



Introduction
Major depressive disorder (MDD) is the leading cause of disability worldwide, with an estimated annual attributable financial loss of $210.5 billion in the United States.1, Reference Greenberg, Fournier, Sisitsky, Pike and Kessler2 Though antidepressant medications are effective for a considerable proportion of patients, large studies suggest that up to 50% of patients do not achieve remission with standard treatments.Reference Hollon, Shelton and Wisniewski3-Reference Rizvi, Grima and Tan5 A probable explanation for this variability in treatment outcomes is that patients with MDD are a heterogeneous group.Reference Akil, Gordon and Hen6-Reference Williams8 To achieve higher rates of remission, a “precision medicine” approach requires validated biomarkers to delineate subgroups of patients who are more likely to respond to specific treatments.
There is converging evidence to support an association between MDD and altered profiles of circulating inflammatory markers.Reference Köhler, Freitas and Maes9-Reference Osimo, Pillinger, Rodriguez, Khandaker, Pariante and Howes11 Based on evidence from longitudinal observational studies, high plasma pro-inflammatory cytokine levels precede, and thus potentially induce depressive symptoms.Reference Chu, Stochl, Lewis, Zammit, Jones and Khandaker12 In addition, there are reports that higher levels of circulating inflammatory mediators are associated with greater severity of illness, and are more prominent in people who are resistant to antidepressants.Reference Grosse, Carvalho and Birkenhager13-Reference Liu, Wei and Strawbridge15 Despite this, clinical trials of repurposed anti-inflammatory agents (eg, NSAIDs, cytokine inhibitors, minocycline) for MDD report inconsistent results, leading to calls for more targeted approaches to address the heterogeneity of the disorder.Reference Jones, Daskalakis and Carvalho16 Specifically, low-grade inflammation is more frequently associated with atypical subtypes of MDD than melancholic subtypes, informing the development of stratified clinical trials of repurposed anti-inflammatory agents for these specific symptom subtypes.Reference Sen, Danyeli and Woelfer17-Reference Zwiep, Bet and Rhebergen19
Previous studies and meta-analyzes have shown associations between pre-treatment levels of, and changes in, levels of inflammatory cytokines and chemokines in patients with MDD who are treated with antidepressants.Reference Hannestad, DellaGioia and Bloch20-Reference Wang, Wang, Liu, Qiao, Baldwin and Hou24 However, there is a high degree of heterogeneity among the studies published to date. Controlling for different covariates across studies provides at least a partial explanation for this heterogeneity. This is particularly important as protein-based inflammatory markers are influenced by multiple factors including body mass index (BMI), physical health, medications, exercise, diet, and substance use; all of which are difficult to account for and poorly reported in studies. Despite these caveats, current evidence supports an association between altered inflammatory mediators and treatment response, in at least a subset of individuals with MDD.
One explanation—at least in part—for the antidepressant action of selective serotonin reuptake inhibitors (SSRIs) is their immunomodulating properties. A meta-analysis of 22 studies including 827 participants with MDD found that SSRI treatment decreased levels of pro-inflammatory markers IL-6, TNF-α, and IL-1β.Reference Wang, Wang, Liu, Qiao, Baldwin and Hou24 Conversely, others reported elevation in inflammatory markers post-treatment with both tricyclic antidepressants (TCAs) and serotonin noradrenaline reuptake inhibitors (SNRIs).Reference Hamer, Batty, Marmot, Singh-Manoux and Kivimäki25, Reference Vogelzangs, Duivis and Beekman26 The diverse effects of antidepressants on levels of inflammatory makers may be due to differences in mechanisms of action: there is evidence that noradrenaline has pro-inflammatory effects on innate immune cells and thus potentiates cytokine production.Reference Thayer and Sternberg27
Atypical antipsychotics (AAPs) are recommended as augmentation treatments for MDD when patients do not respond to first-line SSRIs or SNRIs.Reference Kennedy, Lam and McIntyre28 Pre-clinical studies of AAPs (olanzapine and aripiprazole) demonstrate a decrease in the production of inflammatory cytokines in murine microglial cells and in healthy human blood cells.Reference Stapel, Sieve, Falk, Bleich, Hilfiker-Kleiner and Kahl29, Reference Kato, Mizoguchi and Monji30 Although there is limited evidence on the effect of AAPs on inflammatory markers in MDD patients, studies in participants with schizophrenia report mixed findings.Reference Zajkowska and Mondelli31, Reference Juncal-Ruiz, Riesco-Dávila and Ortiz-García de la Foz32 For instance, in a large meta-analysis of 85 000 participants with schizophrenia, CRP levels were moderately increased in persons with schizophrenia regardless of the use of antipsychotics.Reference Fernandes, Steiner and Bernstein33 This may be because AAPs exert diverse effects on the immune system, having both a direct anti-inflammatory activity and an indirect pro-inflammatory activity, mediated by their metabolic effects (ie, weight gain and increased adiposity). The lack of consensus on the immunomodulating effects of antidepressant and AAP medications highlights the need for larger clinical trials of longer durations. Investigating the effects of individual SSRI and AAP agents on inflammatory markers in subgroups of MDD patients with differing baseline inflammatory status, as well as in subgroups of treatment responders and non-responders, could help stratify medications and advance the quest for “precision psychiatry.”
