



Parkinson’s disease (PD) is a neurodegenerative disorder characterized by Lewy bodies in the midbrain and the loss of dopaminergic neuron activity, particularly in the substantia nigra (Rodriguez-Oroz et al., Reference Rodriguez-Oroz, Jahanshahi, Krack, Litvan, Macias, Bezard and Obeso2009). PD is the second most common neurodegenerative disease worldwide (de Lau & Breteler, Reference de Lau and Breteler2006; Rodriguez-Oroz et al., Reference Rodriguez-Oroz, Jahanshahi, Krack, Litvan, Macias, Bezard and Obeso2009; van den Eeden et al., Reference van den Eeden, Tanner, Bernstein, Fross, Leimpeter, Bloch and Nelson2003). The increased prevalence in some racial groups might be due to the carrier status of certain genetic variants (Chillag-Talmor et al., Reference Chillag-Talmor, Giladi, Linn, Gurevich, El-Ad, Silverman, Friedman and Peretz2011). Studies suggest that first-degree family members of a person with PD are at 2·7-fold increased risk. Epidemiology studies have reported that there is a 10−30% positive family history for PD, although penetrance may vary (Marder et al., Reference Marder, Levy, Louis, Mejia-Santana, Cote, Andrews, Harris, Waters, Ford, Frucht, Fahn and Ottman2003; Sveinbjornsdottir et al., Reference Sveinbjornsdottir, Hicks, Jonsson, Petursson, Gugmundsson, Frigge, Kong, Gulcher and Stefansson2000).
Until 1997, PD was mostly considered a sporadic disorder with certain environmental contributions (Langston et al., Reference Langston, Ballard, Tetrud and Irwin1983). In 1997, an article identified the SNCA gene loci for PD (Polymeropoulos et al., Reference Polymeropoulos, Lavedan, Leroy, Ide, Dehejia, Dutra, Pike, Root, Rubenstein, Boyer, Stenroos, Chandrasekharappa, Athanassiadou, Papapetropoulos, Johnson, Lazzarini, Duvoisin, Di Iorio, Golbe and Nussbaum1997). The genetic etiology of PD is estimated to account for 5−15% of cases (Kalinderi et al., Reference Kalinderi, Bostantjopoulou and Fidani2016). Studies have shown an increasing correlation between early age diagnosis and genetic background (Alcalay et al., Reference Alcalay, Caccappolo, Mejia-Santana, Tang, Rosado, Ross, Verbitsky, Kisselev, Louis, Comella, Colcher, Jennings, Nance, Bressman, Scott, Tanner, Mickel, Andrews, Waters and Clark2010; Marder et al., Reference Marder, Tang, Mejia-Santana, Rosado, Louis, Comella, Colcher, Siderowf, Jennings, Nance, Bressman, Scott, Tanner, Mickel, Andrews, Waters, Fahn, Ross, Cote and Clark2010). Increasing amounts of data suggest that specific genetic alleles exhibit a Mendelian inheritance pattern for PD. SNCA, LRRK2, VPS35, PRKN, PINK1, GBA, and DJ-1 genes are the leading candidates for suggested monogenic PD, although other genes have also been linked to Mendelian forms and sporadic cases of PD (Bandres-Ciga et al., Reference Bandres-Ciga, Diez-Fairen, Kim and Singleton2020; Yan et al., Reference Yan, Tang, Zhou, Lei, Li, Sun, Xu, Yan, Guo and Liu2017). For sporadic cases of PD, which comprise a significant proportion of cases, a small number of genetic loci have been attributed to the etiology. Sporadic cases of PD are mostly explained by the combined effects of genetic and nongenetic factors (Kalinderi et al., Reference Kalinderi, Bostantjopoulou and Fidani2016). In this study, we reanalyzed the genetic data of 43 subjects and found 14 different heterozygous variants, including two novel variants, emphasizing the importance of reanalysis.
Materials and Methods
Ethical Issues
The ethical committee of Canakkale Onsekiz Mart University Faculty of Medicine reviewed the study for the cohort, and permission was obtained (No: 2020-12/September 23-2020). All subjects enrolled in this study were informed, and a signed consent form was obtained from subjects or legal family members. Patient privacy and confidentiality were protected when nonidentifiable cohort data were used.
