


Throughout the lifespan, large individual differences are evident in the expression of aggressive behavior. In children, aggressive behavior is regarded to represent, up to a certain extent, a ‘normal’ behavior that decreases in frequency when children learn to express themselves in more socially preferable ways (Tremblay, Reference Tremblay2008). Longitudinal studies have shown that aggressive behavior emerges in early childhood with a peak between 2 and 4 years of age, but decreases during childhood in most individuals (Alink et al., Reference Alink, Mesman, van Zeijl, Stolk, Juffer, Koot, Bakermans-Kranenburg and van Ijzendoorn2006; Cote et al., Reference Cote, Vaillancourt, LeBlanc, Nagin and Tremblay2006, Reference Cote, Boivin, Nagin, Japel, Xu, Zoccolillo and Tremblay2007; Tremblay et al., Reference Tremblay, Nagin, Seguin, Zoccolillo, Zelazo, Boivin and Japel2004). Abnormal levels of aggression in childhood and adulthood are a characteristic of several psychiatric disorders (Coccaro Reference Coccaro2012; Coie et al., Reference Coie, Lochman, Terry and Hyman1992; Nestor, Reference Nestor2002) and may be caused by traumatic brain injury (Rao et al., Reference Rao, Rosenberg, Bertrand, Salehinia, Spiro, Vaishnavi and Miles2009; Tateno et al., Reference Tateno, Jorge and Robinson2003), substance abuse (Boles & Miotto, Reference Boles and Miotto2003; Moore et al., Reference Moore, Stuart, Meehan, Rhatigan, Hellmuth and Keen2008), and neurodegenerative diseases (Lai et al., Reference Lai, Tsang, Francis, Esiri, Keene, Hope and Chen2003; Moechars et al., Reference Moechars, Lorent, De Strooper, Dewachter and Van Leuven1996; Volicer & Hurley, Reference Volicer and Hurley2003).
Twin studies illustrate that in the general population, a large part of variation in aggressive behavior between individuals is explained by genetic differences. For example, in 3- to 12-year-old children, the heritability ranges between 51% and 72% (Hudziak et al., Reference Hudziak, van Beijsterveldt, Bartels, Rietveld, Rettew, Derks and Boomsma2003), and a study of adults reported heritability estimates between 37% and 57% for subtypes of aggressive behavior (Yeh et al., Reference Yeh, Coccaro and Jacobson2010). Besides genetic influences, it is clear that the conditions under which a person grows up are important. In the twin study of 3- to 12-year-old children, 13–27% of the variation in aggressive behavior was explained by the family environment (Hudziak et al., Reference Hudziak, van Beijsterveldt, Bartels, Rietveld, Rettew, Derks and Boomsma2003), and a range of adverse early life conditions are known to predict high levels of aggression in childhood, such as low parental income, maternal smoking during pregnancy, separation from a parent, family dysfunction, and coercive-hostile parenting by the mother (Tremblay et al., Reference Tremblay, Nagin, Seguin, Zoccolillo, Zelazo, Boivin and Japel2004).
Throughout life, the activity of gene expression is coordinated by epigenetic mechanisms such as DNA methylation at CpG dinucleotides and histone modifications (Bird, Reference Bird2007). These epigenetic marks are not fixed: they change, for example, to switch different genes on and off during development (Reik, Reference Reik2007). There is also increasing evidence from human and animal studies that DNA methylation may change in response to external conditions, including early life events (McGowan et al., Reference McGowan, Sasaki, D’Alessio, Dymov, Labonte, Szyf and Meaney2009; Weaver et al., Reference Weaver, Cervoni, Champagne, D’Alessio, Sharma, Seckl and Meaney2004) and prenatal exposures such as maternal smoking and diet (Bakulski et al., Reference Bakulski, Lee, Feinberg, Wells, Brown, Herbstman and Fallin2015; Breton et al., Reference Breton, Byun, Wenten, Pan, Yang and Gilliland2009; Richmond et al., Reference Richmond, Simpkin, Woodward, Gaunt, Lyttleton, McArdle and Relton2015; Suter et al., Reference Suter, Ma, Harris, Patterson, Brown, Shope and Aagaard-Tillery2011; Tobi et al., Reference Tobi, Slieker, Stein, Suchiman, Slagboom, van Zwet and Lumey2015). Therefore, the study of epigenetic marks may identify genes that are differentially regulated in individuals with higher, compared with lower levels of aggression. This idea is illustrated by a study of rhesus macaques, which showed that monkeys who were separated from their mother in early life and became highly aggressive exhibit numerous methylation differences in the prefrontal cortex and T-cells compared with monkeys reared by their mother (Provencal et al., Reference Provencal, Suderman, Guillemin, Massart, Ruggiero, Wang and Szyf2012). The effects were largely tissue-specific, with only a very small subset of genes and individual methylation sites being significantly differentially methylated in both tissue types (Provencal et al., Reference Provencal, Suderman, Guillemin, Massart, Ruggiero, Wang and Szyf2012). One of such overlapping sites was a CpG site upstream of A2D681, which is the homolog of the human glucocorticoid receptor gene NR3C1. Thus far, epigenetic studies of aggression or related phenotypes in humans have been scarce. A recent candidate gene study reported differences between individuals with antisocial personality disorder from a prisoner population and controls in methylation of the monoamine oxidase A (MAOA) gene in blood (Checknita et al., Reference Checknita, Maussion, Labonte, Comai, Tremblay, Vitaro and Turecki2015). Another candidate gene study reported higher methylation at several CpG sites in the serotonin transporter gene (SLC6A4) promoter in T-cells and monocytes from adult men who exhibited high levels of physical aggression in childhood (Wang et al., Reference Wang, Szyf, Benkelfat, Provencal, Turecki, Caramaschi and Booij2012). Third, methylation differences have been reported in candidate genes encoding cytokines and their transcription factors in a study of eight adult men who were classified as being on a chronic physical aggression trajectory between 6 and 15 years of age (Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013). In the same group of men (Provencal et al., Reference Provencal, Suderman, Guillemin, Vitaro, Cote, Hallett and Szyf2014), and in a group of 25 women with childhood aggression (Guillemin et al., Reference Guillemin, Provencal, Suderman, Cote, Vitaro, Hallett and Szyf2014), differences in methylation were also identified in an analysis targeting genome-wide promoters in T-cells and monocytes. Hence, DNA methylation differences associated with aggressive behavior are observed in blood cells and may reflect changes that are either the cause or consequence of aggressive behavior. Thus far, a large majority of these previous findings have not been replicated, and the role of DNA methylation in aggressive behavior remains inconclusive. Furthermore, no study of aggression has examined genome-wide methylation beyond gene promoters, or studied DNA methylation in relation to continuous variation in aggressive behavior in a general adult population.
