo and in vitro (Gao et al. Reference Gao, Gnutt, Orban, Appel, Righetti, Winter, Narberhaus, Müller and Ebbinghaus2016). The addition of 30% (w/v) PEG of varying sizes and Ficoll70 was shown to modify the thermodynamics of the hairpin, and higher molecular weight polymers were found to have similar effects on the RNA as the in vivo environment. The in vivo data had a very broad distribution of melting temperatures and free energy between both different cells and different cellular compartments, leading to some uncertainty about how the cellular environment is affecting RNA folding.
3.2.2 Cosolutes
While molecular crowding agents generally facilitate the folding of functional RNAs, small cosolutes have varying and complicated effects on RNA thermostability and folding cooperativity. This arises in part because the effect on stability depends strongly on the interactions between the particular cosolute and RNA considered. Cosolutes, also known as osmolytes, regulate osmotic pressure in cells (Record et al. Reference Record, Courtenay, Cayley and Guttman1998; Yancey et al. Reference Yancey, Clark, Hand, Bowlus and Somero1982). Effects of cosolutes on RNA folding were not significantly investigated until the last decade. Studies on RNAs with either secondary and/or tertiary structures report that cosolutes such as betaine, proline, and methanol, almost always destabilize secondary structures, while having mixed effects on tertiary structure (Lambert & Draper, Reference Lambert and Draper2007, Reference Lambert and Draper2012; Lambert et al. Reference Lambert, Leipply and Draper2010; Soto et al. Reference Soto, Misra and Draper2007). Several osmolytes have been shown to interact with the nucleobase, sugar, and phosphate of RNAs, with examples of both favorable and unfavorable interactions (Lambert & Draper, Reference Lambert and Draper2007). Stabilizing osmolytes have unfavorable interactions with the unfolded state of RNA, resulting in RNA compaction that buries functional groups and stabilization of the native state, while destabilizing osmolytes have favorable interactions with the unfolded state of RNA, driving unfolding (Holmstrom et al. Reference Holmstrom, Dupuis and Nesbitt2015; Lambert & Draper, Reference Lambert and Draper2007; Lambert et al. Reference Lambert, Leipply and Draper2010).
There are a limited number of studies available for the effect of cosolutes on RNA function. The hammerhead ribozyme was shown to have increased rates of cleavage in 20% cosolutes, such as glycerol and 1,2-dimethoxyethane, in the presence of physiological Mg2+, which was attributed to enhanced electrostatic interactions with Mg2+ (Nakano et al. Reference Nakano, Kitagawa, Yamashita, Miyoshi and Sugimoto2015). The secondary and tertiary structures of the hammerhead ribozyme were destabilized in the presence of several cosolutes (Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009). In crowded conditions, ribozyme activity also increased, while secondary structure was destabilized and tertiary structure was stabilized (Nakano et al. Reference Nakano, Karimata, Kitagawa and Sugimoto2009).
The influence of the cosolute TMAO on RNA secondary and tertiary structure, as well as on the phosphate backbone, has been studied (Denning et al. Reference Denning, Thirumalai and Mackerell2013; Lambert & Draper, Reference Lambert and Draper2007; Lambert et al. Reference Lambert, Leipply and Draper2010). TMAO is unusual in having almost no effect on secondary structure stability, while generally stabilizing tertiary structure. A small 58 mer rRNA was found to exhibit cooperative two-state folding in the presence of TMAO, observed by a single transition in an optical melting experiment (Lambert et al. Reference Lambert, Leipply and Draper2010).
3.2.3 Protocells and synthetic membranes
There are several groups focusing on how to model RNA function and structure in early Earth conditions, which also relate to compartmentalization in modern cells. Coacervates and synthetic membranes are often used to mimic early Earth protocells, and RNA function in these protocells is often studied through ribozyme cleavage. Our laboratory studied the activity of a two-piece hammerhead ribozyme in aqueous two-phase systems (ATPS) made of polyethylene glycol and dextran (Strulson et al. Reference Strulson, Molden, Keating and Bevilacqua2012). The system forms a dextran-rich phase droplet in which the ribozyme preferentially localizes at a concentration up to 3000 times that of the aqueous phase, resulting in a 70-fold increase in the rate of catalysis. This study suggested that RNA catalysis in the early Earth environment could have arisen from compartmentalization increasing the local concentration of RNA, possibly accelerating very slow reactions, so that they could occur on a biologically relevant timescale.
Similar to these droplets, mononucleotides will form microdroplets when mixed with cationic peptides in water (Koga et al. Reference Koga, Williams, Perriman and Mann2011). Inside these droplets, nucleotides and peptides can reach concentrations as high as 1·6 M and 400 mM respectively, which is much more concentrated than in the aqueous phase. Cationic and anionic dyes and certain nanoparticles were shown to partition into the droplets, indicating that the droplets are permeable to charged molecules (Koga et al. Reference Koga, Williams, Perriman and Mann2011). These droplet phases are another indicator that early life could have arisen in non-membranous compartments. More recently we made coacervates from nucleotides and poly(allylamine) that contain molar concentrations of Mg2+ and nucleotides, which could facilitate RNA catalysis in an early life scenario (Frankel et al. Reference Frankel, Bevilacqua and Keating2016).
