


Introduction
Alzheimer’s disease (AD) is the most common form of dementia,Reference Wong, Gilmour and Ramage-Morin 1 affecting 1 in 10 North American seniors (aged ≥65). 2 , Reference Qiu, Kivipelto and von Strauss 3 Although the search for a disease-modifying agent has been intense, 99.6% of compounds entering clinical trials have failed.Reference Cummings, Morstorf and Zhong 4 , Reference Becker, Greig and Giacobini 5 Typically, these failures are ascribed to an inadequate understanding of the multifactorial etiology of AD; studies have therefore urged renewed investigation of the underlying neuropathologies. However, the emerging complexity and prevalence of AD may suggest that broader systemic or environmental factors, not routinely considered in neurology, may also be relevant to the onset and outcomes of disease.
In early studies, environmental factors, like metals, toxins and diet, were thought to be the definitive causes of AD. Although the etiology has since proven more complex, this environmental hypothesis regained credence in the 1990s when studies of AD prevalence among Japanese migrants in the United States revealed rates assimilative to local populations, and significantly higher than in native Japanese.Reference White, Petrovitch, Ross, Masaki, Abbott and Teng 6 Similar trends were discerned among African-American and Hispanic-American populations, which cumulatively suggest a role for environment in AD pathogenesis.Reference Hendrie, Ogunniyi and Hall 7 – Reference Tang, Cross and Andrews 9
Significant changes in AD prevalence were also emerging among static populations; of note again is Japan, which witnessed a dramatic rise in prevalence from 1985 to 2010, as reported by Grant.Reference Grant 10 This loosely correlated with a period of westernization, in which dietary habits underwent a transformation from traditional cuisines, rich in fish and fresh vegetables, to an appreciably more North-American diet, heavier in fats and processed foods.Reference Tanaka, Shimabukuro and Shimabukuro 11 , Reference Grant 12 Although purely speculative, these loose associations revived some interest in diet as a causative factor in AD.
At present, extensive studies have identified various mitigating and aggravating nutrients, in addition to regional diets and metabolic states, which may play a role in AD development and progression. This review first examines the major nutritional influences on AD, as currently understood, and catalogs the relevant lipids, carbohydrates, vitamins, minerals and broader dietary patterns associated with AD. In addition, we observe and report on a relatively unexplored reciprocal association, wherein AD may alter dietary preference and nutritional intake. This review also considers the implications of these interactions to therapeutic agents and clinical trials. Specifically, we present an argument that the multifaceted interactions among diet, drugs and AD may be sufficient to confound or obscure the efficacy of a clinical agent in a drug trial, and suggest strategies to abate such an influence.
Alzheimer’s Disease Pathology
Alzheimer’s disease is characterized by three dominant neuropathologies: oligomerization of β-amyloid (Aβ), aggregation of tau into neurofibrillary tangles and brain atrophy.Reference Perl 13 The etiology of these pathologies remains unclear; however, it is well established that no recognized singular dysfunction directly precedes the deposition of plaques and tangles. Rather, a complex interlay of proteopathic and immunopathic changes culminate in the observed pathologies.Reference Nelson, Braak and Markesbery 14 – Reference Swerdlow 16
Aβ aggregates originate from transmembrane amyloid-precursor proteins (APP). APP is typically associated with essential signaling roles in cellular physiology,Reference van der Kant and Goldstein 17 but when cleaved by α/γ-secretase or β-site APP cleaving enzyme (BACE), they can misfold and aggregate into toxic oligomers.Reference Murphy and LeVine 18 This is attended by an inflammatory cascade, which causes further damage to the neural parenchyma, as well as elevate oxidative stress.Reference Akiyama, Barger and Barnum 19 Within neurons, AD is characterized by the accumulation of fibrillary tangles composed of microtubule-associated tau protein.Reference Iqbal, Liu, Gong and Grundke-Iqbal 20 Ordinarily, tau participates in intracellular transport and structural functions. In AD, it becomes hyper-phosphorylated and disassociates from the microtubules. Once free, tau aggregates to form a helical fiber, which impedes intracellular transport and communication.Reference Iqbal, Liu, Gong and Grundke-Iqbal 20 Together, Aβ and tau oligomerization lead to neuronal death and cerebral atrophy. This manifests in the deterioration of memory and cognition, as well as progressive declines of reasoning, language, and executive function. Alzheimer’s disease eventually evolves to an end-stage disease, typically resulting in mortality 4-8 years post diagnosis.Reference Helzner, Scarmeas, Cosentino, Tang, Schupf and Stern 21
Diet and Alzheimer’s Disease
Early associations between diet and AD were assumed to be ancillary to other primary disorders, like cerebrovascular disease. However, recent data have suggested a direct role for diet in modulating oxidative stress, inflammation, synaptic plasticity, amyloidogenesis and neurogenesis—processes fundamental to AD pathogenesisReference Wu, Ying and Gomez-Pinilla 22 – Reference Gomez-Pinilla 24 and major treatment strategies.Reference Folch, Ettcheto and Petrov 25 – Reference Vina, Lloret, Orti and Alonso 28 This has implicated diet as contributory in AD, and sparked numerous nutrient-based therapeutic trials. In this section, we review the interaction of major dietary elements, both protective and detrimental, with AD pathologies.
