


Venlafaxine extended-release (ER) is a serotonin–noradrenaline reuptake inhibitor with little or no affinity for muscarinic, histaminergic or adrenergic receptors (Reference Muth, Haskins and MoyerMuth et al, 1986). Its efficacy, safety and tolerability in treating depression (Reference Smith, Dempster and GlanvilleSmith et al, 2002), generalised anxiety disorder (Reference Gelenberg, Lydiard and RudolphGelenberg et al, 2000) and social anxiety disorder (Reference Allgulander, Mangano and ZhangAllgulander et al, 2004) have been well established. Small open-label and double-blind studies with venlafaxine immediate release (IR) have suggested that it is effective in the treatment of panic disorder (Reference Pollack, Worthington and OttoPollack et al, 1996). This randomised, double-blind, placebo-controlled trial further examines the safety and efficacy of venlafaxine ER in out-patients with panic disorder.
METHOD
Study design
This was a randomised, double-blind, parallel-group study conducted at 50 sites in Canada, Europe and South Africa comparing flexible-dose treatment with venlafaxine ER and placebo in adult outpatients who met the criteria for panic disorder with or without agoraphobia according to the Diagnostic and Statistical Manual of Mental Disorders (4th edn, DSM–IV; American Psychiatric Association, 1994). The institutional review board for each site and the regulatory agencies for each country approved the protocol and written informed consent was received from all participants before randomisation.
Sample selection
Eligible out-patients were men and women at least 18 years of age who met DSM–IV criteria for panic disorder for at least 6 months before study day 1. Confirmation of the primary diagnosis of panic disorder and exclusion of other psychiatric diagnoses such as major depressive disorder and generalised anxiety disorder was made using the DSM–IV and the modified Mini-International Neuropsychiatric Interview (Reference Sheehan, Lecrubier and SheehanSheehan et al, 1998). Participants were required to have a Clinical Global Impression – Severity (CGI–S) (Reference GuyGuy, 1976) score ≥4, a minimum of four or more full-symptom panic attacks during the 4 weeks before the screening visit, and at least two full-symptom panic attacks during the 14±3-day placebo lead-in period between the screening visit and baseline (study day –1) to be eligible for the study. For the Panic and Anticipatory Anxiety Scale (PAAS; Reference SheehanSheehan, 1983), participants were given diary cards to record daily details regarding their panic attack frequency (including symptoms, situational or unexpected) and anticipatory anxiety, and the investigator then completed the scale after interview to verify the information in the diary.
Individuals were excluded if they met diagnostic criteria for any clinically important Axis I or Axis II disorder, current or predominant, within 6 months of study day 1, or if they had a history of alcohol dependence or misuse (as defined in DSM–IV) within 1 year of study day 1. Those with a 17-item Hamilton Rating Scale for Depression (HRSD17; Reference HamiltonHamilton, 1960) total score ≥15, an HRSD17 item 1 (depressed mood) score >2, a Covi Anxiety Scale (Reference LipmanLipman, 1982) total score less than or equal to their Raskin Depression Scale (Reference LipmanLipman, 1982) total score, or a Raskin Depression Scale total score >9 or single-item score >3 at screening were also excluded.
Individuals were excluded if they had received treatment with venlafaxine IR or ER within 6 months of study day 1, if they had taken investigational drugs, antipsychotics or fluoxetine, or had regularly used benzodiazepines or triptans within 30 days of study day 1; had taken other antidepressants, monoamine oxidase inhibitors, non-benzodiazepine anxiolytics, or other psychopharmacological drugs (including lithium, stimulants, or sedative-hypnotics or herbal products intended to treat anxiety or depression) within 14 days of study day 1; had undergone investigational procedures within 30 days of study day 1 or electroconvulsive therapy within 60 days; had taken non-psychopharmacological drugs with psychotropic effects unless they had been maintained at a stable dose for at least 3 months before study day 1 and the patient was expected to continue taking the drug without dose changes throughout the study; or had initiated or changed the intensity of formal psychotherapy or cognitive–behavioural therapy within 30 days of study day 1.
