The chromosome 17q21.31 microdeletion syndrome (Koolen-de Vries syndrome, MIM 610443) is a genomic disorder characterized by intellectual disability (ID), friendly behavior, hypotonia and distinct facial features with thin long face, large pear-shaped nose and prominent chin (Koolen et al., Reference Koolen, Vissers, Pfundt, de Leeuw, Knight, Regan and de Vries2006; Sharp et al., Reference Sharp, Hansen, Selzer, Cheng, Regan, Hurst and Eichler2006; Shaw-Smith et al., Reference Shaw-Smith, Pittman, Willatt, Martin, Rickman, Gribble and Carter2006). The typical facial phenotype is usually less apparent in the infancy and becomes remarkable during adolescence (Koolen et al., Reference Koolen, Sharp, Hurst, Firth, Knight, Goldenberg and de Vries2008; Slavotinek, Reference Slavotinek2008). Major anomalies, seizures, joint hyperlaxity and eye anomalies can be also present, but are less common. The prevalence of the syndrome is estimated to 1 in 16,000 (Koolen et al., Reference Koolen, Sharp, Hurst, Firth, Knight, Goldenberg and de Vries2008).
The syndrome is caused by a recurrent 600 kb deletion of 17q21.31. The region is predisposed to rearrangement by its specific genome architecture. The deletion breakpoints map to large clusters of low copy repeats (LCRs) predisposing to non-allelic homologous recombination (NAHR). The 17q21.31 region is known for its inversion polymorphism of about 900 kb and the presence of two highly divergent SNP haplotypes designated H1 and H2. H2 is associated with the inversion and is found at a frequency of 20% in the European population (Stefansson et al., Reference Stefansson, Helgason, Thorleifsson, Steinthorsdottir, Masson, Barnard and Stefansson2005). H2 differs from the non-inverted H1 allele by the arrangement of LCRs, which makes H2 prone to NAHR events (Koolen et al., Reference Koolen, Sharp, Hurst, Firth, Knight, Goldenberg and de Vries2008; Steinberg et al., Reference Steinberg, Antonacci, Sudmant, Kidd, Campbell, Vives and Eichler2012). At least one of the parents of deletion patients always carried at least one H2 allele, which seems to be necessary for the deletion formation. The deletion encompasses several genes, among which haploinsufficiency of KANSL1 has recently been shown to be responsible for the syndrome (Koolen et al., Reference Koolen, Kramer, Neveling, Nillesen, Moore-Barton, Elmslie and de Vries2012b; Zollino et al., Reference Zollino, Orteschi, Murdolo, Lattante, Battaglia, Stefanini and Marangi2012).
Herein we present the first report of monozygotic twins carrying the 17q21.31 microdeletion and showing only slightly different phenotypes. Analysis on high-resolution arrays did not reveal any genetic differences between the twins. The subtle clinical differences can probably be explained by different perinatal history of the twins or by the variable expressivity of the disorder.
Materials and Methods
Patients
The girls were born from a twin pregnancy to healthy, non-consanguineous parents of Czech origin. The age of the mother and father were 22 and 25 years, respectively. The delivery was in the 38th week of gestation by cesarean section due to hypoxia in Twin B.
Twin A was born with a weight of 1980 g and length of 43 cm (both below the 3rd centile). The Apgar score was 3-7-7 (Apgar, Reference Apgar1953). Partial exchange transfusion had to be administered due to polyglobulia and hyperviscosity syndrome. The newborn suffered from left-side hypotonic hydronephrosis with reflux. Twin B was born with a weight of 1910 g and length of 43 cm (both below the 3rd centile). The Apgar score was 3-7-7. Perinatal hypoxia followed by intracranial hemorrhage occurred during the delivery. Right-side hydronephrosis, strabismus and horizontal nystagmus were noted in the newborn.
Postaxial polydactyly of toes and fingers, congenital hip dysplasia, delay in motor milestones and speech delay were observed in both twins. Psychological examination at the age of 10 years showed moderate ID in both twins, but Twin A performed slightly better than Twin B (Twin A was assessed as functioning in the upper range of moderate ID and being slightly more diligent and adaptable, and less anxious). At the examination at 19 years of age both twins had disproportionately short stature (Twin A 153.3 cm, the 1st centile; Twin B 157.7 cm, the 6th centile) with shortening of upper and lower limbs, thoracic hyperkyphosis, low-pitched voice and similar facial expression (Figure 1), and with very long, thin and coarse face, coarse hair, thick eyebrows, large nose, bulbous nasal tip, smooth broad philtrum, thick lips, mandibular prognathism, and hirsutism. Twin A had a high palate. Twin B had wide-spaced teeth and diastema, and slightly more coarse facial features compared to Twin A. However, especially with respect to their age, the overall clinical picture of both twins was remarkably similar. None of them showed other symptoms often described in the 17q21.31 microdeletion syndrome, such as seizures, joint hypermobility, cleft lip/palate, heart defects, or pectus excavatum (Koolen et al., Reference Koolen, Kramer, Neveling, Nillesen, Moore-Barton, Elmslie and de Vries2012b).