Lipids: Cholesterol and Fatty Acids
In the 1990s, a relationship between lipids and AD was suggested when mutations in the cholesterol carrier apolipoprotein E were identified as a major genetic predisposition for late-onset Alzheimer’s.Reference Strittmatter, Saunders and Schmechel 29 , Reference Strittmatter and Roses 30 Thereafter, cholesterol was shown to have a regulatory role on the proteolytic enzymes cleaving APP, where elevated membrane cholesterol increases both γ-secretase and BACE1 activity, ultimately upregulating Aβ production.Reference Xiong, Callaghan and Jones 31 , Reference Thirumangalakudi, Prakasam and Zhang 32 Cholesterol may also facilitate the aggregation of Aβ from its monomeric form to the neurotoxic oligomer.Reference Liu and Zhang 33 , Reference Di Scala, Chahinian, Yahi, Garmy and Fantini 34 In a mechanistic study, Kakio et al demonstrated that cholesterol elicits the binding of Aβ to cellular membranes, which can then facilitate a conformational change from a helical-rich Aβ structure to an aggregation prone β-pleated sheet.Reference Kakio, Nishimoto, Yanagisawa, Kozutsumi and Matsuzaki 35 Cholesterol may therefore seed the oligomerization of Aβ via membrane binding.Reference Kakio, Nishimoto, Yanagisawa, Kozutsumi and Matsuzaki 35
This cholesterol hypothesis, reviewed by Liu and Zhang,Reference Liu and Zhang 33 has yielded contradictory clinical outcomes. Certain statin-based therapies,Reference Lin, Chuang and Hsieh 36 , Reference Shepardson, Shankar and Selkoe 37 and the inhibition of cholesterol biosynthesis,Reference Kim, Kim, Jang and Mook-Jung 38 have offered protection against Aβ accumulation and AD development. However, cholesterol is known to decrease with AD progression, and a gradual decline of serum cholesterol was identified as a risk factor for dementia in a large, 32-year investigation.Reference Mielke, Zandi and Shao 39 , Reference Presecki, Muck-Seler and Mimica 40 Studies are further suggesting vital structural and functional roles for cholesterol in neurons.Reference Qin, Liao, Baudry and Bi 41 , Reference Mathews and Appel 42 Consequently, as an independent therapeutic strategy for AD, cholesterol management alone may prove challenging.
The role of fatty acids has garnered similar attention. These are among the best-studied lipids, and have shared conclusive associations to various diseases, including AD.Reference Liu and Zhang 33 Moreover, following early work by Foot et al, which demonstrated that diet can directly alter the fat composition of the brain, fatty acid management has long been purported to possess therapeutic potential.Reference Foot, Cruz and Clandinin 43
Dietary fats are classified by length, degree of saturation and isomerism (cis or trans). Saturated fatty acids and trans-unsaturated fatty acids have been associated with an adverse risk of AD. An observational study by Morris et al discerned that diets high in these fats could more than double the risk of AD over an average of 4 years.Reference Morris, Evans and Bienias 44 Unlike cholesterol, a clear interaction with AD pathologies has not been discerned. Rather, saturated and trans-fats have been linked to altered gene expression, elevated inflammatory signaling, disrupted metabolism, protein pathologies and higher oxidative stress, all of which have been linked to cognitive decline and behavioral changes.Reference Freeman, Haley-Zitlin, Rosenberger and Granholm 45 – Reference Julien, Tremblay and Phivilay 48 Prominent associations between these fats and traditional AD risk factors such as cerebrovascular disease, obesity and diabetes may also mediate the observed correlations.Reference Takechi, Galloway, Pallebage-Gamarallage, Lam and Mamo 49 , Reference Pugazhenthi, Qin and Reddy 50 Although occasional studies have revealed null or insignificant associations after extended follow-up,Reference Engelhart, Geerlings and Ruitenberg 51 the link between saturated and trans-fats with AD appears conclusive and detrimental.
Conversely, some polyunsaturated fatty acids (PUFAs) have revealed protective associations. Omega(ω)-3 PUFAs in particular, typically obtained from fish, are associated with beneficial outcomes in both AD and dementia.Reference Hooijmans, Pasker-de Jong, de Vries and Ritskes-Hoitinga 52 , Reference Cole, Ma and Frautschy 53 Studies have repeatedly demonstrated that consumption of these fats (such as docosahexaenoic acid) are inversely associated with cognitive decline and AD.Reference Yurko-Mauro 54 – Reference Siegel and Ermilov 58 Furthermore, an exhaustive review by Cole et al identified 11 pleiotropic mechanisms by which ω-3 fats may confer this protection, ranging from regulating apoptosis to diminishing local pro-inflammatory markers.Reference Cole, Ma and Frautschy 53 Yet, the effect is not without contradiction, particularly among carriers of the ApoE4 allele, as well as otherwise well-nourished subjects, where no effect has been observed.Reference Freund-Levi, Hjorth and Lindberg 59 – Reference Araya-Quintanilla, Gutierrez-Espinoza and Sanchez-Montoya 63 Clinical trials and meta-analyses have also failed to demonstrate a significant benefit in AD, although other cognitive parameters may be improved in unrelated cohorts.Reference Araya-Quintanilla, Gutierrez-Espinoza and Sanchez-Montoya 63 , Reference Mazereeuw, Lanctot, Chau, Swardfager and Herrmann 64
Omega-6 PUFAs, however, derived from meat, and often more abundant in a western diet, may be deleterious to an AD brain.Reference Creegan, Hunt, McManus and Rainey-Smith 65 – Reference Baierle, Vencato and Oldenburg 67 Specifically, the phospholipid product of ω-6 fatty acids, arachidonic acid, is implicated in upregulating inflammation and promoting Aβ aggregation.Reference Otsuka, Yamaguchi and Ueki 66 , Reference Amtul, Uhrig, Wang, Rozmahel and Beyreuther 68 This claim has been occasionally disputed, and studies have suggested that a therapeutic ratio between ω-6 and ω-3 PUFAs may exist. Hosono et al showed that in a 2:1 dietary ratio of arachidonic acid to docosahexaenoic acid, Tg2576 mice had lower deposition of amyloid plaques and altered APP processing;Reference Hosono, Nishitsuji and Nakamura 69 ω-6 and ω-3 fats have also been shown to preserve neural stem cells in rats.Reference Tokuda, Kontani, Kawashima, Kiso, Shibata and Osumi 70 This suggests PUFAs may operate under a dynamic synergy. However, without further study it remains unclear whether combinations of these fats are simply confounding the observation of each other’s effects, or whether there is a genuine additive association.