Other reasons for exclusion were the presence of clinically significant abnormal findings on laboratory tests, electrocardiogram (ECG), vital signs or physical examination; a history or presence of clinically important medical conditions; and, in women of childbearing potential, pregnancy, lactation or not using a medically acceptable form of contraception. A positive test result for any drug of misuse at the screening visit required either immediate exclusion or discussion with the medical monitor as to whether a negative result of a retest was acceptable for inclusion in the study. All concomitant treatments prohibited before study day 1 were also prohibited during the study.
Drug administration
After a 14±3-day single-blind, placebo lead-in period, participants were randomly assigned to receive double-blind venlafaxine ER or placebo for up to 10 weeks, followed by a taper period of up to 14 days (which could be omitted or prolonged if clinically indicated). Participants returned for a post-study evaluation 4–10 days after taking the last dose of study medication. A 37.5 mg dose was used for the first 4 days of treatment and then increased to 75 mg. If clinically indicated, the dose could be increased to 150 mg venlafaxine ER (or two placebo capsules) after day 14 and to a maximum of 225 mg venlafaxine ER (or three placebo capsules) after day 21. Dosage decreases were permitted at any time during the study to improve tolerance at the discretion of the investigator, but after study day 7, the minimum daily dose allowed was one capsule in the morning (75 mg venlafaxine ER or placebo). Those unable to tolerate 75 mg venlafaxine ER were withdrawn from the study.
Efficacy assessments
The primary efficacy assessment, the PAAS, was administered at the screening visit and on study days –1, 7, 14, 21, 28, 42, 56 and 70, along with the CGI–S and the Phobia Scale (Reference SheehanSheehan, 1983). The Clinical Global Impression – Improvement (CGI–I; Reference GuyGuy, 1976) scale was assessed starting on study day 7 and at each time point thereafter. The final on-therapy evaluation for each measure was performed within 3 days of the last full dose of study medication (before taper).
The primary outcome measure was the percentage of participants who were free from full-symptom panic attacks (≥4 symptoms) at the on-therapy evaluation. The post-baseline full-symptom panic attack frequency from the PAAS was evaluated in 2-week (14-day) periods, and for the PAAS, the on-therapy evaluation included the last 14 days of data collected during the on-therapy period.
Secondary outcome measures comprised the change from baseline in fullsymptom panic attack frequency (based on the PAAS), response and remission. Response to treatment was defined as a CGI–I score of 1 (very much improved) or 2 (much improved). Remission was defined using the CGI–I and CGI–S. Patients were considered to be in remission at a given evaluation evaluation using the CGI–I if they had zero full-symptom panic attacks on the PAAS and a CGI–I score of 1 (very much improved) at any 2-week evaluation and at the on-therapy evaluation. Using the CGI–S, patients were considered to be in remission at a given evaluation if they had zero full-symptom panic attacks on the PAAS and a CGI–S score of 1 or 2 (1=normal, not at all ill; 2=borderline mentally ill) at any 2-week evaluation and at the on-therapy evaluation. Other secondary efficacy measures analysed at each 2-week time point or at the time of discontinuation included anticipatory anxiety and limited-symptom panic attacks (both measures from the PAAS), the CGI–S and CGI–I, the Phobia Scale fear and avoidance factors, and the Covi Anxiety Scale.
At baseline and on study day 70, participants were evaluated on the Quality of Life Enjoyment and Satisfaction Questionnaire (Q–LES–Q; Reference Endicott, Nee and HarrisonEndicott et al, 1993), the Sheehan Disability Scale (Reference SheehanSheehan, 1983) and an exploratory resource utilisation in panic disorder (RUPD) assessment comprising a series of single-item responses measuring unscheduled use of various healthcare services.