The Canadian Biomarker Integration Network in Depression (CAN-BIND) was developed to take a consistent integrated approach to biomarker discovery during multiple treatment trials.Reference Kennedy, Downar and Evans34 The overall goal of CAN-BIND is to identify predictors, moderators, and mediators of treatment response and non-response in people with MDD that may guide clinical decision-making. CAN-BIND-1 is a multisite clinical trial with sequential pharmacotherapy over 16 weeks. The design includes assessment of clinical, molecular, genomic, electrophysiological, and neuroimaging parameters.Reference Hollon, Shelton and Wisniewski3 Reference Rizvi, Grima and Tan5, Reference Kennedy, Lam and Rotzinger36 Using inflammatory marker data obtained during CAN-BIND-1, we conducted a secondary analysis to assess associations between individual pro-inflammatory chemokines and cytokines, and response to sequential treatment with escitalopram and adjunctive aripiprazole in MDD patients. We selected those inflammatory markers with established pro-inflammatory activity as replicated evidence has suggested that an activated inflammatory response is associated with unfavorable response to antidepressant medications Reference Grosse, Carvalho and Birkenhager13, Reference Liu, Wei and Strawbridge15, Reference Chamberlain, Cavanagh and de Boer37
Methods
Study overview
The study protocol and clinical outcomes for CAN-BIND-1, including details of eligibility criteria and study procedures, have been previously reported.Reference Kato, Mizoguchi and Monji30-Reference Zajkowska and Mondelli31 Participants were the outpatients of the age group between 18 and 60 years old, meeting Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV-TR criteria for a major depressive episode (without psychosis) in MDD, with a baseline Montgomery-Åsberg Depression Rating Scale (MADRS)Reference Montgomery and Asberg38 score ≥ 24. Individuals with schizophrenia-spectrum disorders, bipolar disorders, and major neurological disorders, head trauma, or other unstable medical conditions were excluded.
The trial was registered on ClinicalTrials.gov (Identifier: NCT01655706) on August 2, 2012, and is a multisite initiative involving six Canadian academic health centers working collaboratively with other universities and research centers. During the first 8 weeks, participants received escitalopram 10–20 mg daily: responders (≥50% reduction in MADRS score) continued escitalopram for another 8 weeks, while non-responders received adjunctive aripiprazole 2–10 mg for the remaining 8 weeks. Escitalopram and aripiprazole were chosen since they are, respectively, evidence-based first-line mono and adjunctive pharmacotherapies for MDD. Both drugs are endorsed in international guidelines on treatment for MDD, including the Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines.Reference Kennedy, Lam and McIntyre28, 39
Ethics approval and consent to participate
Research Ethics Boards (REB) at each institution: University of British Columbia Clinical (UBC) Research Ethics Board (Vancouver); University of Calgary Conjoint Health Research Ethics Board (Calgary); University Health Network (UHN) Research Ethics Board (Toronto; primary site); Centre for Addiction and Mental Health Research (CAMH) Ethics Board (Toronto); Hamilton Integrated Research Ethics Board (Hamilton); Queen’s University Health Sciences (QNS) and Affiliated Teaching Hospitals Research Ethics Board (Kingston), approved the trial. The UHN REB approval number is 11-0917. Participants provided written, informed consent for all study procedures.
Clinical measures
The primary symptomatic outcome measure to assess antidepressant response was the MADRS.Reference Montgomery and Asberg38 A full list of secondary clinical measures has been published previously.Reference Lam, Milev and Rotzinger35 A full assessment of physical health history including a history of immune-related conditions (eg, diabetes mellitus, asthma, ischemic heart disease, inflammatory bowel disease, rheumatoid arthritis, and autoimmune disease) was conducted with each participant.
Measurement of pro-inflammatory markers
Peripheral venous whole blood samples were collected in EDTA tubes at baseline, weeks 2, 8, and 16, and centrifuged at 1 500g at 4 °C for 15 min. Following plasma extraction, aliquots were stored at −80 °C and subsequently transported to UT Southwestern on dry ice and stored at −80 °C until immediately prior to cytokine assays. Chemokine/cytokine levels were measured using the Microarray Core at UT Southwestern Medical Center using a Bioplex Pro human cytokine standard 27-plex kit (Bio-Rad Laboratories, Hercules, CA, USA) with a Bioplex 200 instrument that was equipped with Bioplex Manager software, version 6.0 (Bio-Rad Laboratories, Hercules, CA, USA). This assay has been used in previous studies.Reference Jha, Minhajuddin, Gadad, Greer, Mayes and Trivedi21 High-sensitivity C-reactive protein (hsCRP) levels were measured in a separate plasma aliquot using Beckman DxC600 at McMaster University, Hamilton, ON. The present secondary analysis focussed on seven chemokines and cytokines with established pro-inflammatory activity: high sensitivity C-reactive protein (CRP), interleukin (IL)-1, IL-6, IL-17, interferon (IFN)-Γ, tumor necrosis factor (TNF)-α, and Chemokine C–C motif ligand-2 (CCL-2).Reference Zhang40, Reference Turner, Nedjai, Hurst and Pennington41 Immune marker levels were calculated in mg/L using the standards provided in the kit. Interplate controls were used to monitor for batch effects.
Statistical analysis
To explore differences in pre-treatment inflammatory marker levels between responders and non-responders, independent samples t-tests were performed as a preliminary analysis. A series of logistic regression models were then employed to assess associations between individual pro-inflammatory markers and response to escitalopram at week 8 and to adjunctive aripiprazole at week 16, with age (>40, ≤40), sex (male and female), history of immune-related illness (eg, autoimmune disease), and study site controlled as covariates. The present analysis did not assess associations between individual pro-inflammatory markers and specific depressive symptom subtypes (eg, atypical symptoms).