Study Population
This retrospective study examined the genetic data of 43 patients who were referred to our clinic between 2018 and 2019 with indications of clinically diagnosed PD (based on the International Parkinson and Movement Disorder Society criteria,) parkinsonism without a clear diagnosis (at least one cardinal symptom of PD for more than one year), family history, and incidental genetic findings. The data were collected and re-evaluated in late 2020. Clinical history, pedigree analysis, age, sex, history of drug use, and exposure to pesticides were obtained during clinical observation. Phone calls were made to gather missing data due to the COVID-19 pandemic. The patients exhibited a wide range of symptoms, including rigidity, tremor, dyskinesia, postural instability, dysphagia, axial deformities, sleep disturbances, memory deficits, reduced cognitive function, dementia, hallucinations, mood disorders, autonomic dysfunctions (mostly orthostatic hypotension, urogenital dysfunction, constipation, excessive sweating, etc.), sensory symptoms (decreased olfactory senses), and pain disorders. Any imaging studies involving probands were also noted.
Genetic Analyses
We conducted genetic analyses on 43 patients in this study using a custom-designed gene panel containing 18 genes associated with PD. The 18 genes, their transcript numbers, and related clinical OMIM numbers are summarized in Table 1. The gene panel was specifically designed to detect any single nucleotide variation and copy number variation changes in exons, exon-intron junctions, and splicing regions (+-10bp). DNA was extracted from peripheral blood using the PureLinkTM Genomic DNA Mini Kit (Invitrogen, Germany), and genes were sequenced on the Ion S5TM System (Thermo Fisher Scientific, USA) after library preparation. The sequence reading results were aligned to the reference human genome (hg19/GRCh37.p13) via Torrent Suite™ Software to obtain the Binary Alignment/Map (BAM)-Variant Calling Format (VCF). The Franklin Genoox database was used for annotation in the reanalysis process. All variants were classified and reclassified according to American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants as pathogenic (P), likely pathogenic (LP), variants of unknown significance (VUS), likely benign (LB), or benign (B). The ClinVar database was used for annotation of variants in addition to variant frequency in the gnomAD database. Clinically significant variants were then reported in three categories based on their pathogenicity: pathogenic variants, likely pathogenic variants, and VUS. We compared the first and second interpretations of the variants, and confirmation of variants was achieved using the IGV genome browser 2.9.4.
Table 1. Custom designed NGS gene panel

Note: OMIM, Online Mendelian Inheritance in Man (catalog); Ref Seq, Reference Sequence (database).
Results
A total of 43 patients from 39 families were enrolled in this study, consisting of 22 females and 21 males, with a median age of 53.4 years. Only three cases showed consanguinity in the pedigree analysis. We identified 14 different clinically significant reportable variants (classified as pathogenic, likely pathogenic, or VUS) in 14 individuals from nonconsanguineous families. Eight (57%) of the probands were considered late onset (50 years of age or after), and 4 (28%) of the probands were considered early onset (before 50 years of age) when considering probands with a clinical PD diagnosis or having at least one long-term cardinal parkinsonism symptom. Probands who had a family member diagnosed with PD or showed at least one long-term parkinsonism symptom constituted 20 (46%) of the cohort. Of this subgroup, 8 (40%) had clinically significant variants. Only one proband had an incidental genetic test result regarding PD-related genes. None of the three juvenile onset probands (20 years of age and before) had any clinically significant variant. Table 2 summarizes the clinical and demographic characteristics of the patients with clinically significant variants. Out of the 14 (32%) clinically significant variants, 3 (7%) were classified as P or LP, while 11 (26%) were VUS. Two variants were not reported in databases and were defined as novel variants. There was no homozygosity or compound heterozygosity. We have summarized the details in Table 3.