Here we describe the first epigenome-wide association study (EWAS) for aggressive behavior in whole blood performed in a population-based sample from the Netherlands Twin Register (NTR). The aim was to identify genomic locations where DNA methylation level is associated with aggressive behavior.
Methods
Subjects and Samples
The subjects in this EWAS participated in longitudinal survey studies from the NTR and participated in the NTR biobank project. Venous blood samples were drawn in the morning after an overnight fast, and multiple ethylenediaminetetraacetic acid (EDTA) tubes were collected for isolation of DNA and RNA and assessment of hematological profiles. Phenotyping through longitudinal surveys and blood sampling procedures have been described in detail previously (Boomsma et al., Reference Boomsma, de Geus, Vink, Stubbe, Distel, Hottenga and Willemsen2006; van Beijsterveldt et al., Reference van Beijsterveldt, Groen-Blokhuis, Hottenga, Franic, Hudziak, Lamb and Boomsma2013; Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks, Estourgie-van Burk and Boomsma2010, Reference Willemsen, Vink, Abdellaoui, den Braber, van Beek, Draisma and Boomsma2013).
In total, 3,264 blood samples from 3,221 NTR participants were assessed for genome-wide methylation, of which 3,089 samples from 3,057 subjects passed quality control (QC). For a small group of subjects, methylation was measured in two longitudinal blood samples. The genome-wide methylation dataset is described in Van Dongen et al. (under review). In the current EWAS, we included individuals for whom the following data were available: aggression score, good quality DNA methylation data, and data on white blood cell counts, leaving 2,059 samples from 2,029 subjects. This cohort included 1,247 monozygotic (MZ) twins (479 complete MZ pairs), 663 dizygotic (DZ) twins (199 complete DZ pairs), 48 fathers of twins, 56 mothers of twins, 13 siblings, and 2 spouses of twins. For 30 subjects, longitudinal methylation data (two samples) were included in the EWAS. A separate discordant twin analysis was performed on data from a subset of 40 MZ twins (20 pairs) highly discordant for aggressive behavior.
All subjects provided written informed consent, and the study protocols were approved by the Central Ethics Committee on Research Involving Human Subjects of the VU University Medical Centre, Amsterdam, an Institutional Review Board certified by the US Office of Human Research Protections (IRB No. IRB-2991 under Federal-wide Assurance-3703; IRB/Institute codes, NTR 03-180).
Aggression
Data on aggression were collected in multiple NTR surveys. Here data from NTR Survey 8 (data collection in 2009) and Survey 10 (data collection in 2013) were analyzed. Aggressive behavior was rated with the ASEBA Adult Self-Report (ASR; Achenbach & Rescorla, Reference Achenbach and Rescorla2003). The aggression score was computed following the ASR guidelines as the sum of 15 items (Table 1). For individuals who completed Surveys 8 and 10, the aggression score that was assessed closest to the moment of blood draw was selected. The aggression data were adjusted for sex and age at the moment of survey completion. Residuals were analyzed in the EWAS.
TABLE 1 ASR Survey Items Used to Score Aggressive Behavior, Based on the ASEBA Manual

ASR = Adult Self-Report (Achenbach & Rescorla, Reference Achenbach and Rescorla2003).
Aggression Discordance in MZ Twins
To identify MZ twins discordant for aggressive behavior, the within-pair differences in aggression scores at Surveys 8 and 10 were computed. In total, aggression scores were available from at least one survey for 461 MZ pairs (18 pairs were excluded, of which one twin completed only Survey 8 and the other twin completed only Survey 10). For 205 pairs, longitudinal aggression scores were available from two time points (Surveys 8 and 10) and for 256 pairs, one aggression score was available. Discordance was defined as a within-pair difference in aggression score ≥7 at the closest available survey to the moment of blood draw. This threshold is equivalent to >2 times the SD of aggressive behavior in the entire NTR cohort. For pairs with longitudinal aggression data, the difference had to be at ≥5 (equivalent to >1.5 SD) at the other time point. This selection yielded 20 discordant MZ pairs, of which discordance was supported by longitudinal data for six pairs and by one time point for 14 pairs.