In the first set of analyzes, associations between individual inflammatory marker levels at baseline and response status at week 8 were examined using the logistic regression models described above. Next, focusing on non-responders to escitalopram at week 8, associations between inflammatory marker levels at week 8 and response status at week 16 were examined using similar logistic regression models.
We then investigated the relationship between response status at weeks 8 and 16, and changes in inflammatory marker levels. First, we examined associations between response status at week 8 and change in levels of each inflammatory marker from baseline to week 2, as well as change from baseline to week 8 using logistic regression analysis. After restricting our analytical sample to those who did not respond to escitalopram at week 8 and received adjunct aripiprazole, the association between the changes in levels of each inflammatory marker from week 8 to week 16, and response status at week 16 was examined. Adjusted Odds Ratios (ORs), with their 95% confidence intervals and associated p-values for all predictors are reported. All statistical analyzes were conducted in SAS Enterprise Guide 7.1 (SAS Institute, Cary, NC).
Results
Recruitment took place between April 2012 and January 2017, during which 211 participants with MDD were recruited (Figure 1). Clinical and demographic details of participants evaluated for inflammatory markers are summarized in Table 1. Inflammatory marker levels at each time point are summarized in Tables 2 and 3.
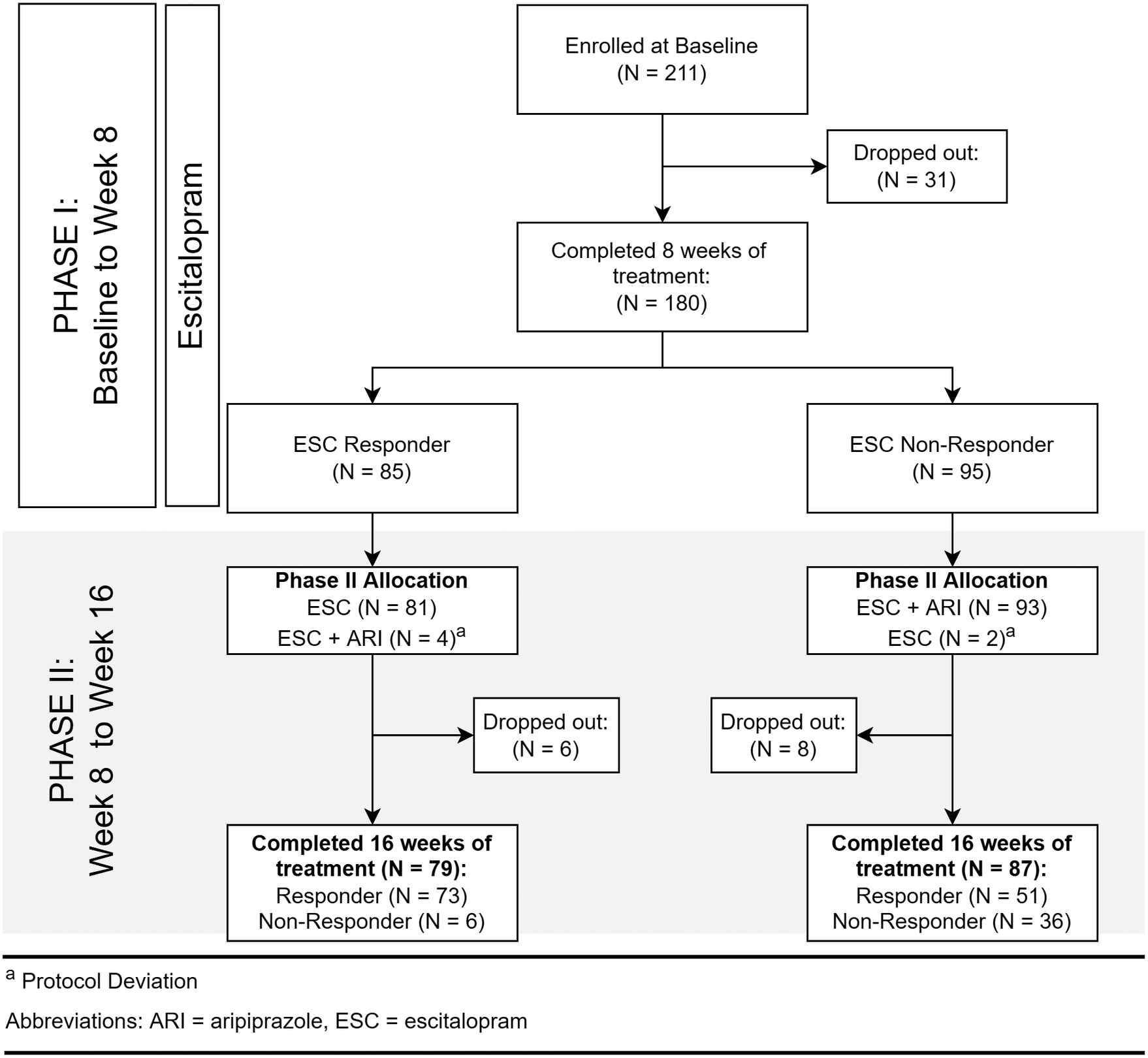
Figure 1. Flow of participants.