Broadly, cholesterol, saturated fats, trans-fats and ω-6 fatty acids are likely deleterious in the context of AD, while ω-3 fatty acids have demonstrated some potential benefits. Although in all cases associations have been obscured by clinical inefficacy and contradictory findings, dietary fats may alter Aβ dynamics, influence neural inflammation and present numerous additional mechanisms of interaction with AD.
Carbohydrates: Sugars and Insulin
High sugar and high calorie diets are well-established risk factors for dementia and AD.Reference Otsuka 71 – Reference Luchsinger, Tang, Shea and Mayeux 74 Further, as contributors to diabetes and other metabolic syndromes, which themselves are risk factors for AD, the association between elevated sugar levels and Alzheimer’s is strong.Reference Mushtaq, Khan, Kumosani and Kamal 75 – Reference Peila, Rodriguez, Launer and Honolulu-Asia Aging 78 Paradoxically, AD-afflicted neurons are typically starved of glucose with marked reductions in glucose metabolism (up to 44%) across brain regions most associated with AD pathology.Reference Ishii, Sasaki and Kitagaki 79 – Reference Hoyer, Oesterreich and Wagner 81 This pattern has become so robust that topographic patterns of impaired glucose metabolism can serve as an early marker of AD, and act as a criterion for staging progression.Reference Wattamwar and Mathuranath 82
One explanation for this apparent paradox implicates glucose transport across the blood-brain barrier (BBB). AD brains have lower levels of the glucose transporters GLUT1 and GLUT3, and therefore diminished shuttling of glucose across the BBB.Reference Szablewski 83 Winkler et al demonstrated this transporter-mediated glucose starvation can aggravate AD neuropathologies, vascular degeneration and impair cognition.Reference Iadecola 80 , Reference Winkler, Nishida and Sagare 84 An alternative theory implicates insulin resistance for the observed glucose dysregulation. Insulin resistance occurs when tissues loose sensitivity to insulin signaling after protracted exposure, triggered by chronically elevated blood sugar. The breakdown of signaling prevents glucose from entering cells, leading simultaneously to hyperglycemia, and diminished glucose within cells. Insulin resistance has also been implicated in triggering inflammation, which may exacerbate AD pathologies.Reference Creegan, Hunt, McManus and Rainey-Smith 65 In addition, insulin signaling is integral to neural function, maintenance, plasticity and is increasingly associated with cognition (in the context of diabetes) all of which have been compromised when insulin signaling is perturbed.Reference McNay and Recknagel 85 – Reference Akintola and van Heemst 87 More recently, components of insulin signaling were implicated in regulating tau phosphorylation and tangling,Reference Hong and Lee 88 – Reference Planel, Tatebayashi and Miyasaka 90 as well as Aβ synthesis and degradation.Reference Vekrellis, Ye and Qiu 91 – Reference Craft 93 This has led to speculation as to whether altered glucose metabolism may be causative in AD. Evidence of glucose dysregulation preceding clinical symptoms, as well as its high prevalence among AD cohorts provide some support for this notion.Reference Mosconi, Pupi and De Leon 94
Micronutrients: Vitamins and Minerals
The rise of commercial vitamins and dietary supplements reflects a growing appreciation of the importance of micronutrients in health maintenance. The accessibility of these supplements also offers a practical strategy to manage AD on a large scale. However, in spite of exhaustive investigations, including several well-powered clinical trials, no isolated micronutrient has yet been definitively demonstrated as an effective therapy for AD.Reference Luchsinger, Noble and Scarmeas 95 Yet, deficiencies of various vitamins and minerals have been associated with an elevated risk of AD, cognitive impairment and other dementias.Reference Creegan, Hunt, McManus and Rainey-Smith 65 ,95
The B vitamin family is critical to the metabolism of most macronutrients, including carbohydrates and lipids, and has therefore garnered some interest in the context of AD.Reference Huskisson, Maggini and Ruf 96 Vitamin B1 or thiamine was among the first nutrients associated with dementia when it was conclusively linked to Wernicke’s encephalopathy in long-term alcoholics.Reference Butterworth 97 , Reference Rodríguez, Qizilbash, López-Arrieta and Rodríguez 98 Earlier studies have noted conspicuous similarities between the symptoms of Wernicke’s and Alzheimer’s, including shared deficiencies of thiamine-dependent processes, cognitive decline and aberrant glucose metabolism.Reference Gibson, Hirsch, Cirio, Jordan, Fonzetti and Elder 99 – Reference Zhang, Yang and Li 101 In vitro studies have since shown that in a state of acute thiamine insufficiency, neurons produce markers of stress, including reactive oxygen species, and begin apoptotic processes, implying a physiologic role for thiamine in neuronal function.Reference Ba 102 , Reference Wang, Wang, Fan, Shi, Ke and Luo 103 This has been corroborated by Zhang et alReference Zhang, Yang and Li 101 and othersReference Karuppagounder, Xu and Shi 104 who demonstrated that thiamine deficiency promoted β-secretase activity, Aβ accumulation and oxidative stress. In clinical trials, however, thiamine supplementation has not borne significant outcomes. An early systematic review did not identify a single trial with significant positive associations between thiamine and AD.Reference Rodríguez, Qizilbash, López-Arrieta and Rodríguez 98 Koh et al also demonstrated that thiamine deficiency lost its associations with dementia when subjects were otherwise well nourished.Reference Koh, Charlton and Walton 105 This may suggest that the initial correlations were in fact between malnourishment, in which thiamine deficiency is often concomitant, and AD.