Infinium HumanMethylation450 BeadChip Data
DNA methylation was assessed with the Infinium HumanMethylation450 BeadChip Kit (Illumina, Inc.; Bibikova et al., Reference Bibikova, Barnes, Tsan, Ho, Klotzle, Le and Shen2011). Genomic DNA, 500 ng, from whole blood was bisulfite-treated using the ZymoResearch EZ DNA Methylation kit (Zymo Research Corp, Irvine, CA, USA) following the standard protocol for Illumina 450K micro-arrays, by the Department of Molecular Epidemiology from the Leiden University Medical Center (LUMC), the Netherlands. Subsequent steps (i.e., sample hybridization, staining, and scanning) were performed by the Erasmus Medical Center micro-array facility, Rotterdam, the Netherlands. QC and processing of the blood methylation dataset have been described in detail previously (Tobi et al., Reference Tobi, Slieker, Stein, Suchiman, Slagboom, van Zwet and Lumey2015; Van Dongen et al., under review). In short, a number of sample- and probe-level quality checks were performed. Sample-level QC was performed using MethylAid (van Iterson et al., Reference van Iterson, Tobi, Slieker, den Hollander, Luijk, Slagboom and Heijmans2014). Probes were set to be missing in a sample if they had an intensity value of exactly zero, or a detection p > .01, or a bead count of <3. After these steps, probes that failed based on the above criteria in >5% of the samples were excluded from all samples (only probes with a success rate ≥0.95 were retained). Probes were also excluded from all samples if they mapped to multiple locations in the genome (Chen et al., Reference Chen, Lemire, Choufani, Butcher, Grafodatskaya, Zanke and Weksberg2013), or if they had a single nucleotide polymorphism (SNP) within the CpG site (at the C or G position) irrespective of minor allele frequency in the Dutch population (Genome of the Netherlands Consortium, 2014). Only autosomal methylation sites were analyzed in the EWAS. The methylation data were normalized with functional normalization (Fortin et al., Reference Fortin, Labbe, Lemire, Zanke, Hudson, Fertig and Hansen2014) and normalized intensity values were converted into beta (β) values. The β-value represents the methylation level at a site, ranging from 0 to 1, and is calculated as follows:
 $$\begin{equation*}
\beta = \frac{M}{{M + U + 100}}
\end{equation*}$$
$$\begin{equation*}
\beta = \frac{M}{{M + U + 100}}
\end{equation*}$$
where M = methylated signal, U = unmethylated signal, and 100 represents a correction term to control the β-value of probes with very low overall signal intensity.
Principal component analysis was conducted on genome-wide methylation sites (after QC and normalization), including those on sex chromosomes.
Covariates
White blood cell percentages were included as covariates in the EWAS to account for variation in cellular composition between whole blood samples. The following subtypes of white blood cells were counted in blood samples: neutrophils, lymphocytes, monocytes, eosinophils, and basophils (Willemsen et al., Reference Willemsen, Vink, Abdellaoui, den Braber, van Beek, Draisma and Boomsma2013). Lymphocyte percentage was not included in models because it was strongly correlated with neutrophil percentage(r = -0.93), and basophil percentage was not included because it showed little variation between subjects, with a large number of subjects having 0% of basophils. Inspection of principal components (PCs) from the genome-wide methylation data indicated that PC1 reflected sex, PC2 correlated strongly with lymphocyte percentage (r = -0.8), and neutrophil percentage (r = 0.79). PC3 correlated moderately with age (r = -0.41) and weakly with measured white blood cell percentages (absolute r ~ 0.1), but we hypothesize that this PC may potentially be reflective of unmeasured white blood cell subtypes and included this PC in the model. PC4 and PC5 were included to account for technical variability between samples, as these PCs were correlated with several indices related to the lab procedure, such as sample plate and order of processing. PCs 3, 4, and 5 were not correlated with aggression (absolute r < 0.08).
Epigenome-Wide Association Analyses
To examine the association between aggressive behavior and DNA methylation level, we performed two analyses: (1) An EWAS on aggressive behavior in the entire NTR cohort (N = 2,029 individuals), and (2) an analysis in MZ twin pairs (N = 20 MZ pairs) highly discordant for aggression, where we tested for differences in methylation levels between high- and low-scoring twins.
In the entire NTR cohort, we tested for each methylation site whether DNA methylation level was associated with aggressive behavior using generalized estimation equation (GEE) models, with DNA methylation β-value as outcome and the following predictors: aggression residual score, sex, age at blood sampling, monocyte, eosinophil, and neutrophil percentage values, HM450k array row, and PCs 3, 4, and 5. GEE models were fitted with the R package GEE, with the following specifications: Gaussian link function (for continuous data), 100 iterations, and the ‘exchangeable’ option to account for the correlation structure within families and within persons.
In the discordant MZ twin analysis, paired t-tests were employed to test for methylation differences between the twin scoring higher on aggressive behavior and the twin scoring lower on aggressive behavior. Paired t-tests were performed on residual methylation levels, which were obtained by adjusting the DNA methylation β-values for the same covariates as used in the EWAS on all subjects (sex, age at blood sampling, monocyte, eosinophil, and neutrophil percentage values, HM450k array row, and PCs 3, 4, and 5).
False discovery rate (FDR) q-values were computed with the R package q-value with default settings. The genomic inflation factor (λ) was calculated with the default regression method from the R package GenABEL. In all analyses, an FDR q-value < 0.05 was considered statistically significant. Follow-up analyses, including a test for enrichment of genomic locations and gene ontologies (GOs), were performed based on the output from the EWAS on the entire NTR cohort.