Table 1. Clinical and demographic details of participants
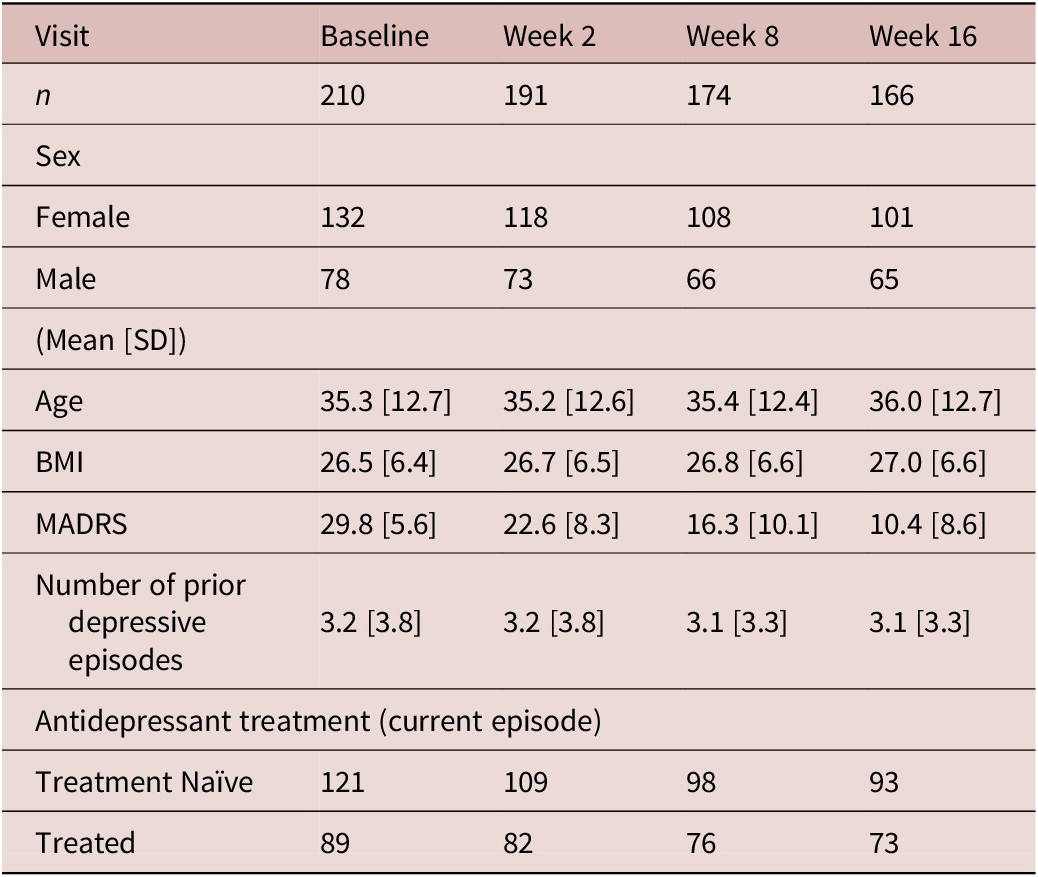
BMI, body mass index; MADRS, Montgomery-Asberg Depression Rating Scale; SD, standard deviation.
Table 2. Inflammatory marker levels (mg/L) at each time point by week 8 response
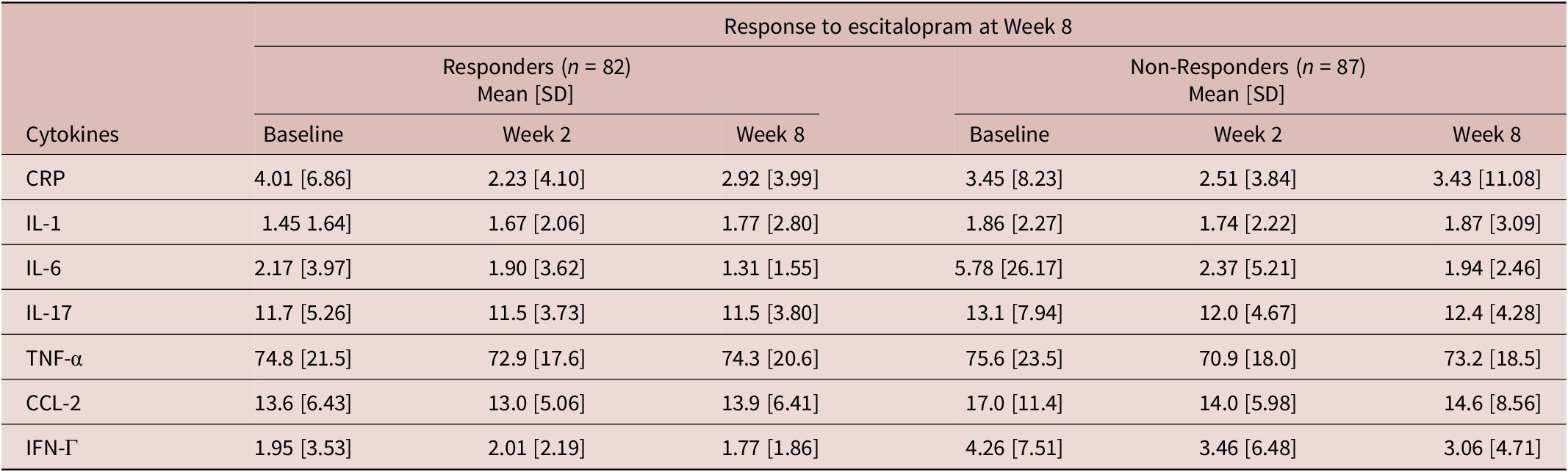
CRP, C-reactive protein; CCL-2, Chemokine C–C motif ligand-2; IFN, interferon; IL, interleukin; SD, standard deviation; TNF, tumor necrosis factor.