FIGURE 1 Facial photographs of the patients at the age of 19 (top) and 23 years (bottom). Twin A is on the left, Twin B on the right. Features typical for the 17q21.31 microdeletion syndrome (long, narrow and coarse face, coarse hair, large nose with bulbous nasal tip, broad philtrum, thick lips, mandibular prognathism) and subtle differences between the twins (slightly more coarse facial features in Twin B) can be observed.
Laboratory Analyses
Informed consent for genetic analyses was obtained from the parents of the patients. Genomic DNA of both twins and the parents was extracted from blood lymphocytes using the Gentra Puregen Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Conventional cytogenetic analysis was performed using standard G-banding. The FMR1 gene testing used the Fragile X PCR Kit (Abbot, Abbot Park, IL, USA). The BAC array comparative genome hybridisation (CGH; BlueGnome, Cambridge, UK) analysis of Twin A was performed according to the manufacturer's instructions. The FISH analysis with the BAC clone RP11-111L23 (BlueGnome) was used to independently confirm the deletion in the twins and to test for its presence in the parents. Diagnostic alleles of single nucleotide polymorphisms (SNPs) rs1800547 (G) and rs9468 (C) and the presence of the 238 bp deletion in intron 9 of the MAPT gene characteristic for the H2 allele (Koolen et al., Reference Koolen, Sharp, Hurst, Firth, Knight, Goldenberg and de Vries2008) were analysed in the family using DNA sequencing and gel electrophoresis, respectively (PCR primer sequences are available upon request). The high-resolution SNP array analysis of both twins using the HumanCytoSNP-12 BeadChip (~300 K; Illumina, San Diego, CA, USA) and direct array CGH comparison of their genomes using the Nimblegen 2.1M Whole-Genome CGH Array (Roche NimbleGen, Madison, WI, USA) were used for confirmation of monozygosity and for a more detailed analysis of potential differences in copy number variants (CNVs) in the genomes of both twins. Data were analysed using GenomeStudio (Illumina), QuantiSNP (Colella et al., Reference Colella, Yau, Taylor, Mirza, Butler, Clouston and Ragoussis2007) and SignalMap (Roche NimbleGen). Multiplex ligation-dependent probe amplification (MLPA) analysis was performed using custom synthetic probes and the P200 Human DNA Reference Probemix (MRC Holland, Amsterdam, The Netherlands; probe sequences are available upon request). All analyses used genome build hg18/NCBI36.
Results
The cytogenetic analysis revealed normal female karyotypes, and the FMR1 gene testing excluded the fragile X syndrome in both twins. The BAC array CGH analysis of Twin A identified a deletion characteristic for the 17q21.31 microdeletion syndrome with breakpoints between bases 40,740,861-41,074,265 and 41,679,148-42,178,065. The FISH analysis confirmed the deletion in both twins but in neither of their parents. The haplotype analysis revealed homozygosity for the inverted H2 allele in the mother, homozygosity for the non-inverted H1 allele in the father, and hemizygosity for H1 in both twins. Thus the deletion was de novo in the twins and it affected one of the maternal chromosomes 17.