4. Future directions
The majority of what is known about RNA folding and structure comes from studies that were performed in vitro on small model systems and highly structured RNAs. In contrast, little is known about how RNA folds and functions in vivo. Current in vivo methods probe the RNA structure ensemble. While providing a benchmark for new prediction parameters, ensemble methods cannot themselves generate thermodynamic parameters.
The current thermodynamic parameters for RNA structure prediction were established in 1 M NaCl. However, several-transcript-specific and genome-wide studies have shown that certain RNAs do not fold into the same structures in vivo and in vitro (Kwok et al. Reference Kwok, Ding, Tang, Assmann and Bevilacqua2013; Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014; Tyrrell et al. Reference Tyrrell, Mcginnis, Weeks and Pielak2013, Reference Tyrrell, Weeks and Pielak2015), so improved software and prediction parameters are needed to model in vivo structure. In particular, genome-wide in vivo structure probing datasets (Ding et al. Reference Ding, Tang, Kwok, Zhang, Bevilacqua and Assmann2014; Rouskin et al. Reference Rouskin, Zubradt, Washietl, Kellis and Weissman2014) contain a wealth of information that has not yet been completely realized or understood. One barrier to taking full advantage of the data is that most of the available in silico methods assume that RNAs fold to a single structure (Cordero et al. Reference Cordero, Kladwang, Vanlang and Das2012; Deigan et al. Reference Deigan, Li, Mathews and Weeks2009; Hajdin et al. Reference Hajdin, Bellaousov, Huggins, Leonard, Mathews and Weeks2013; Ouyang et al. Reference Ouyang, Snyder and Chang2013; Wu et al. Reference Wu, Shi, Ding, Liu, Hu, Yip, Yang, Mathews and Lu2015), while probing data averages across all structures populated by sequences for the duration of the experiment.
Modeling a single structure works well for ncRNA sequences that function with a single structure, such as ribosomal RNAs, but there are many RNAs for which this assumption is not correct, such as RNA switches and open reading frames. A key challenge is developing methods to use the probing data to model ensembles of relevant structures. Three recent papers highlight work to address this challenge. Cordero and Das report an in silico method (M2-REEFFIT) that models complex mixtures of multiple structures, aided by in vitro SHAPE mapping of the wild-type sequence and also a set of mutant sequences, which reveal nucleotide interactions (Cordero & Das, Reference Cordero and Das2015). Multiple structures for the 5′ UTR of an mRNA were modeled using in vitro SHAPE mapping of a mixture of structures (Kutchko et al. Reference Kutchko, Sanders, Ziehr, Phillips, Solem, Halvorsen, Weeks, Moorman and Laederach2015). The multiple conformations were modeled in silico using stochastic sampling, restrained using the standard SHAPE restraints expressed as free energy terms (Deigan et al. Reference Deigan, Li, Mathews and Weeks2009). A third in vitro approach separated multiple conformations of HIV RNA using native gel electrophoresis, and mapped the structures with SHAPE in the gel (Sherpa et al. Reference Sherpa, Rausch, Le Grice, Hammarskjold and Rekosh2015). This simplified the in silico analysis because the SHAPE mapping data were acquired for each conformation independently.
Another key challenge for mapping studies is determining the best way to discover or model interactions of RNAs with proteins or other RNAs. In vivo, all RNAs can interact with macromolecules and metabolites. These interactions generally result in protection from probing agents. Deconvoluting in silico whether a nucleotide is unreactive because of intramolecular structure or intermolecular interactions is a grand challenge that will likely require new types of experimental information to address. Modeling and predicting 3D RNA structures in vitro is an ongoing challenge. A recent RNA puzzle tested blind 3D folding predictions by providing research teams with RNA sequences and chemical probing data for those RNAs (Miao et al. Reference Miao, Adamiak, Blanchet, Boniecki, Bujnicki, Chen, Cheng, Chojnowski, Chou, Cordero, Cruz, Ferré-D'amaré, Das, Ding, Dokholyan, Dunin-Horkawicz, Kladwang, Krokhotin, Lach, Magnus, Major, Mann, Masquida, Matelska, Meyer, Peselis, Popenda, Purzycka, Serganov, Stasiewicz, Szachniuk, Tandon, Tian, Wang, Xiao, Xu, Zhang, Zhao, Zok and Westhof2015). The structures that the teams modeled were compared with crystal structures, and most teams could predict Watson–Crick base pairs, but struggled in predicting non-canonical WC base pairing and stacking interactions. A long range of goal is to predict relevant RNA 3D structures in vivo to understand the biologically relevant confirmation(s).
The study of RNA under in vivo-like conditions is relatively young. To better mimic the cellular environment, more complex cytoplasm mimics should be developed. To date, artificial cytoplasms have focused on synthetic polymers and cosolutes, but more accurate ionic conditions, biopolymers and even cell extracts need to be applied. In addition, studies under in vivo-like conditions have focused on single transcripts in synthetic crowding and cosolute conditions. Genome-wide comparisons of RNA folding under in vivo and in vivo-like conditions are needed. Lastly, methods that can probe the thermodynamics and kinetics of RNA folding under complex in vivo-like conditions will enhance our understanding of in vivo RNA folding. Overcoming the challenges outlined herein will allow the field to accomplish the ultimate goal, to understand how RNA folds in the cell.
Acknowledgements
The authors would like to thank the NIH for funding under R01-GM110237 and the NSF for funding under IOS-1339282.