Table 3. Inflammatory marker levels (mg/L) at each time point by week 16 response
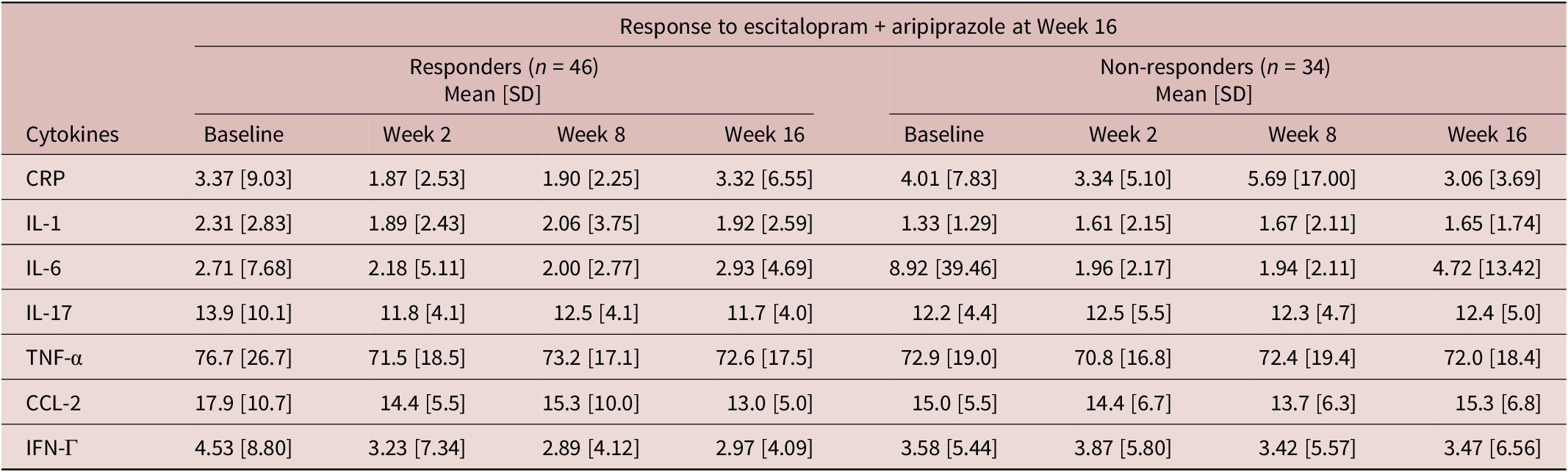
CRP, C-reactive protein; CCL-2, chemokine C–C motif ligand-2; IFN, interferon; IL, interleukin; SD, standard deviation; TNF, tumor necrosis factor.
Relationship between pre-treatment IFN-Γ and CCL-2 levels and response to escitalopram
Baseline levels of pro-inflammatory markers, IFN-Γ (DF 167, t = 2.53, p = 0.012) and CCL-2 (DF 167, t = 2.39, p = 0.016) were significantly higher in escitalopram non-responders. There were also significant effects of pre-treatment pro-inflammatory markers on response status at week 8 (Table 4). Specifically, higher levels of pre-treatment IFN-Γ were significantly associated with lower odds of response to escitalopram treatment (odds ratio [OR] = 0.900, 95% confidence interval [CI] = [0.815, 0.990], p = 0.018) and higher levels of pre-treatment CCL-2 were also associated with lower odds of response to escitalopram at week 8 (OR = 0.941, 95% CI = [0.895, 0.990], p = 0.039). There were no significant associations between pro-inflammatory marker levels at week 8 and response to adjunctive aripiprazole at week 16 (Table 4).