Other B vitamins, including pyridoxine (B6), cobalamin (B12), folate (B9) and homocysteine, have also come under scrutiny for potential links to AD. Homocysteine is a metabolic precursor of the amino acids methionine and cysteine; however, before metabolism, homocysteine is a potent neurotoxin.Reference Obeid and Herrmann 106 Multiple studies have demonstrated a persistent correlation between dietary homocysteine and AD, with physiological levels exceeding 14 µmol/L estimated to double the risk of AD.Reference Baierle, Vencato and Oldenburg 67 , Reference Seshadri, Beiser and Selhub 107 – Reference Li and Pratico 109 Further, in 3xTg transgenic mice, homocysteine was directly associated with cognitive decline, as well as promoting Aβ and tau pathologies.Reference Li, Chu, Barrero, Merali and Pratico 110
As an upstream metabolite, homocysteine levels do not necessarily have to be controlled by reducing dietary intake. Instead, increasing consumption of vitamins B6, B12 and folate can perpetuate the conversion of homocysteine into its amino acid products. Wang et al evidenced this by demonstrating that when vitamin B12 and folate levels were impaired, subjects exhibited a higher risk of AD (over a 3-year follow-up).Reference Wang, Wahlin, Basun, Fastbom, Winblad and Fratiglioni 111 Yet, the association has been repeatedly proven insignificant and current evidence on the role of these vitamins has been deemed insufficient.Reference Morris, Evans, Schneider, Tangney, Bienias and Aggarwal 112 – Reference Nelson, Wengreen, Munger and Corcoran 114
Additional B vitamins including riboflavin (B2) and niacin/nicotinamide (B3) have also received attention owing to their metabolic roles in the brain.Reference Creegan, Hunt, McManus and Rainey-Smith 65 Niacin, in particular, has shown promise with high doses demonstrating some protective and even curative capacity in mouse models.Reference Green, Steffan and Martinez-Coria 115 , Reference Morris, Evans and Bienias 116 Green et al demonstrated that specific phosphorylated species of tau were reduced and proteins associated with microtubule stability were elevated upon exposure to a high dose of vitamin B3.Reference Green, Steffan and Martinez-Coria 115 This rescued cognition in their murine model (3xTg) raising the possibility of a curative treatment, rather than a typical arresting strategy. However, clinical trials have not demonstrated any tangible improvements to cognition from vitamin B3, particularly when given as supplements rather than in their natural form.Reference Phelan 117 , Reference Morris 118 The optimal dosage of vitamins is further controversial. Studies have often examined high doses, well in-excess of established guidelines. It has been suggested that this may exceed a therapeutic benefit, if any, and potentially expose patients to toxic side effects. The excretion of supplemental vitamins may also diminish a therapeutic efficacy. As such, a re-examination of the optimal form, dose and functional levels may be required.
Nutrients with antioxidant properties have also been an area of intense study. Oxidative damage, elevated production of reactive oxygen species and impeded clearance of oxidative stress markers are well recognized in AD neurons.Reference Akiyama, Barger and Barnum 19 , Reference Verdile, Keane and Cruzat 119 , Reference Wyss-Coray and Rogers 120 Furthermore, accumulating evidence suggests that this oxidative damage may precede the deposition of Aβ or tau.Reference Verdile, Keane and Cruzat 119 Accordingly, antioxidants have been proposed as a potential preventative and therapeutic strategy. Common dietary antioxidants include vitamin C, vitamin E, polyphenols and carotenes.Reference Luchsinger, Noble and Scarmeas 95 The Cache County Study involving over 3000 participants demonstrated that in combination, vitamins C and E supplements were associated with a lower incidence of AD.Reference Zandi, Anthony and Khachaturian 121 Several other studies of comparable scope demonstrated similar associations,Reference Luchsinger, Noble and Scarmeas 95 though randomized control trials have largely failed to demonstrate any therapeutic utility for antioxidants.Reference Polidori and Nelles 122 Rather, a double-blind trial by Galasko et al noted that cognitive decline appeared accelerated in AD subjects given a combination of antioxidants, in spite of diminished oxidative stress markers.Reference Galasko, Peskind and Clark 123 As with other vitamins, these inconsistencies may arise from the form of antioxidant under consideration, where supplements, as opposed to dietary antioxidants, may prove less effective; though further study is required to ascertain the validity of this supposition.