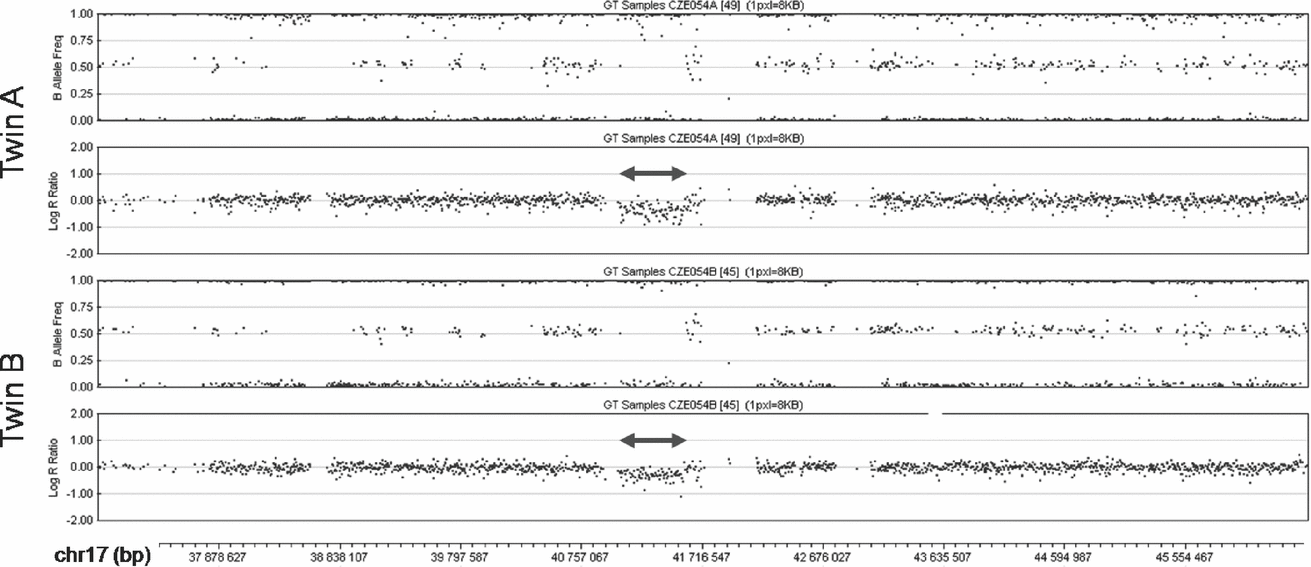
The SNP array analysis confirmed the monozygosity of the twins. This high-resolution analysis found no differences in the extent of the 17q21.31 microdeletion between the patients (chr17:41,041,709-41,560,151; Figure 2). Both twins shared two additional CNVs, a 0.1 Mb long duplication in 10q26.3 (chr10:135,102,337-135,215,135) encompassing CYP2E1, and a 1.7 Mb long deletion in 16p11.2 (chr16:31,977,497-33,704,396) involving TP53TG3. Both these CNVs were located in highly polymorphic copy-number variable regions. The analysis with the highest resolution used (2.1M array CGH) did not detect any obvious CNV differences between the genomes of the twins. In several small regions copy number differences between the patients could not be excluded (chr18:14,184,640-15,370,613 and chr21:13,302,864-14,139,384 being most suspicious), but most of these segments coincided with complex segmental duplications, where the validity of the findings was questionable, impossible to confirm using standard methods and of uncertain clinical impact even if they were confirmed. The analysis of three of these regions where unique sequences could be targeted with custom MLPA probes (chr14:18,127,587-19,272,166; chr16:32,082,491-34,128,024 and chr22:49,414,658-49,584,579) failed to confirm any copy number differences between the twins in these regions.
FIGURE 2 SNP array analysis of the middle part of 17q in the patients. The deletions are marked by double arrows. In the diagrams of the B Allele Frequency (top panel in each twin) deletions are indicated by the absence of dots around the value of 0.5 (absence of heterozygous genotype AB). Concurrently, in the diagrams of Log R Ratio (bottom panel in each twin) the deletions are indicated by dots clustering below the value of 0.0 (decreased intensity of the signal).
Discussion
To our knowledge this is the first description of monozygotic twins with the 17q21.31 microdeletion syndrome. The deletion was de novo on a maternal chromosome 17, although a low-level somatic and gonadal mosaicism could not be excluded (Koolen et al., Reference Koolen, Dupont, de Leeuw, Vissers, van den Heuvel, Bradbury and Parker2012a). The twin sisters showed only a subtle phenotypic discordance. Generally, discordant monozygotic twins are a valuable resource for the analysis of genetic, epigenetic or environmental variation contributing to the disease. The 17q21.31 microdeletion syndrome is one of a few clinically recognizable new syndromes with well-defined clinical outcome, and rare instances of affected twins could contribute to understanding the variability of this disorder.
The phenotypes of our patients were very similar and fully corresponded to the typical picture of the syndrome (Koolen et al., Reference Koolen, Sharp, Hurst, Firth, Knight, Goldenberg and de Vries2008). The subtle phenotypic differences between the twins included a slightly more severe cognitive impairment and more coarse facial features with strabismus and horizontal nystagmus in Twin B. These differences prompted us to search for possible genomic differences. The 17q21.31 microdeletion was of the same size in both twins, and also the two other CNVs detectable at the 300K level were present in common and were unlikely to contribute to the phenotype. The 10q26.3 duplication encompassing CYP2E1 is a common polymorphism possibly associated with alcohol addiction (Deng & Deitrich, Reference Deng and Deitrich2008). The 16p11.2 deletion around TP53TG3 affected a very variable gene-poor pericentric region. Also, the direct comparison of both genomes using an even higher resolution (2.1M) did not yield any findings. Several suspicious CNV differences were located in highly polymorphic regions of segmental duplications, the structure of which made the confirmation of these aberrations difficult or impossible, and analysis of three of these regions failed to confirm any differences between the two genomes. However, it should be noted that these regions are susceptible to de novo events, and that any genomic differences between the twins could be expected to be in a mosaic state, further complicating their detection. In any case, due to the paucity of genes, these potentially differential CNVs were unlikely to affect the phenotype.