Table 4. Associations between baseline inflammatory markers on treatment response at weeks 8 and 16
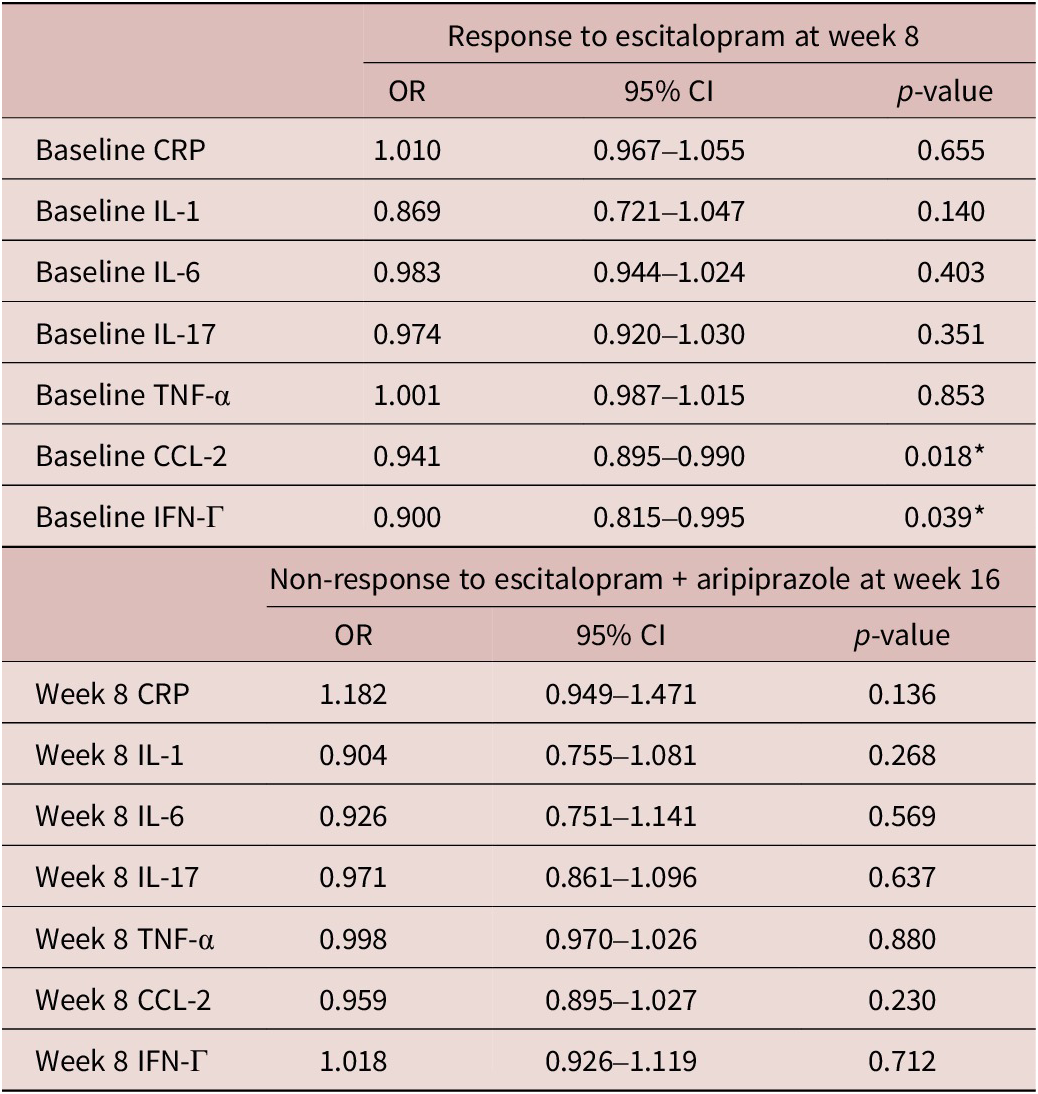
CI, confidence interval; CRP, C-reactive protein; CCL-2, chemokine C–C motif ligand-2; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor; OR, odd ratio.
* p < 0.05.
Change in pro-inflammatory markers are associated with response to adjunctive aripiprazole
Changes in pro-inflammatory markers during treatment appeared to associate with response to adjunctive aripiprazole (Table 5). Specifically, an increase in CCL-2 levels between week 8 and 16 in the adjunctive aripiprazole group was significantly associated with higher odds of non-response to adjunctive aripiprazole at week 16 (OR = 1.128, 95% CI = [1.020, 1.249], p = 0.020). None of the other pro-inflammatory markers appeared to associate with responses to either treatment in this sample (Table 5).
Table 5. Associations between changes in inflammatory markers on treatment Response at weeks 8 and 16
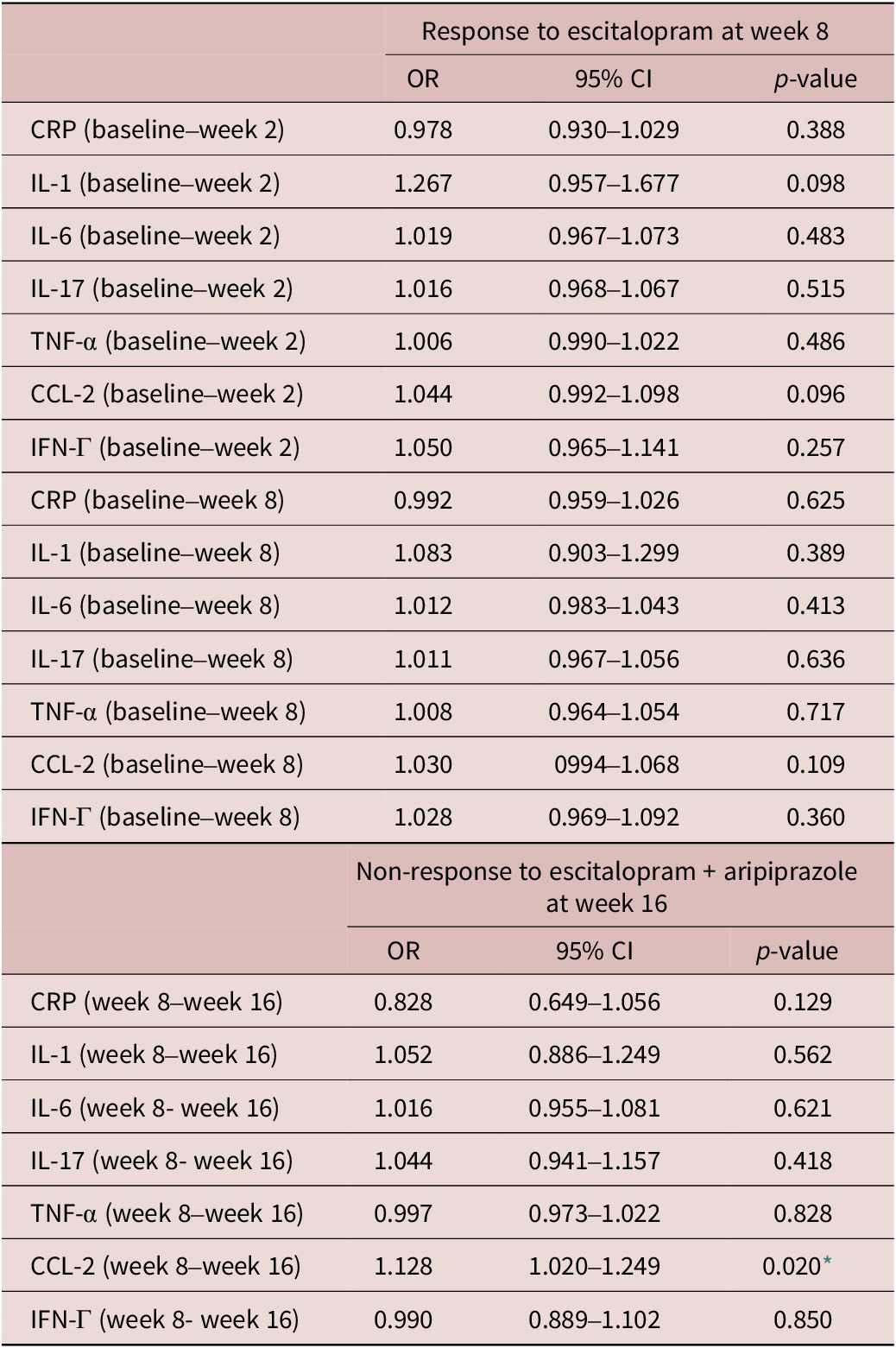
CI, confidence interval; CRP, C-reactive protein; CCL-2, chemokine C–C motif ligand-2; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor; OR, odd ratio.