Table 2. Clinical and demographical characteristics of probands with clinically significant variant
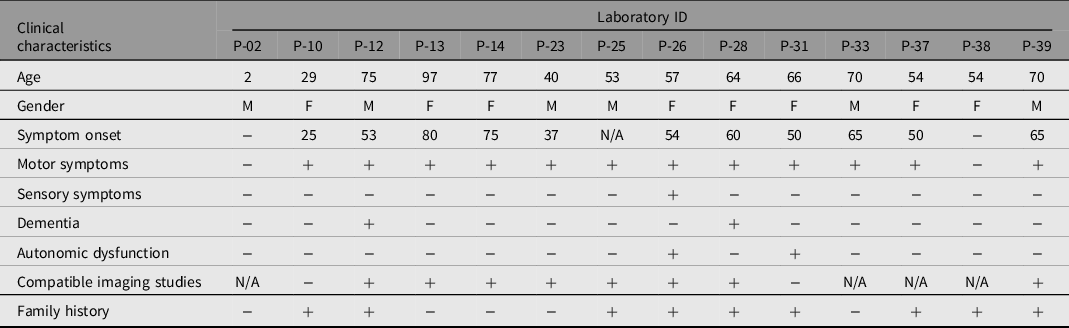
Table 3. Laboratory IDs of probands, reference number, codon, mutation type, allele frequency and clinical significance according to different databases of variants detected in the cohort

Note: VUS, variants of unknown significance; ACMG, American College of Medical Genetics and Genomics standards and guidelines.
Reanalysis Findings
In the reanalysis process, 14 variants were found to be benign or likely benign, despite being classified as VUS in the first interpretation. Only one variant, PRKN(NM_004562.3):c.136G>A, was originally classified as benign or likely benign and later reclassified as VUS. Table 4 shows a comparison between the first interpretation and reanalysis results for the variants, which occurred 12−24 months apart.
Table 4. Comparison of the variants according to first evaluation and reanalysis

Note: VUS, variants of unknown significance; ACMG, American College of Medical Genetics and Genomics standards and guidelines. Bold type indicates the only variant showing reverse evolution.
Discussion
Genotype-Phenotype Correlation
In this study, we aimed to contribute to the literature by making phenotype correlations and emphasizing the importance of reanalysis of VUS by using a targeted gene panel with next-generation sequencing (NGS). We performed genetic testing on 43 patients and procured 14 different variants from 14 different subjects. To date, this study is the widest cohort with the largest gene panel study conducted with the NGS technique in Turkey, where approximately 150,000 patients have been diagnosed with PD, according to the Turkish Neurology Society.
We designed this study with 18 genes that are related to PD. A targeted gene panel with the NGS method gives us the possibility to study multiple genes with high confidence. The percentage of P or LP variants found in the cohort was 7%, which is consistent with the literature. The detected variants were all heterozygous, and the total percentage of clinically significant variants remained consistent with the literature (Blauwendraat et al., Reference Blauwendraat, Nalls and Singleton2020; Gasser, Reference Gasser2009; Kalinderi et al., Reference Kalinderi, Bostantjopoulou and Fidani2016).
The c.3142A>G (p. Ser1048Gly) variant in EIF4G1 was found in a patient with clinically diagnosed PD in her 70s. A study from France concluded that EIF4G1 variants are the etiological cause for late-onset PD (Chartier-Harlin et al., Reference Chartier-Harlin, Dachsel, Vilarino-Guell, Lincoln, Lepretre, Hulihan, Kachergus, Milnerwood, Tapia, Song, Le Rhun, Mutez, Larvor, Duflot, Vanbesien-Mailliot, Kreisler, Ross, Nishioka, Soto-Ortolaza and Farrer2011). Another study found that the R1205H variant was detected in familial cases but cited one of the relatives who was also carrying the variant even though he/she had no symptoms, which suggests reduced penetrance (Nuytemans et al., Reference Nuytemans, Bademci, Inchausti, Dressen, Kinnamon, Mehta, Wang, Zuchner, Beecham, Martin, Scott and Vance2013). Last, a wide European cohort of PD patients concluded that EIF4G1 variants cause late-onset PD with low penetrance (Huttenlocher et al., Reference Huttenlocher, Kruger, Capetian, Lohmann, Brockmann, Csoti, Klein, Berg, Gasser, Bonin, Riess and Bauer2015). This patient shows compatibility with these reports.