The most controversial debate in the realm of antioxidants has been relating to the minerals zinc and copper. With some variability, serum zinc deficiency has been repeatedly correlated with AD.Reference Tully, Snowdon and Markesbery 124 – Reference Ventriglia, Brewer and Simonelli 127 The zinc cation (Zn2+) is integral to neural enzymes mediating clearance of Aβ, APP processing and the neutralization of reactive oxidative species, all of which imply a protective role for zinc in AD.Reference Nuttall and Oteiza 125 , Reference Watt, Whitehouse and Hooper 128 However, Zn2+ is often elevated in AD brains and has also been directly implicated in potentiating the neurotoxicity of Aβ and tau; further, by binding to Aβ, Zn2+ may promote oligomerization.Reference Watt, Whitehouse and Hooper 128 – Reference Huang, Wu, Cao, Lang, Lu and Zhou 130 This inconsistency has brought the role of Zn2+ into dispute, and studies have speculated on a potential dual role in the context of AD.Reference Huang, Cuajungco, Atwood, Moir, Tanzi and Bush 131
However, it is more likely that Zn2+ levels are elevated in the brain as a physiological countermeasure against the oxidative and inflammatory stress of AD. This would decrease serum Zn2+ concentrations, as observed, but may elevate cerebral Zn2+ to toxic levels, perpetuating the observed pathological functions. This phenomenon, termed zinc flooding, implies that dietary zinc should be reduced, in spite of the observed zinc deficiency among Alzheimer’s patients. The copper ion (Cu2+) has similarly been implicated in AD, sharing much the same associations as Zn2+. Studies have shown that binding of Cu2+ to Aβ may lead to the creation of neurotoxic products and that it may have further roles in mediating the characteristic oxidative signature of AD.Reference Mathys and White 132 – Reference Syme, Nadal, Rigby and Viles 134 However, as with zinc, Cu2+ deficiencies have also been linked to AD.Reference Xu, Church and Patassini 135 As reported by Quinn et al, these contradictions have led to the investigation of both copper-sequestering and copper-supplementing agents in clinical trials.Reference Quinn, Crane, Harris and Wadsworth 136
In general, inconsistencies are ubiquitous when considering the role of individual micronutrients in AD. It is likely that variations in dosing, disease stage and the form of nutrient may influence any perceived utility on AD risk or progression. Furthermore, few studies have attempted to consider any broad synergistic effects among differing nutrients, preferring instead to focus on a single cause-and-effect relationship. As diets seldom provide nutrients in isolation, traditional trials may fail to capture the inherent interactions among different dietary elements.
Dietary Patterns
Analysis of larger dietary patterns may alleviate some of the uncertainties raised by single-nutrient trials. In particular, they offer the ability to examine the role of nutrients in situ or as components of a larger system rather than distinct entities. Among the best studied has been the Mediterranean diet (MeDi). In studies of other diseases, the MeDi has demonstrated significant reductions in mortality and morbidity.Reference Martinez-Gonzalez and Bes-Rastrollo 137 , Reference Grosso, Mistretta and Frigiola 138 The MeDi may have benefit in AD as it features many nutrients suggested to have beneficial impacts on AD risk and progression. This includes a high intake of fruits and vegetables, rich in natural antioxidants and ω-3 unsaturated fatty acids (in the form of fresh fish and olive oil). Moreover, the diet is low in saturated fats, cholesterol, high-fat dairy products and red meats.Reference Huhn, Kharabian Masouleh, Stumvoll, Villringer and Witte 139 It therefore excludes many of the known foods associated with AD risk, and includes several protective nutrients.Reference Gu, Nieves, Stern, Luchsinger and Scarmeas 140 It is thus unsurprising that in numerous studies and meta-analyses, adherence to the MeDi was correlated with a reduced risk of AD.Reference Scarmeas, Stern, Tang, Mayeux and Luchsinger 141 – Reference Sofi, Cesari, Abbate, Gensini and Casini 143 Scarmeas et al were further able to demonstrate that in addition to AD onset, the MeDi also reduced the risk of mortality after diagnosis.Reference Scarmeas, Luchsinger, Mayeux and Stern 144
Other diets, such as the “Dietary Approaches to Stop Hypertension” (DASH) diet were associated with improved cognition in hypertensive cohorts.Reference Smith, Blumenthal and Babyak 145 Observational studies have also correlated general diet quality to cognition in non-demented cohorts.Reference Correa Leite, Nicolosi, Cristina, Hauser and Nappi 146 , Reference Wengreen, Neilson, Munger and Corcoran 147 It is noteworthy that when attempting to resolve this effect, no single element of the diet could be independently associated with the purported benefits. Even supplementation of multiple discrete nutrients have proved clinically insignificant, as was the case in a recent trail of Souvenaid (branded Fortasyn Connect) which provided a preformulated combination ω-3 PUFAs, synaptic membrane precursors, cofactors and vitamins.Reference Ritchie, Bajwa and Coleman 148 , Reference Soininen, Solomon and Visser 149 Therefore, it is likely that a complex additive relationship may sustain the observed benefits of the MeDi and other diets.