Several other studies focused on monozygotic twins with microdeletion syndromes and a different degree of phenotypic discordance. Ghebranious et al. (Reference Ghebranious, Giampietro, Wesbrook and Rezkalla2007) presented monozygotic twins with a 16p11.2 microdeletion and no other CNV differences, who showed similar phenotypes but severe aortic stenosis developed only in one twin. Most monozygotic twin pairs reported with 22q11 deletions were also phenotypically discordant. Singh et al. (Reference Singh, Murphy and O’Reilly2002) reviewed five such pairs in whom no high-resolution whole genome analyses were performed to uncover potential genomic differences. The discordance in a recently identified monozygotic twin pair with a 22q11 microdeletion was explained by size differences of the deletions (Halder et al., Reference Halder, Jain, Chaudhary and Varma2012), which however have not been confirmed using an independent method and are thus questionable. Rio et al. (Reference Rio, Royer, Gobin, de Blois, Ozilou, Bernheim and Malan2013) reported a phenotypically and genetically discordant monozygotic twin pair carrying a 2p25.3 deletion in one twin and mosaicism with one third of cells with the 2p25.3 deletion, one third with a 2p25.3 duplication, and one third of normal cells in the other one. Other recent studies of monozygotic twin pairs discordant for breast cancer (Lasa et al., Reference Lasa, Cajal, Llort, Suela, Cigudosa, Cornet and Baiget2010), schizophrenia (Ono et al., Reference Ono, Imamura, Tasaki, Kurotaki, Ozawa, Yoshiura and Okazaki2010) or congenital heart defect (Breckpot et al., Reference Breckpot, Thienpont, Gewillig, Allegaert, Vermeesch and Devriendt2012) also identified no CNV differences explaining the discordance.
In the absence of genetic differences, the twin discordance can be explained by epigenetics or environment (Czyz et al., Reference Czyz, Morahan, Ebers and Ramagopalan2012). The study of DRD2 methylation in two pairs of monozygotic twins, one discordant and one concordant for schizophrenia, showed that the affected twin from the discordant pair was epigenetically ‘closer’ to the affected concordant twins than to his unaffected co-twin (Petronis et al., Reference Petronis, Gottesman, Kan, Kennedy, Basile, Paterson and Popendikyte2003). Similarly, the affected twin from a monozygotic pair discordant for caudal duplication anomaly showed higher methylation of the AXIN1 promoter than the unaffected twin, whose AXIN1 methylation was higher than that of normal controls (Oates et al., Reference Oates, van Vliet, Duffy, Kroes, Martin, Boomsma and Chong2006). An epigenome-wide approach found that approximately one third of monozygotic twins had epigenetic differences in DNA methylation and histone modification (Fraga et al., Reference Fraga, Ballestar, Paz, Ropero, Setien, Ballestar and Esteller2005). Epigenetic marks were more distinct in twins who were older, had different lifestyles, and had spent less of their lives together, underlining the significant role of environmental factors in the process (Fraga et al., Reference Fraga, Ballestar, Paz, Ropero, Setien, Ballestar and Esteller2005; Kaminsky et al., Reference Kaminsky, Tang, Wang, Ptak, Oh, Wong and Petronis2009). Environmental factors could include the differences in the intrauterine environment and in perinatal and postnatal history, and the twinning process itself could play a role as well as stochastic factors can do (Czyz et al., Reference Czyz, Morahan, Ebers and Ramagopalan2012). Mosaicism resulting from later postzygotic genomic rearrangements or epigenetic changes can be difficult to detect, and it can differentially affect specific tissues (e.g., the brain) that are not accessible to testing. Another limitation of twin studies, including ours, which are using blood as the source of DNA, is blood chimerism, which can mask genetic or epigenetic discordance (Erlich, Reference Erlich2011).
In the case of our patients who show no CNV differences, all other factors mentioned above could contribute to their subtle phenotypic discordance. The currently emerging whole exome and whole genome sequencing approaches could identify possible genetic variation on the nucleotide level not addressed in our study, and epigenetic differences could also play a role. However, the simplest and likely sufficient explanation of the slightly discordant phenotype of the twins is in their perinatal history, which was clearly more severe in Twin B (perinatal hypoxia followed by intracranial hemorrhage). The differences in the clinical picture of our patients can also be the consequence of stochastic factors acting in the common inter-individual variability, and the variable expressivity of the 17q21.31 microdeletion syndrome.
Acknowledgments
We thank the family of the patients for cooperation. Supported by grants CHERISH 223692 and CZ.2.16/3.1.00/24022 from the European Commission, DRO UH Motol 00064203 and NT/14200 from the Czech Ministry of Health, and SF0180027s10 from the Estonian Ministry of Education and Research.