Annotation
Methylation sites were mapped to genomic features and DNase I hypersensitive sites (DHS) as described by Slieker et al. (Reference Slieker, Bos, Goeman, Bovee, Talens, van der Breggen and Heijmans2013). Genomic features included five gene-centric regions: intergenic region (>10 kb from the nearest transcription start site [TSS]), distal promoter (-10 to -1.5 kb from the nearest TSS), proximal promoter (-1.5 kb to +500 bp from the nearest TSS), gene body (+500 bp to 3′ end of the gene), and downstream region (3′ end to +5 kb from 3′ end). Next, CpGs were mapped to CpG islands (CGIs) (CG content > 50%, length > 200 bp, and observed/expected ratio of CpGs > 0.6; locations were obtained from the UCSC genome browser [44]), CGI shore (2-kb region flanking CGI), CGI shelf (2-kb region flanking CGI shore), or non-CGI regions. The locations of DHS, mapped by the ENCODE project (ENCODE Project Consortium, 2012), were downloaded from the UCSC genome browser (Kent et al., Reference Kent, Sugnet, Furey, Roskin, Pringle, Zahler and Haussler2002). Finally, we annotated methylation sites to genes of which the methylation level was previously reported to be associated with aggression in humans: MAOA (Checknita et al., Reference Checknita, Maussion, Labonte, Comai, Tremblay, Vitaro and Turecki2015), SLC6A4 (Wang et al., Reference Wang, Szyf, Benkelfat, Provencal, Turecki, Caramaschi and Booij2012), cytokine genes and their transcription factors (IL6, IL1A, IL8, IL4, IL10, NFKB1, NFAT5, STAT6; Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013), and all genes significant in men and women from Guillemin et al. (Reference Guillemin, Provencal, Suderman, Cote, Vitaro, Hallett and Szyf2014). For each annotation category, a variable with two levels (0 and 1) was created to indicate whether a methylation site mapped to the category (1) or not (0).
Enrichment of Genomic Locations
We developed a custom enrichment analysis method to examine whether certain genomic locations on average showed a stronger association between DNA methylation and aggressive behavior, based on the EWAS test statistics from all genome-wide methylation sites. The following nine categories were tested for being enriched among sites more strongly associated with aggression: gene body, proximal promoter, distal promoter, downstream region, CGI, CGI shore, CGI shelf, DHS, and loci previously associated with aggression (all annotation categories described above, minus two to allow for identification of the model):
 $$\begin{eqnarray}
&&\left( {\frac{{\beta _{{\rm EWAS}} }}{{se_{{\rm EWAS}} }}} \right)_{{\rm CpGi}}^{\rm 2} = {\rm Intercept}\nonumber\\
&&\quad + \beta _{{\rm category}\,{\rm 1}\;} {\bm X}\;{\rm Category}\,1_{{\rm CpGi}} \; \ldots \nonumber\\
&&\quad + \beta _{{\rm category}\,x} \;{\bm X}\;{\rm Category}\,x_{{\rm CpGi}} ,\end{eqnarray}$$
$$\begin{eqnarray}
&&\left( {\frac{{\beta _{{\rm EWAS}} }}{{se_{{\rm EWAS}} }}} \right)_{{\rm CpGi}}^{\rm 2} = {\rm Intercept}\nonumber\\
&&\quad + \beta _{{\rm category}\,{\rm 1}\;} {\bm X}\;{\rm Category}\,1_{{\rm CpGi}} \; \ldots \nonumber\\
&&\quad + \beta _{{\rm category}\,x} \;{\bm X}\;{\rm Category}\,x_{{\rm CpGi}} ,\end{eqnarray}$$
where
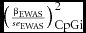 $\big( {\scriptstyle\frac{{\beta _{{\rm EWAS}} }}{{se_{{\rm EWAS}} }}} \big)_{{\rm CpGi}}^{\rm 2}$
represents the squared standardized effect size from the EWAS for methylation site i; βEWAS is the estimate for the association between aggression and methylation level at site i, and se
EWAS is its standard error. βcategoryx
represents the estimate for category x, that is, the change in the EWAS test statistic associated with a one unit change in category x (e.g., being located within a proximal promoter area), and Categoryx
CpGi is a dichotomous variable indicating whether methylation site i maps to category x. To account for differences in variability between methylation sites, we also included the mean and SD of DNA methylation level in the model as covariates. As an additional check, we repeated the analysis restricting to the most variable methylation sites only (SD of the methylation proportion >0.03), which resulted in the same conclusions (results not shown).
$\big( {\scriptstyle\frac{{\beta _{{\rm EWAS}} }}{{se_{{\rm EWAS}} }}} \big)_{{\rm CpGi}}^{\rm 2}$
represents the squared standardized effect size from the EWAS for methylation site i; βEWAS is the estimate for the association between aggression and methylation level at site i, and se
EWAS is its standard error. βcategoryx
represents the estimate for category x, that is, the change in the EWAS test statistic associated with a one unit change in category x (e.g., being located within a proximal promoter area), and Categoryx
CpGi is a dichotomous variable indicating whether methylation site i maps to category x. To account for differences in variability between methylation sites, we also included the mean and SD of DNA methylation level in the model as covariates. As an additional check, we repeated the analysis restricting to the most variable methylation sites only (SD of the methylation proportion >0.03), which resulted in the same conclusions (results not shown).
The regression is weighted by the reciprocal of the fitted values of the variance function (i.e., the predicted values of regressing the squared residuals of regression Equation 1 on the predictors in Equation 1). To account for the fact that the errors in this regression are not normally distributed, Jackknife standard errors were computed. Since the results based on Jackknife standard errors (see Supplementary material) led to the same conclusions as the normal linear regression standard errors, only the standard linear regression estimates are presented in the main text.
In contrast to commonly used enrichment strategies (e.g., chi-square and Fisher's exact test), in which it is tested whether certain genomic annotation groups are enriched within a selected group of methylation sites (e.g., sites significantly associated with a phenotype), our approach uses information from all genome-wide methylation sites to test whether there is a stronger overall signal in certain genomic annotation groups (thus including information from all methylation sites, rather than limiting the analysis to sites that are significantly associated with the phenotype).