Perspectives on the Microbiota
Although the human microbiota has recently emerged as a major contributor to various disease states, it remained largely unexplored in the field of neurology until the recognition of the gut-brain axis, which revealed the existence of complex bilateral communication between the brain and the intestines.Reference Carabotti, Scirocco, Maselli and Severi 150 To date, investigations have catalogued a variety of signaling molecules originating from the gut as modulators of pain, stress and possibly emotions.Reference Mayer, Tillisch and Gupta 151 – Reference Cryan and Dinan 153 Murine models have also demonstrated significant behavioral and biochemical differences in animals raised with a sterile gut, which can be restored after colonization with probiotic bacteria.Reference Sampson and Mazmanian 154
The role of microbiota in AD is an emerging area of study. From preliminary data, we glean three gross mechanisms by which the microbiota may influence AD. The first involves the well-studied ability of the microbiota to affect nutrient absorption and activation.Reference Moco, Martin and Rezzi 155 Jandhyala et al report that absorption and activation of polyphenols, a class of organic antioxidants with protective associations in AD, is contingent on enteric microbial enzymes.Reference Jandhyala, Talukdar, Subramanyam, Vuyyuru, Sasikala and Nageshwar Reddy 156 Likewise, the absorption of ω-3 fatty acidsReference Menni, Zierer and Pallister 157 and the bioavailability of macronutrients may also depend on the state and composition of the microbiome.Reference Krajmalnik-Brown, Ilhan, Kang and DiBaise 158 It is therefore conceivable that the microbiota may mediate the physiological levels of the nutrients implicated in AD. Thus, the microbiota may cause identical quantities of administered nutrients to result in substantially different concentrations within the body. The failure of most trials to account for this may therefore offer some explanation for the inability of nutrient-based trials to demonstrate associations on AD. However, this remains speculative pending definitive quantification of the potential influence and variability imposed by the microbiota.
A second emerging mechanism lies in the capacity of the microbiota to modulate inflammation. The gut is known to be a source for various oxidative stressors and signals,Reference Hakansson and Molin 159 , Reference Boulange, Neves, Chilloux, Nicholson and Dumas 160 and recent data by Bruce-Keller et al and others have demonstrated that varying the microbiota can directly alter inflammation, both in the brain and systemically.Reference Bruce-Keller, Salbaum and Luo 161 , Reference Mu, Yang and Zhu 162 Studies have narrowed this tendency to specific bacterial species, which can secrete or otherwise generate pro-inflammatory cytokines; others may counteract this signaling by producing anti-inflammatory agents.Reference Hakansson and Molin 159 , Reference Boulange, Neves, Chilloux, Nicholson and Dumas 160 , Reference Mu, Yang and Zhu 162 Notably, it was microbes thriving in a high-fat environment that were directly associated with elevated neuroinflammation.Reference Bruce-Keller, Salbaum and Luo 161 Mice enriched with these bacteria further demonstrated significant behavioral changes in several domains, in comparison to healthy controls.Reference Bruce-Keller, Salbaum and Luo 161 As high fat consumption and inflammation are directly associated with AD, it is conceivable that the microbiota may, at least in part, moderate the pathogenic effects of fats in AD.
Data have also suggested a role for the microbiota in amyloidogenesis. Bacteria may perpetuate the leakage of dietary amyloids into the blood, where they may eventually enter the nervous system and seed further amyloid transformations.Reference Walker and Jucker 163 , Reference Pistollato, Sumalla Cano, Elio, Masias Vergara, Giampieri and Battino 164 The gut may therefore facilitate Aβ oligomerization in a manner similar to transmissible encephalopathies or prion diseases.Reference Walker and Jucker 163 However, this theory is again speculative and has yet to be proven in patients. It is also unlikely any pathogenic amyloids derived from diet may preserve their unique misfolded conformations as they pass through the gastrointestinal tract. Nonetheless, it is now known that in transgenic murine models (3xTg) of AD, a probiotic formula can slow cognitive decline and decrease amyloid plaques in the brain.Reference Bonfili, Cecarini and Berardi 165 A role for the microbiota in AD is therefore possible. In future, this should be a field of study, and trials, particularly in nutrition should begin to consider the state of the microbiota as a potential disrupting variable.
Alzheimer’s Disease, Drugs and Diet
We have thus far summarized how diet may mediate the onset and progression of AD. Although this has been the principal focus of nutritional studies, the reciprocal relationship, wherein AD may alter diet, is also an emerging area of investigation. As early as 1990, Mungas et al demonstrated that patients with dementia displayed significantly altered dietary preferences when compared with healthy controls.Reference Mungas, Cooper, Weiler, Gietzen, Franzi and Bernick 166 Specifically, they preferred high-fat and/or high-sugar foods, which may arise from diminished inhibition of cravings.Reference Mungas, Cooper, Weiler, Gietzen, Franzi and Bernick 166 More recent data suggest that various forms of dementia are associated with broader changes in appetite, food preference, eating habits and swallowing, with up to 80% of AD patients displaying some dietary disturbance (although data have varied heavily by form and stage of dementia considered).Reference Ikeda, Brown, Holland, Fukuhara and Hodges 167 – Reference Cipriani, Carlesi, Lucetti, Danti and Nuti 170 These conclusions posit an intriguing scenario: AD may drive patients toward a diet that may worsen outcomes and prognoses. The fact that sizable fractions of AD cohorts are routinely observed with hyperphagia,Reference Hwang, Yang, Tsai and Liu 171 – Reference Tsang, Keene and Hope 175 and poorer adherence to beneficial diets,Reference Gardener, Gu and Rainey-Smith 176 add suggestive evidence to this hypothesis.