* p < 0.05.
Discussion
Using a panel of established pro-inflammatory cytokines and chemokines in a large cohort of depressed patients, we identified two pro-inflammatory markers, IFN-Γ and CCL-2 that were associated with response to pharmacotherapy with escitalopram. Higher pre-treatment levels of these pro-inflammatory markers were associated with an unfavorable response after 8 weeks of escitalopram treatment. Increase in CCL-2 during treatment was also associated with an unfavorable response to 8 weeks of adjunctive aripiprazole in previous non-responders to escitalopram monotherapy.
Our results are in keeping with previous reports that elevated pro-inflammatory markers are associated with a poorer response to escitalopram. In a 12-week study of 71 participants with MDD treated with escitalopram, elevated CRP levels were associated with an unfavorable response.Reference Zhou, Zhou and Sun42 Similarly, in a previous trial, MDD participants receiving escitalopram showed higher pre-treatment levels of pro-inflammatory cytokine TNF-α in non-responders. The authors did not assess other pro-inflammatory cytokines like IFN-Γ and CCL-2.Reference Eller, Vasar, Shlik and Maron43 The differences in sample size and clinical populations may explain why the CRP and TNF-α findings were not replicated in CAN-BIND-1.
IFN-Γ is produced by several immune cells including natural killer (NK) cells, CD4+ T cells, and macrophages. It has also been implicated in the pathophysiology of MDD.Reference Litteljohn, Nelson and Hayley44 We are not aware of previous studies assessing associations between IFN-Γ and response to escitalopram monotherapy. However, in a meta-analysis of clinical trials involving other antidepressants, including several SSRIs, there were no significant differences in baseline IFN-Γ levels between responders (n = 223) and non-responders (n = 221).Reference Liu, Wei and Strawbridge15 In the same meta-analysis, there were no significant treatment effects on IFN-Γ levels in either responders or non-responders to antidepressant treatment after pooling evidence from 10 clinical trials.Reference Liu, Wei and Strawbridge15 Again, differences in sample size, clinical population, or choice of antidepressant, may account for the divergence from our findings.
CCL-2, also known as monocyte chemoattractant protein 1 (MCP-1), is a key mediator of neuroinflammation, neurogenesis, neuroplasticity, and synaptic transmission. It is considered an essential mediator in the link between peripheral and central inflammation due to its role in cellular migration and immune coordination. There is support from animal and human studies that CCL-2 is involved in the pathophysiology of MDD, Reference Curzytek and Leśkiewicz45 although there are mixed findings on CCL-2 and associations with antidepressant response. In a small pilot study involving participants—with dysthymia and MDD who received escitalopram 10 mg, those with dysthymia but not MDD, had higher pre-treatment CCL-2 levels. There were no changes however in CCL-2 levels in either group during the 4 weeks of escitalopram.Reference Ho, Yen, Chen, Huang and Liang46 Another study assessed changes in CCL-2 during treatment with higher escitalopram doses (20 to 40 mg) in MDD over 12 weeks. In this study, higher pre-treatment levels of CCL-2 were identified in MDD participants compared with healthy controls, but there were no changes in CCL-2 following escitalopram treatment.Reference Halaris, Myint and Savant47
There are limited data on the effect of aripiprazole on cytokine measures in MDD. Notwithstanding this, associations between inflammatory markers and adjunctive aripiprazole in our sample complement pre-clinical and clinical literature on the anti-inflammatory effects of aripiprazole in schizophrenia, where response to aripiprazole was associated with a reduction in inflammatory cytokines including IL-1β.Reference Stapel, Sieve, Falk, Bleich, Hilfiker-Kleiner and Kahl29, Reference Sobiś, Rykaczewska-Czerwińska, Świętochowska and Gorczyca48 In our sample, non-responders to aripiprazole augmentation showed increases in CCL-2 over 8 weeks of treatment. The absence of an aripiprazole treatment effect on pro-inflammatory marker levels in CANBIND-1 responders may in part be explained by the small sample sizes, short duration of follow-up, or differences in clinical populations (ie, schizophrenia vs MDD) in previous studies.
Although we controlled for potential confounders including age, sex, and history of comorbid immune-related illness, a limitation of our study is the lack of control for other potential confounders such as cigarette smoking, and socioeconomic status. As this secondary analysis was exploratory in nature, we did not correct the results for multiple comparisons. An additional limitation is the applicability of our findings to other antidepressants or atypical antipsychotic augmentation treatments with different mechanisms of action. The results of the present study suggest that responders and non-responders to both escitalopram and aripiprazole show similar post-treatment inflammatory profiles, which may suggest regression to the mean. Despite these limitations, the relatively large sample size, broad inclusion criteria, and serial assessments of a comprehensive panel of established pro-inflammatory markers are strengths of the present study.