Enrichment of Gene Ontology Terms
To test for enrichment of GO terms among methylation sites with a stronger association with aggression, all methylation sites tested were ranked by EWAS p-value, and the resulting ranked gene list (keeping the highest rank for each gene for which multiple CpG sites were measured) was supplied to the online software tool GOrilla (Eden et al., Reference Eden, Navon, Steinfeld, Lipson and Yakhini2009). This tool performs GO enrichment analysis based on gene rank, thus not requiring a p-value cut-off for defining the input list of genes. The background in this analysis consists of all genes for which methylation sites were analyzed in the EWAS. In all analyses, we accounted for multiple testing by controlling FDR. An FDR q-value < 0.05 was considered statistically significant.
Results
Characteristics of NTR Cohort
Data on aggressive behavior scored with the ASR were available for 14,173 individuals who filled out Survey 8 and for 15,535 individuals who filled out Survey 10. The EWAS was performed on 2,059 blood samples from 2,029 subjects (mean age at blood sampling = 36.4 years, SD = 12.4, females = 69.2%), for whom the aggression score closest to the moment of blood draw (from which DNA was extracted) was selected: For 1,906 samples, aggression was assessed after blood draw (range: 0–10 years, mean value = 3.6) and for 153 samples, aggression was assessed before blood draw (range: 0–8 years, mean value = 2.1). The characteristics of the aggression data and EWAS cohort are described in Table 2. The average aggression score of the EWAS cohort was similar to the averages in the entire NTR cohort (mean EWAS cohort = 2.8, mean NTR cohort in survey 8 = 2.9, mean NTR cohort in survey 10 = 2.8). On average, women scored 0.15 SD higher on aggressive behavior compared with men (p survey 8 = 9.6 × 10−19; p survey 10 = 2.3 × 10−20) and with each one-year increase in age, aggression scores decreased on average 0.01 SD (Supplementary Table S1, p survey 8 = 4.2 × 10−81 and P survey 10 = 2.4 × 10−72). The EWAS was performed on the residuals derived after correcting aggression scores for sex and age at completion of survey (for a distribution of aggression data, see Supplementary Figure S1).
TABLE 2 Characteristics of the NTR Aggression Data and the EWAS Group

aIncludes all individuals with 450k methylation data and data on white blood cell counts who were included in the EWAS of the entire NTR cohort. The descriptives are given for samples. For each blood sample, the aggression data closest to blood sampling was selected.
ASR = Adult Self-Report. EWAS = Epigenome-Wide Association Study.
EWAS in the Entire NTR Cohort
For 411,169 autosomal sites in the genome, we tested the association between DNA methylation and (age- and sex-adjusted) aggression score, while correcting for white blood cell counts, age at blood sampling, sex, array row, and three PCs from the methylation data. The quantile–quantile (QQ) plot is shown in Figure 1; λ was 1.047.

FIGURE 1 Quantile-quantile (QQ) plot from the EWAS of aggressive behavior in the entire NTR cohort. The observed p-values (y-axis) are plotted against the p-values expected under the null hypothesis (x-axis). The straight diagonal line denotes the pattern expected under the null hypothesis, with 95% confidence intervals indicated by the shaded grey area.
None of the p-values was below the genome-wide significance threshold of p < 1 × 10−7 (Bonferroni) or FDR q-value < 0.05 (Figure 2). The two highest ranking methylation sites, cg01792876 (p = 7.6 × 10−7, FDR = 0.18) and cg06092953 (p = 9.0 × 10−7, FDR = 0.18), located on chromosomes 8 and 18, respectively, showed a positive relationship between DNA methylation level and aggressive behavior (Supplementary Figure S2). At these CpG sites, an increase of one SD in aggressive behavior (3.1 points) was associated with a methylation change of 0.1% (cg01792876) and 0.9% (cg06092953), respectively, corresponding to 9.1% of the SD of methylation level at cg01792876 and 9.7% of the SD of methylation level at cg06092953. Characteristics of all methylation sites with an EWAS p-value < 1 × 10−5 are provided in Table 3.

FIGURE 2 Manhattan plot showing p-values for the association between aggression score and DNA methylation level in the entire NTR cohort at genome-wide autosomal sites. The horizontal gray line represents the Bonferroni-adjusted p-value threshold.
TABLE 3 Top-Ranking CpG Sites From the EWAS of Aggression in the Entire NTR Cohort

Top hits from the EWAS of aggression (p-value < 1 × 10−5 for the association between methylation level and aggression).
aMean and SD of the methylation proportion (β-value) in the entire NTR cohort.
bEstimate from the regression of methylation proportion on aggression score.
cRobust standard error of the estimate (accounting for the clustering of observations within families).
FDR = False Discovery Rate.
Discordant MZ Twin Analysis
We next performed an alternative EWAS approach, in which we tested for DNA methylation differences at 411,169 sites within 20 MZ pairs discordant for aggressive behavior (Table 2, mean age at blood draw = 34.5 years, SD = 12.0, N [females] = 17). At the survey closest to blood draw, aggressive behavior was on average 9.5 points higher (range: 7–15) in the high-scoring twins compared with the low-scoring twins (Figure 3).

FIGURE 3 Aggression scores of discordant MZ twins. The ASR aggression score at the survey closest to the moment of blood draw is plotted for low-scoring and high-scoring twins of 20 discordant MZ twin pairs. The scores of co-twins are connected by lines. ASR = Adult Self-Report (Achenbach & Rescorla, Reference Achenbach and Rescorla2003).
The within-pair difference in DNA methylation across the genome is plotted for each aggression-discordant pair in Supplementary Figure S3. Screening for large DNA methylation differences, we observed that each aggression-discordant MZ pair had on average 24 methylation sites (range: 0–311), where the within-pair methylation difference was larger than 30% (methylation β-value difference > 0.30), and 291 sites with a difference larger than 15% (range: 39–793; highlighted in Supplementary Figure S3); however, these highly discordant methylation sites were generally twin pair-specific, as has been previously observed in autism-discordant MZ twins (Wong et al., Reference Wong, Meaburn, Ronald, Price, Jeffries, Schalkwyk and Mill2014).