A patient carrying the c.4915del (p. Arg1639GlyfsTer15) variant in LRRK2 was diagnosed with PD in her early fifties. The patient had a history of multiple relatives diagnosed with PD in her family and two relatives with long-term nonintentional tremor without any particular diagnosis. LRRK2 is one of the most researched genes linked with PD, and it has been related to dominant Mendelian inheritance as well as risk factors for sporadic PD (Dachsel & Farrer, Reference Dachsel and Farrer2010). Several familial PD studies report that different heterozygous LRRK2 variants show dominant inheritance (Nichols et al., Reference Nichols, Pankratz, Hernandez, Paisan-Ruiz, Jain, Halter, Michaels, Reed, Rudolph, Shults, Singleton and Foroud2005; Paisan-Ruiz et al., Reference Paisan-Ruiz, Jain, Evans, Gilks, Simon, van der Brug, Lopez de Munain, Aparicio, Gil, Khan, Johnson, Martinez, Nicholl, Marti Carrera, Pena, de Silva, Lees, Marti-Masso, Perez-Tur and Singleton2004; Zimprich et al., Reference Zimprich, Biskup, Leitner, Lichtner, Farrer, Lincoln, Kachergus, Hulihan, Uitti, Calne, Stoessl, Pfeiffer, Patenge, Carbajal, Vieregge, Asmus, Muller-Myhsok, Dickson, Meitinger and Glasser2004). In addition, different groups of studies concluded that single nucleotide changes in LRRK2 are a risk factor for sporadic PD (Di Fonzo et al., Reference Di Fonzo, Wu-Chou, Lu, van Doeselaar, Simons, Rohe, Chang, Chen, Weng, Vanacore, Breedveld, Oostra and Bonifati2006; Tan et al., Reference Tan, Zhao, Skipper, Tan, Di Fonzo, Sun, Fook-Chong, Tang, Chua, Yuen, Tan, Pavanni, Wong, Kolatkar, Lu, Bonifati and Liu2007). This frameshift mutation causes termination of the reading frame. A history of multiple PD cases and other relatives showing cardinal symptoms of the disease without diagnosis in the family might be consistent with the literature. Unfortunately, we did not have the chance to test the other family members. This variant has not previously been mentioned in the literature before according to our knowledge and is therefore considered a novel variant.
The c.2859G>A (p. Thr953=) variant in ATP13A2 was found in a patient who had a history of typical PD symptoms with advanced age. One study pointed out that two cases of early-onset PD with Italian descent heterozygous mutations in ATP13A2 might be related to an increased risk of PD (Di Fonzo et al., Reference Di Fonzo, Chien, Socal, Giraudo, Tassorelli, Iliceto, Fabbrini, Marconi, Fincati, Abbruzzese, Marini, Squitieri, Horstink, Montagna, Libera, Stocchi, Goldwurm, Ferreira, Meco and Bonifarti2007). Another study in a Chinese Han population stated that the heterozygous A746T variant was seen more frequently in the early-onset PD group than in controls (Lin et al., Reference Lin, Tan, Chen, Tan, Lim, Chen and Wu2008). In contrast, Podhajska et al. (Reference Podhajska, Musso, Trancikova, Stafa, Moser, Sonnay, Glauser and Moore2012) reported that a cellular study showed heterozygous variants in ATP13A2 related to PD in previous literature as risk factors do not alter protein stability or subcellular localization but instead impair the ATPase activity of microsomal ATP13A2, which might not be evaluated as sufficient to link with PD. It is fair to say that the accurate correlation of heterozygous ATP13A2 mutations and PD seems controversial.
Variants of uncertain significance need more complicated and long processes to assess their real contribution to complex diseases such as PD. According to the ACMG, VUS is a class for which we do not have enough data or evidence to make any certain implications about pathogenicity (Richards et al., Reference Richards, Aziz, Bale, Bick, Das, Gastier-Foster, Grody, Hegde, Lyon, Spector, Voelkerding and Rehm2015). Due to the excessive data we gather from gene panels, we are facing a substantial number of variants with uncertain significance, which mostly leads to obscurity. Studies of cancer genetics, which is the most common indication for sequencing, report 34−41% VUS, and this fact might give us a projection (Esterling et al., Reference Esterling, Wijayatunge, Brown, Morris, Hughes, Pruss, Manley, Bowles and Ross2020; Frey et al., Reference Frey, Kim, Bassett, Martineau, Dalton, Chern and Blank2015). We often benefit from in silico databases to evaluate these variants and perform segregation testing if possible to conclude any verdict. It would be better to consider that every database shapes and changes our understanding of these variants’ contribution to any disease, and by nature, these databases are being updated continuously. It is reasonable to deduce that reanalysis and re-evaluation are vital for making clear curations for these variants; likewise, several studies have concluded the same idea (Eccles et al., Reference Eccles, Copson, Maishman, Abraham and Eccles2015). Variants that have been interpreted as VUS in reanalysis from our clinical experience are shown in Table 3.