An alternate scenario suggests that AD and dementia, particularly in its latter stages, may be associated with malnutrition and nutrient deficiencies.Reference Tombini, Sicari, Pellegrino, Ursini, Insarda and Di Lazzaro 177 , Reference Sun, Wen, Zhou and Ding 178 Both are near endemic among elderly and AD populations, and are well associated with deleterious outcomes and prognosis.Reference Sparre-Sorensen and Kristensen 179 , Reference Yildiz, Buyukkoyuncu Pekel, Kilic, Tolgay and Tufan 180 It is currently unclear whether AD may be more strongly associated with malnourishment or higher fat and sugar consumption, or a complex mix of both. However, given that both dietary states are known to impact AD, either case implies that AD and diet may exert a self-modifying interaction, wherein each may modulate the other.
The impact of these associations may also be significant in the context of drug metabolism. Nutrition has long shared associations to the efficacy and bioactivity of drugs, and it is well known that diet is an important determinant of drug metabolism.Reference Walter-Sack and Klotz 181 A review by Walter-Sack and Klotz summarized many known interactions, including changes in excretion, enzyme function and clearance that have been demonstrated by alterations in diet.Reference Walter-Sack and Klotz 181 In the context of AD, perhaps the most relevant observation is that a high-fat diet may moderate the clearance of drugs.Reference Walter-Sack and Klotz 181 – Reference Gupta and Benet 183 Studies have also indicated that malnutrition may decrease drug efficacy by impeding requisite biotransformation and activation reactions.Reference Krishnaswamy 184 , Reference Speerhas 185 States of elevated inflammation may similarly hamper crucial drug activation and metabolism systems.Reference Morgan 186 , Reference Renton 187 As evidenced previously, inflammation as well as both high-fat diets and malnutrition are endemic in AD, and may be partly perpetuated by AD itself. Though the reported associations have been largely from isolated studies for unrelated disease and drug models—donepezil, rivastigmine and memantine—three major drugs used to manage AD symptoms, are all associated with gastrointestinal reactions and/or appetite changes.Reference Farlow, Grossberg, Sadowsky, Meng and Somogyi 188 – Reference Popik, Kos, Zhang and Bisaga 190 As such, drugs and diet may conceivably interact in yet another multifaceted, reciprocal interaction, where drugs can modify diet and diet can in turn modify the efficacy of drugs. Although it is premature to know whether this interaction may have any role in AD, these relationships underlie a definitive web of dynamic interactions between diet, drugs and disease (Figure 1).
Diet in Alzheimer’s Clinical Trials
An ideal clinical trial would administer a singular intervention, in isolation, and discern the effect on a discrete therapeutic outcome. However, the complexity of AD inevitably means that numerous interceding factors may modulate outcome measures, irrespective of the action of a potential therapy. In cataloguing the interaction of diet and AD, this review has identified various dietary elements as potential intervening factors (Table 1).
Table 1 Summary of mechanistic interactions between major nutrients and AD pathologies

AD=Alzheimer’s disease; PUFAs=polyunsaturated fatty acids.
To the best of authors’ knowledge, the role of diet has not been systematically considered in the context of a non-nutritional therapeutic agent in AD. Although an array of extensive interactions has now been established, along with a potentially reciprocal self-modifying interactions, studies have largely relied on the control group to mitigate any variability induced by diet. Yet, in raising the variability of cohorts, studies can compromise statistical power, and hinder the observance of the efficacy of a therapy. We therefore suggest that trials in AD proactively consider strategies to account for diet in their study design and analysis.
Shim et al have reviewed approaches to assess diet in the context of epidemiology.Reference Shim, Oh and Kim 191 They note that diet may be tracked by objective observation or subjective reports;Reference Shim, Oh and Kim 191 however, in AD both may prove challenging. Alzheimer’s disease trials require extended follow-up to observe the manifestation of symptoms; thus consistent observations of diet by study personnel is impractical—although it should be considered the gold standard. Likewise, patients with advancing dementia cannot reliably be asked to track food intake or adhere to a prescribed diet. Caregivers may be a potential substitute for self-reporting, yet typical concerns about recall, bias and overburdening may be raised.
A more feasible strategy may be to track baseline dietary preferences in domains like fat, sugar, vitamin and mineral status. In secondary or sensitivity analyses, this data could stratify patients in the exposure and control cohorts into subgroups of comparable diet. Similar results could be attained by multivariate modeling for studies of sufficient size, and appropriate design. Alternatively, for pair-matched control trials, diet may be a factor of consideration when matching subjects and controls. Either strategy may strain study resources, and stratification is known to diminish statistical power. However, these measures may be performed as sensitivity analyses, and accompany primary data, rather than detract from it.
Studies should further consider substantial changes in these nutrients’ levels during the study period as a potential confounding interaction. For example, a dramatic rise of cholesterol, an ostensible Aβ pro-aggregate, or perturbations in micronutrients (including the use of nutritional supplements) may combat the efficacy of an anti-aggregative agent. Likewise, intra-study variations of ω-6 fatty acid levels, or the development of glucose/insulin dysregulation may alter neuroinflammation, thereby confounding anti-inflammatory therapies. Data on these parameters may serve to refine participant inclusion and exclusion criteria, or shape data analysis and the selection of control subjects as outlined previously.
Conclusions
A comprehensive analysis of the repeated failure of AD clinical trials must consider secondary factors which may diminish the apparent efficacy of these drugs. Since diet may be a major factor in regulating the onset and progression of AD, as well as influencing the ultimate bioavailability of drugs, we suggest that diet may be an important uncontrolled variable in conventional AD clinical trial design.