We tested for consistent differences in DNA methylation levels between aggression-discordant MZ twins by applying paired t-tests to genome-wide autosomal methylation sites. The QQ plot from this analysis is shown in Figure 4 (λ = 0.98). No genome-wide significant methylation differences were identified between discordant twins (Figure 5). Characteristics of the top methylation sites from the discordant twin analysis (p-values < 1 × 10−5) are provided in Table 4, and their methylation levels in the high- and low-scoring twins from discordant pairs are plotted in Figure 6.

FIGURE 4 Quantile-Quantile (QQ) plot from the comparison of aggression-discordant MZ twins. The observed p-values (y-axis) are plotted against the p-values expected under the null hypothesis (x-axis). The straight diagonal line denotes the pattern expected under the null hypothesis, with 95% confidence intervals indicated by the shaded gray area.

FIGURE 5 Manhattan plot showing the p-values from paired t-tests comparing DNA methylation level at genome-wide autosomal sites between aggression-discordant twins. The horizontal gray line represents the Bonferroni-adjusted p-value threshold.

FIGURE 6 Methylation levels at the three most significant differentially methylated sites in aggression-discordant MZ twins. The residual DNA methylation β-values (adjusted for covariates) are plotted for low- and high-scoring twins of 20 discordant MZ twin pairs. The DNA methylation levels of co-twins are connected by lines.
TABLE 4 Top-Ranking CpG Sites From the Comparison of Aggression-Discordant MZ Twins

Top hits from the comparison of aggression-discordant MZ twins (p-value < 1 × 10−5 from a paired t-test comparing methylation level in the higher-scoring versus the lower-scoring twins).
aMean and SD of the methylation proportion (β-value) in the entire NTR cohort.
bMean difference in residual methylation level between aggression-discordant twins (aggression high-scoring twin minus aggression low-scoring twin).
FDR = False Discovery Rate.
Enrichment of Signal in Proximal Promoter Regions and DHS
We performed further follow-up analyses based on the results from the EWAS in the entire NTR cohort. Regression of the EWAS test statistics on annotation categories across all genome-wide sites (Table 5 and Supplementary Table S2) revealed a small but significant enrichment of signal in proximal promoter regions (p = 1.5 × 10−7) and DNAse hypersensitivity sites (DHS, p = 1.0 × 10−5). Based on their test-statistic for the association with aggressive behavior, methylation sites in proximal promoters and DHS on average ranked higher than expected (median rank proximal promoter = 203,439, median rank DHS = 203,382, expected median rank under the null hypothesis = 205,585). Sites with a lower mean methylation level also on average showed stronger evidence for being associated with aggressive behavior (p = 9.0 × 10−5). These findings indicate that methylation sites associated with aggressive behavior are enriched in promoter areas and other regions of regulatory active DNA.
TABLE 5 Results From the Regression of EWAS Test Statistics on Genomic Annotation Categories

aMean methylation level of the site.
bStandard deviation of methylation level.
cLoci where DNA methylation level was previously reported to be associated with aggression. Results based on Jackknife are presented in Supplementary Table S2.
Gene Ontology Analysis
Gene ontology enrichment analysis based on EWAS p-value rank (from the analysis in the entire NTR cohort) identified a large number of GO terms that were significantly enriched among higher ranking methylation sites (Supplementary Table S3). The strongest enriched GO term was single-organism developmental process (GO:0044767, p = 4.5 × 10−23), and many other GO terms related to development and metabolism were enriched. The list of significantly enriched GO processes also included many brain or central nervous system processes, such as regulation of neurogenesis (GO:0050767, p = 3.9 × 10−13), axon guidance (GO:0007411, p = 8.0 × 10−10), excitatory post-synaptic potential (GO:0060079, p = 6.9 × 10−6), cognition (GO:0050890, p = 6.0 × 10−4), and behavior (GO:0007610, p = = 6.0 × 10−4); for a complete list of significant GO terms, see Supplementary Table S3. In line with the enrichment of genes connected to brain processes, significantly enriched GO components included many neuron-specific components; for example, axon part (GO:0033267, p = 2.3 × 10−6), dendrite (GO:0030425, p = 1.6 × 10−5), post-synaptic density (GO:0014069, 9.5 × 10−5), and pre-synaptic active zone (GO:0048786, p = 5.5 × 10−4). The most strongly enriched GO component was cell junction (GO:0030054, p = 7.4 × 10−13).
Discussion
We performed an EWAS to identify genomic locations where DNA methylation level is associated with aggressive behavior in a population-based cohort of adults. No genome-wide significant methylation hits were identified after correction for multiple testing. GO analysis, in which categories of genes rather than single methylation sites are tested, highlighted that genes involved in developmental and central nervous system processes are enriched among higher-ranking genes from our EWAS. Here we describe our two top-ranked CpGs based on the entire NTR cohort, which have an FDR of 0.18. Our top CpG, cg01792876 (genomic location: chr8; 116,684,801) is located near the trichorhinophalangeal syndrome I (TRPS1) gene. This gene encodes a zinc finger transcription factor that represses GATA-regulated genes, and mutations in this gene are linked to tricho-rhino-phalangeal syndromes I–III, which are characterized by distinctive craniofacial and skeletal malformations (ONIM 190350, ONIM 150230, and ONIM 190351). Interestingly, the region also harbors a suggestive SNP association for major depressive disorder (rs2721937, chr8: 116,702,874) based on the genome-wide association study meta-analysis from the PGC consortium published in 2013 (Ripke et al., Reference Ripke, Wray, Lewis, Hamilton, Weissman, Breen and Sullivan2013). TRPS1 is also linked to a whole range of other complex traits based on genome-wide association studies (UCSC genome browser, accessed on August 17, 2015). ENCODE transcription factor binding site (TFBS) ChIP-seq data (Mar 2012 Freeze, accessed through the UCSC genome browser) indicate that our top CpG, cg01792876, is located within the binding sites of transcription factors FOXA1, GATA3, and EP300.