The c.1546G>C p. Asp516His variant in FBXO7 has been reported in the literature before, and two articles about this variant are not consistent with each other; comments on this particular variant are controversial (Ghani et al., Reference Ghani, Lang, Zinman, Nacmias, Sorbi, Bessi, Tedde, Tartaglia, Surace, Sato, Moreno, Xi, Hung, Nalls, Singleton, St George-Hyslop and Rogaeva2015; Gorostidi et al., Reference Gorostidi, Marti-Masso, Bergareche, Rodriguez-Oroz, Lopez de Munain and Ruiz-Martinez2016). This variant was an incidental finding, and the proband was asymptomatic.
The c.499G>C (p. Val167Leu) variant in FGF20 was found in a female patient with early-onset PD diagnosis, and her father also had parkinsonism without a definitive diagnosis and carried the same variant in addition to the c.245C>A (p. Ala82Glu) variant in PRKN. Although the c.245C>A (p. Ala82Glu) variant in PRKN was not found in the ClinVar and GnomAD databases, it is classified as VUS by ACMG criteria. In addition to these data, previous articles on this specific variant show no agreement on pathogenicity (Erer et al., Reference Erer, Egeli, Zarifoglu, Tezcan, Cecener, Tunca, Ak, Demirdogen, Kenangil, Kaleagasi, Dogu, Saka and Elibol2016; Gorostidi et al., Reference Gorostidi, Marti-Masso, Bergareche, Rodriguez-Oroz, Lopez de Munain and Ruiz-Martinez2016).
The c.700G>A (p. Ala234Thr) variant in SYNJ1 was found in a patient who had been clinically diagnosed with PD. The patient also had multiple family members with dementia. c.3863C>T (p. Pro1288Leu) in SYNJ1 was found in a patient who had been suffering from atypical symptoms of unilateral bradykinesia, chorea and hemiparesis for more than two years. The pathogenicity of these SYNJ1 variants has been evaluated according to ACMG criteria mostly linked with in silico databases.
The c.506+6T>C variant in VPS35 has been classified as a VUS because of the variant’s low frequency in public genome databases. This variant shows the possibility of affecting splicing. A patient carrying this variant showed early onset unilateral bradykinesia and tremor.
A patient carrying the c.136G>A (p. Ala46Thr) variant in PRKN was diagnosed with PD and had multiple family members with PD and dementia separately. This variant has been mentioned in a study enrolled in a Nigerian population and has been found more frequently compared to the control group. A computer-based protein function study stated that there is a small chance that this variant affects protein function (Okubadejo et al., Reference Okubadejo, Britton, Crews, Akinyemi, Hardy, Singleton and Bras2008).
c.3872A>G (p. Glu1291Gly) in DNAJC13 was found in a proband displaying unilateral bradykinesia and tremor who also showed cogwheel rigidity during examination. The proband also had multiple family members with the same symptoms without any clinical diagnosis. A patient carrying c.2816T>C (p. Leu939Pro) in the ATP13A2 variant was asymptomatic and had a family history of PD and dementia. This variant has not been mentioned in the literature before according to our knowledge and is therefore considered a novel variant.
The c.2915A>G (p. Asp972Gly) variant in LRRK2 and the c.1403C>A (p. Ala468Glu) variant in EIF4G1 was found in a patient whose only symptom was unilateral tremor but who had multiple family members with a PD diagnosis. One study from China stated that although the c.1403C>A (p. Ala468Glu) variant in EIF4G1 was found at a low frequency in public genome databases, it showed no difference from the control group (Ma et al., Reference Ma, Zheng and Li2018).
Reanalysis
The main purpose of this study was to emphasize the importance of reanalysis and re-evaluation of variants, particularly VUS. Since the NGS era began, a tremendous amount of genetic data has been generated, and correlating this data with patients’ clinical status can often be challenging. Even experienced clinicians have acknowledged significant problems with this issue (Richards et al., Reference Richards, Aziz, Bale, Bick, Das, Gastier-Foster, Grody, Hegde, Lyon, Spector, Voelkerding and Rehm2015; Vears et al., Reference Vears, Senecal and Borry2017). Reanalysis of variants and patient follow-up by genetic clinics are especially important in these situations. Although there is no international consensus, some societies have published variable opinions about the issue (Carrieri et al., Reference Carrieri, Dheensa, Doheny, Clarke, Turnpenny, Lucassen and Kelly2017a, Reference Carrieri, Dheensa, Doheny, Clarke, Turnpenny, Lucassen and Kelly2017b; Dheensa et al., Reference Dheensa, Carrieri, Kelly, Clarke, Doheny, Turnpenny and Lucassen2017).