Conclusion
This secondary analysis of the CANBIND-1 trial suggests that IFN-Γ and CCL-2 may have utility as biomarkers of treatment response to escitalopram monotherapy and adjunctive aripiprazole in escitalopram non-responders. We encourage validation of these findings in independent studies. Predicting antidepressant treatment response is a clinical challenge for MDD. The inflammatory hypothesis of depression suggests that inflammatory processes play a key role in the pathophysiology of MDD in at least a subgroup of the population and evidence indicates that alterations in peripheral cytokine levels are associated with antidepressant treatment outcome.Reference Osimo, Pillinger, Rodriguez, Khandaker, Pariante and Howes11, Reference Liu, Wei and Strawbridge15 However, results from clinical trials of anti-inflammatory agents in MDD continue to be conflicting and the antidepressant efficacy of anti-inflammatories has yet to be established.Reference Jones, Daskalakis and Carvalho16, Reference Husain, Strawbridge, Stokes and Young49 Further longitudinal studies are required to elucidate sources of heterogeneity in the present literature and to examine the feasibility of using peripheral inflammatory markers as predictive biomarkers for the treatment of MDD. There is emerging evidence that distinct depressive symptom profiles may be associated with an activated inflammatory response.Reference Franklyn, Stewart, Beaurepaire, Thaw and McQuaid50 Moving beyond the search for a single biomarker of disease activity and toward the development of composite biomarkers associated with clinical phenotypes of treatment response may more likely lead to the identification of MDD subtypes that inform personalized treatments including immunomodulatory drugs.
Author contribution
Conceptualization: M.H.T., J.A.F., S.K., W.W., M.I.H.; Methodology: M.H.T., B.L.M., D.M., B.N.F., G.T., J.A.F., K.H., M.J., R.W.L., R.M., S.J.R., S.K., W.W., M.I.H.; Writing – review & editing: M.H.T., B.L.M., D.M., B.N.F., G.T., K.H., R.W.L., R.M., S.R., M.I.H.; Data curation: B.L.M., H.Z., S.C.; Funding acquisition: D.M., G.M., G.T., R.W.L., R.M., S.K.; Project administration: D.M., B.N.F., G.M., G.T., K.H., R.M., S.K., S.R., M.I.H.; Investigation: B.N.F., J.A.F., S.J.R.; Formal analysis: H.Z., J.A.F., M.J., S.C., S.R., W.W.; Writing – original draft: H.Z., M.J., S.J.R., S.C., S.K., W.W., M.I.H.
Funding statement
This work is part-funded by a University of Toronto, Department of Psychiatry Academic Scholar Award to MIH. CAN-BIND is an Integrated Discovery Program carried out in partnership with, and financial support from, the Ontario Brain Institute, an independent non-profit corporation, funded partially by the Ontario government.
Disclosure
This article was prepared while Brittany Mason was employed at the University of Texas Southwestern Medical Center. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
Competing interest
There was no role of the following granting agencies/sponsors in the design, collection of data, analysis, interpretation of data, writing of this paper, and decision to submit the paper for publication. M.I.H. receives research support from the Brain and Behavior Research Foundation, Canadian Institutes of Health Research, CAMH Foundation, the PSI Foundation, and the University of Toronto. He has provided consultancy to Mindset Pharma, PsychEd Therapeutics, and Wake Network. He holds stock/stock options in Mindset Pharma. J.A.F. receives consulting and speaking fees from Takeda Canada and RBH. R.W.L. has received honoraria for ad hoc speaking or advising/consulting or received research funds from: Asia-Pacific Economic Cooperation, BC Leading Edge Foundation, Canadian Institutes of Health Research, Canadian Network for Mood and Anxiety Treatments, Grand Challenges Canada, Healthy Minds Canada, Janssen, Lundbeck, Lundbeck Institute, Medscape, Michael Smith Foundation for Health Research, MITACS, Myriad Neuroscience, Ontario Brain Institute, Otsuka, Pfizer, Sanofi, Unity Health, Vancouver Coastal Health Research Institute, and VGH-UBCH Foundation. Roumen Milev has received consulting and speaking honoraria from AbbVie, Allergan, Eisai, Janssen, KYE, Lallemand, Lundbeck, Neonmind, Otsuka, and Sunovion, and research grants from CAN-BIND, CIHR, Janssen, Lallemand, Lundbeck, Nubiyota, OBI and OMHF. D.J.M.’s research work is supported by the Canadian Institutes of Health Research, the Ontario Brain Foundation, the Alternate Funding Plan of Ontario, and the Centre for Addiction and Mental Health Foundation. S.K. received grants/has pending grants, including Abbott, Allergan, Brain Canada, Canadian Institutes for Health Research, Janssen, Lundbeck, Ontario Brain Institute, Ontario Research Fund, Otsuka, Pfizer, Servier, Sunovion, and Xian-Janssen. He also receives consulting fees or honorarium from Abbott, Alkermes, Allergan, Boehringer Ingelheim, Brain Canada, Canadian Institutes for Health Research, Janssen, Lundbeck, Lundbeck Institute, Ontario Brain Institute, Ontario Research Fund, Otsuka, Pfizer, Servier, Sunovion and Xian-Janssen. He has stock/stock options from Field Trip Health. All other coauthors declare no conflict of interest.







 Open access
Open access