The second-ranked CpG site from our EWAS (cg06092953, genomic location: chr18; 77,905,699) is located between the non-coding RNA PARD6G antisense RNA 1 (PARD6G-AS1) and the activity-dependent neuroprotective protein 2 gene (ADNP2). ADNP2 is ubiquitously expressed in mice tissues, but especially highly expressed in the brain (particularly in the cerebral cortex), and is thought to play a role in cellular survival pathways (Kushnir et al., Reference Kushnir, Dresner, Mandel and Gozes2008). ADNP2 expression has been linked to schizophrenia (Dresner et al., Reference Dresner, Agam and Gozes2011) and to the action of the mood-stabilizing drug lithium that is prescribed in bipolar disorder (McEachin et al., Reference McEachin, Chen, Sartor, Saccone, Keller, Prossin and McInnis2010). Our second-ranked site cg06092953 overlaps with the binding sites of a number of transcription factors (ENCODE TFBS ChIP-seq data Mar 2012 Freeze).
In the comparison of DNA methylation levels within aggression-discordant MZ twin pairs, we also found no genome-wide significant methylation differences, but high-ranking sites may nevertheless be of potential interest. Here we describe the sites with a p-value < 10−5 (FDR = 0.99, N = 3). The effect sizes of these sites range from an average within-pair difference in DNA methylation percentage of 0.8% to 1.8% (Table 4). The top site (cg21557159, chr 11: 107,795,699, p = 5.7 × 10−6) is located upstream of the RAS oncogene family 39 gene (RAB39/RAB39A). RAB39 is involved in cellular endocytosis, and is considered to be ubiquitously expressed in human tissues (Chen et al., Reference Chen, Han, Yang, Zhang, Li, Wan and Cao2003). In a neuronal cell line from mice, this gene was shown to play a role in cell differentiation, with knock-down of Rab39A resulting in a decrease in the number of neurites formed (Mori et al., Reference Mori, Matsui, Omote and Fukuda2013). The second-ranked CpG site, cg08648367 (chr 19; 51,925,472, p = 7.6 × 10−6) overlaps the binding sites of a number of transcription factors near the sialic acid-binding Ig-like lectin 10 gene (SIGLEC10). The encoded Siglec-10 protein is involved in regulating immune responses induced by tissue damage (Chen et al., Reference Chen, Tang, Zheng and Liu2009). The third-ranked CpG site, cg14212412 (chr6; 105,918,992, p = 8.0 × 10−6) is located near the prolyl endopeptidase (PREP) gene. Prolyl endopeptidase is a proteinase that cleaves small neuropeptides and peptide hormones, such as angiotensin, thyrotropin-releasing hormone, gonadotropin-releasing hormone, neurotensin, vasopressin, and oxytocin (Garcia-Horsman et al., Reference Garcia-Horsman, Mannisto and Venalainen2007). A recent study reported significant associations between serum prolyl endopeptidase (protein) level and problem behavior scored by the Child Behavior Check List (CBCL) delinquent, aggressive, externalizing, and internalizing behavior subscales (Frenssen et al., Reference Frenssen, Croonenberghs, Van den Steene and Maes2015). Abnormal PREP activity has been also linked to several neurological and psychiatric conditions, including depression, mania, and schizophrenia (Maes et al., Reference Maes, Goossens, Scharpe, Calabrese, Desnyder and Meltzer1995), autism spectrum disorders (Momeni et al., Reference Momeni, Nordstrom, Horstmann, Avarseji and Sivberg2005), neurodegenerative diseases such as Alzheimer's disease (Mantle et al., Reference Mantle, Falkous, Ishiura, Blanchard and Perry1996), and to the aggregation of α-synuclein in Parkinson's disease (Myohanen et al., Reference Myohanen, Hannula, Van, Gerard, Van, Garcia-Horsman and Lambeir2012).
A cross-comparison of results from different analyses showed that the two top hits based on the entire study showed the same direction of effect in discordant MZ pairs: cg01792876, near TRPS1 showed a mean difference of 0.4% between aggression-discordant MZ twins (p = .09) and cg06092953 (nearest gene PARD6G-AS1), a mean difference of 1.9% (p = .24). The other way around, two of the three most significant sites in discordant MZ pairs showed the same direction of effect in the entire NTR cohort: cg08648367, near SIGLEC10, β EWAS = 2.6 × 10−5, p = .59, and cg14212412, near PREP, β EWAS = 3.3 × 10−4, p = 3.1 × 10−3. Finally, we also looked up the heritability of the top methylation sites in our dataset to gain insight into the extent to which they are influenced by genetic and environmental influences (van Dongen et al., under review). Each of the three most significant sites from the comparison of discordant MZ twins had a low heritability: cg21557159, heritability = 10%; cg08648367, heritability = 7%; and cg14212412, heritability = 0%; indicating that most of the variation at these methylation sites is caused by individual-specific environmental and/or stochastic effects. The top hit based on the overall NTR cohort, cg01792876, also had a heritability of 0% in our data, while the second hit cg0609295 was strongly influenced by genetic effects, with an MZ twin correlation of 0.67, and a DZ twin correlation of 0.15.