One study indicated that reanalysis of variants within 5−6 years has increased clinical benefit from 26% to 47% (Liu et al., Reference Liu, Meng, Normand, Xia, Song, Ghazi, Rosenfeld, Magoulas, Braxton, Ward, Dai, Yuan, Bi, Xiao, Wang, Chiang, Vetrini, He, Cheng and Yang2019). For diseases like PD, which have several candidate genes in addition to candidate variants, reanalysis and clinical follow-up seem inevitable. It has been noted that keeping in touch with patients, scheduling clinical visits at proper intervals, and providing patients with new information about genetic data is vital when making this plan (Carrieri et al., Reference Carrieri, Dheensa, Doheny, Clarke, Turnpenny, Lucassen and Kelly2017a, Reference Carrieri, Dheensa, Doheny, Clarke, Turnpenny, Lucassen and Kelly2017b). Various clinical genetic organizations around the Western world have published different statements, but to generalize, the suggestion of reanalysis of VUS and informing the patients was kept favorable (Boycott et al., Reference Boycott, Hartley, Adam, Bernier, Chong, Fernandez, Friedman, Geraghty, Hume, Knoppers, Laberge, Majewski, Mendoza-Londono, Meyn, Michaud, Nelson, Richer, Sadikovic, Skidmore and Armour2015; Dheensa et al., Reference Dheensa, Carrieri, Kelly, Clarke, Doheny, Turnpenny and Lucassen2017; Matthijs et al., Reference Matthijs, Souche, Alders, Corveleyn, Eck, Feenstra, Race, Sistermans, Sturm, Weiss, Yntema, Bakker, Scheffer and Bauer2016; Richards et al., Reference Richards, Aziz, Bale, Bick, Das, Gastier-Foster, Grody, Hegde, Lyon, Spector, Voelkerding and Rehm2015). Our clinical experience is shown in Table 4. Fourteen variants had been classified as VUS in the initial evaluation and curated as benign or likely benign in the reanalysis process. We observed an inverse evolution in only one variant, PRKN(NM_004562.3):c.136G>A, which had been classified as benign in the initial evaluation and as a VUS in reanalysis. We also added this variant to the final report of the patient. The minimum and maximum re-evaluation times for these data were 12 and 24 months respectively. Similarly, a few reports in the literature have suggested that reanalysis of variants within even a small time gap, such as a 12-month period, provides a clinically significant alteration of data (Ewans et al., Reference Ewans, Schofield, Shrestha, Zhu, Gayevskiy, Ying, Walsh, Lee, Kirk, Colley, Ellaway, Turner, Mowat, Worgan, Freckmann, Lipke, Sachdev, Miller, Field and Roscioli2018; Wenger et al., Reference Wenger, Guturu, Bernstein and Bejerano2017; Wright et al., Reference Wright, McRae, Clayton, Gallone, Aitken, FitzGerald, Jones, Prigmore, Rajan, Lord, Sifrim, Kelsell, Parker, Barrett, Hurles, FitzPatrick, Firth and Study2018).
In conclusion, to our knowledge, this is the first study to use a targeted gene panel with the widest number of genes with NGS method in the largest PD cohort from our country so far. This study has contributed to the genotype-phenotype correlation of PD, demonstrated the benefits of using a targeted gene panel with NGS method for molecular etiology in PD, and, perhaps most importantly, shown that re-evaluating genetic data, particularly VUS, is critical, especially in multifactorial diseases like PD, to arrive at definitive conclusions about variants.
Acknowledgments
The authors thank the family members who agreed to participate in the presented results.
Authors’ contributions
AK, acquisition of data, analysis of the clinical data and design of the clinical experiments, analysis of the sequencing data and writing of the manuscript; AK and FS performed PCR, software, validation; OO, conceptualization, interpretation of data, supervised the study and reviewed the manuscript. All authors read and approved the final manuscript.
Funding
The authors did not receive support from any organization for the submitted work.
Conflict of interest
None.