The role of diet in altering AD risk is well established. Nutrients including fats, sugars, minerals and vitamins have been conclusively associated with varying AD risk and disease progression via a range of mechanisms. Larger dietary patterns have also been definitively linked to AD risk, and the microbiota is emerging as a potent variable in this context. A reciprocal association in which AD modulates diet is also conceivable. Notably, the diets that AD predisposes are those linked with worse prognoses, and impeded drug metabolism. As such, a highly complex set of interactions between diet, AD, and drugs is evident.
This review cannot claim that the failure of experimental drugs in AD is attributable to diet; however, it does suggest that diet may play a role in the outcomes of AD, and the perceived clinical efficacy of novel agents. We therefore suggest that future studies, particularly clinical trials make a concerted effort to monitor, regulate or account for diet among AD patients. This may require significant investment of resources, and may burden patients and caregivers. Studies may also diminish statistical power depending on the mode of analysis undertaken. However, the extent of diet’s interactions with AD and its apparent role modulating outcomes justifies greater scrutiny in clinical trials.
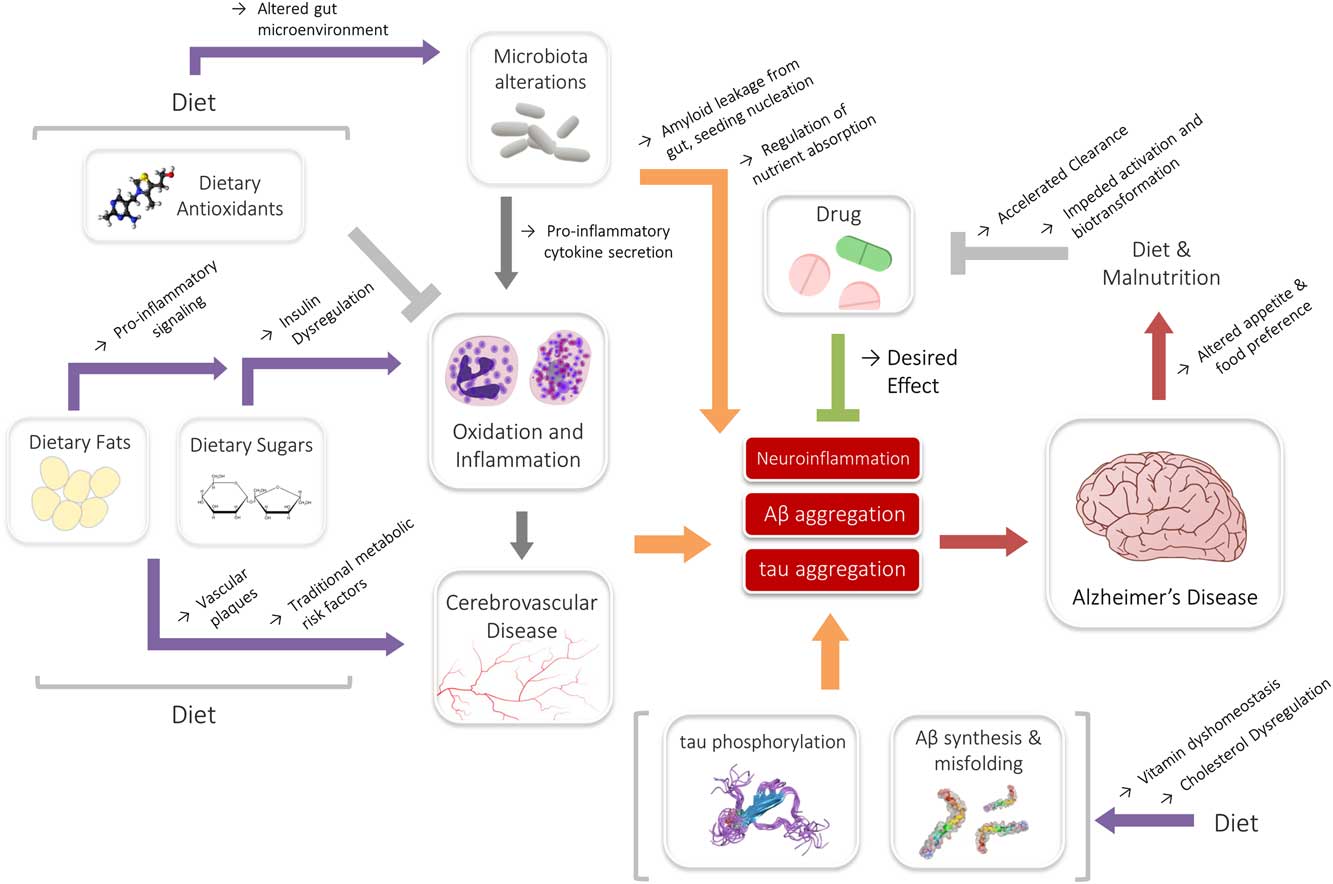
Figure 1 Summary of the interactions of diet, drugs and Alzheimer’s disease (AD). Dietary nutrients (fats or sugars) may medicate inflammatory signaling, or contribute toward underlying metabolic states associated with AD. This interaction may be modified by the actions of the microbiota. Likewise, cholesterol and vitamins may interact directly with Aβ, contributing to AD risk. Antioxidants and drugs may counteract some of the exacerbating signaling, even though further reciprocal interactions among diet and nutritional status may affect their efficacy.
Acknowledgements
DFW acknowledges salary support from a Tier 1 Canada Research Chair.
Disclosures
The authors have no relevant financial disclosures.
Statement of Authorship
All authors contributed equally to the conception, design and drafting of this work.