We limited this study to aggressive behavior only and did not consider other aspects of behavior or psychiatric symptoms. Since aggressive behavior is connected to a range of comorbidities in psychiatric and neurodegenerative disease domains, DNA methylation levels associated with aggressive behavior may to some extent be related to other comorbid conditions of aggression (e.g., depression and substance abuse). Importantly, aggressive behavior as scored on a continuous scale by the ASR by definition overlaps to a certain extent with other psychiatric dimensions. For instance, the ASR aggression scale includes items that score mood-related symptoms (e.g., ‘My moods swing between elation and depression’), which are considered to be part of the aggression phenotype in adults. Yet, for this reason, it is possible that our EWAS results to some extent reflect methylation sites related to mood. To distinguish between methylation sites primarily associated with aggression and methylation sites related to other aspects of psychiatric symptomatology, the ideal psychiatric EWAS approach would involve an elaborate analysis of multiple symptom dimensions of aggressive behavior and comorbidities (e.g., substance abuse, depression). Such an approach would give insight into which methylation sites are common to multiple psychiatric symptoms, which are specific to certain symptoms, and which are mainly reflective of effects of comorbid exposures.
While we did not identify genome-wide significant methylation hits for aggressive behavior in whole blood, a previous much smaller study reported numerous methylation hits for aggressive behavior based on a study of individual cell populations (monocytes and T-cells) extracted from blood samples of adults who had shown a trajectory of chronic physical aggression in childhood (Guillemin et al., Reference Guillemin, Provencal, Suderman, Cote, Vitaro, Hallett and Szyf2014; Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013). Three studies of candidate genes, including MAOA (Checknita et al., Reference Checknita, Maussion, Labonte, Comai, Tremblay, Vitaro and Turecki2015), SLC6A4 (Wang et al., Reference Wang, Szyf, Benkelfat, Provencal, Turecki, Caramaschi and Booij2012), and genes encoding cytokines and their transcription factors (Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013), also reported associations between DNA methylation level and aggression or antisocial personality disorder. We had data on 694 CpGs in or near 35 genes identified in these previous studies (Checknita et al., Reference Checknita, Maussion, Labonte, Comai, Tremblay, Vitaro and Turecki2015; Guillemin et al., Reference Guillemin, Provencal, Suderman, Cote, Vitaro, Hallett and Szyf2014; Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013; Wang et al., Reference Wang, Szyf, Benkelfat, Provencal, Turecki, Caramaschi and Booij2012). After correcting for the number of loci (35), one site was significantly replicated in our EWAS (cg14260773, chr2: 33,226,136, in LTBP1, p = 8.01 × 10−4) and eight additional sites, in or near the genes PCDH8, UCK2, IL4, TGIF1, PCDHB2, TGIF1, AGTR1, and NDUFS2, had a nominal p-value in our EWAS (p < .01, Supplementary Table S4). Possible explanations for differences between our findings and those from previous studies may include differences in the technique used to measure methylation, differences in the examined cell populations, but perhaps most importantly, differences in the study populations. While we examined the relationship between DNA methylation and a continuously scored measure of aggression in an unselected population-based adult cohort of adults, the previous studies focused on much more extreme cohorts. The MAOA gene was studied in a prisoner population (Checknita et al., Reference Checknita, Maussion, Labonte, Comai, Tremblay, Vitaro and Turecki2015) and the other previous studies included individuals who had shown a chronic physical aggression trajectory in childhood, while the relationship between DNA methylation and aggression in adulthood was not examined (Guillemin et al., Reference Guillemin, Provencal, Suderman, Cote, Vitaro, Hallett and Szyf2014; Provencal et al., Reference Provencal, Suderman, Caramaschi, Wang, Hallett, Vitaro and Szyf2013, Reference Provencal, Suderman, Guillemin, Vitaro, Cote, Hallett and Szyf2014; Wang et al., Reference Wang, Szyf, Benkelfat, Provencal, Turecki, Caramaschi and Booij2012). Aggressive behavior scored with the ASR in the NTR cohort is highly left-skewed, with very few people scoring high on aggression, and very few people responding positively to the physical aggression items (e.g., ‘I physically attack people’). Future EWASs on aggression in the general population may benefit from focusing on childhood or adolescent ages, where variation in aggressive behavior is more pronounced. Moreover, longitudinal studies with repeated measures of DNA methylation and aggression, analysis strategies such as Mendelian randomization, and analyses of DNA methylation in relevant brain regions (taking into account genetic variation) are required to unravel whether aggression-associated methylation changes in a peripheral tissue such as whole blood are related to the cause or consequence of aggression (Mill & Heijmans, Reference Mill and Heijmans2013). Once genome-wide association studies of aggressive behavior have identified genetic risk variants for aggression, future studies integrating genetic information (e.g., SNP data), and epigenetic information (e.g., methylation data) are warranted to unravel how the interplay of genetic risk variants and epigenetic mechanisms gives rise to aggressive behavior.
Acknowledgments
We would like to thank the twins and their family members who participated in the NTR studies. We thank Hill Fung Ip for writing the Jackknife software that we used to test for the enrichment of genomic annotations. This work was supported by ACTION. ACTION receives funding from the European Union Seventh Framework Program (FP7/2007–2013) under grant agreement No. 602768. This study was also supported by the BBRMI-NL-financed BIOS Consortium (NWO 184.021.007) and by the European Research Council (ERC): ERC-230374: Genetics of mental illness, a lifespan approach to the genetics of childhood and adult neuropsychiatric disorders and comorbid conditions, and ERC-284167: Beyond the genetics of addiction. MV is supported by Royal Netherlands Academy of Science Professor Award (PAH/6635) to DIB.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/thg.2015.74.