Safety and tolerability assessments
All ‘treatment-emergent’ adverse events were reported, including those not considered to be drug related and those that occurred during the taper/post-study period. Safety assessments were based on reports of adverse events and results of routine physical examinations, measurements of vital signs, laboratory determinations, and ECG. Vital signs (supine and standing blood pressure and supine pulse) were evaluated at screening and baseline, at each visit during the double-blind period, and at the post-study visit. Weight was evaluated at baseline (study day –1) and on study day 70. Electrocardiogram recordings were obtained at screening and on study day 70. Laboratory investigations were performed at screening and on study day 70, and included haematology, blood chemistry, free thyroxine index (screening visit only), urine drug screen, urinalysis and a serum beta-human chorionic gonadotropin pregnancy test for women of childbearing potential (at the screening visit and any time that pregnancy was suspected).
If a participant left the study before the end of the double-blind period (study day 70±3 days), all of the above assessments were performed on the last day on which the individual took the full dose of study medication (before taper) or as soon as possible thereafter. The post-study evaluations were performed 4–10 days after the participant took the last dose of double-blind or taper study medication.
Statistical analyses
Statistical analyses of efficacy measures were performed on the intent-to-treat (ITT) population using last-observation-carried-forward (LOCF) and final on-therapy values. The ITT population included participants who had a baseline PAAS evaluation, took at least one dose of study medication and had at least 7 days of PAAS data during the double-blind period, and had at least one on-therapy double-blind evaluation for the primary efficacy variable during visits on days 7–70 or within 3 days of stopping the study medication, before taper. The safety population consisted of those who completed the pre-study period and took at least one dose of randomly assigned study medication under double-blind conditions.
The percentage of those free from PAAS full-symptom panic attacks was analysed using logistic regression with treatment group and site as factors (sites with five or fewer patients were combined). The median change in panic attack frequency and anticipatory anxiety data from the PAAS were analysed with the Mann–Whitney U-test (Wilcoxon rank sum test). The remission and CGI–I responder data were analysed by the Fisher exact test. The Phobia Scale, Covi–Raskin, HRSD, CGI–I and CGI–S data were analysed by analysis of covariance (ANCOVA) with treatment and site as the factors and the baseline scores as covariates.
The sample size computation was based on estimates from the literature: the percentage of those free from panic attacks was expected to be roughly 50% in the venlafaxine-treated group compared with 30% in the placebo-treated group. It was also estimated that 140 ITT participants per treatment arm would be needed to provide a 90% power for a two-sided test at the 0.05 significance level. To compensate for about 15% of participants expected not to qualify for the ITT criteria, the planned enrolment was 165 per treatment group.
Improvement on each domain of the Sheehan Disability Scale was defined as reduction from baseline and analysed using ANCOVA with treatment as the main effect and centre and baseline score as covariates. The Q–LES–Q was evaluated as improvement (increase) from baseline on each domain using ANCOVA with treatment as the main effect and the baseline score and centre as covariates. The RUPD data were analysed using a χ2-test with treatment group as the exposure variable and any use of a specific type of healthcare service versus no use of that healthcare service as the dichotomous outcome variable.
RESULTS
Disposition of participants
Of 531 individuals screened, 361 were randomly assigned to the venlafaxine ER or placebo groups; of these, 355 were included in safety analyses and 328 were included in efficacy analyses (Fig. 1). A total of 265 participants completed the 10 weeks of the double-blind period. Treatment-emergent adverse events were the most common reason for withdrawal in the venlafaxine ER group (16 participants, 9%) and lack of efficacy was the most common reason for withdrawal in the placebo group (17 participants, 10%). There were no significant differences between treatment groups in primary reasons for withdrawal during the double-blind period. The mean daily doses of venlafaxine ER for those in the ITT population ranged from 114.8±36.9 mg at week 3 to 162.9±60.6 mg at week 10, and no individual received a mean daily dose greater than 200 mg.
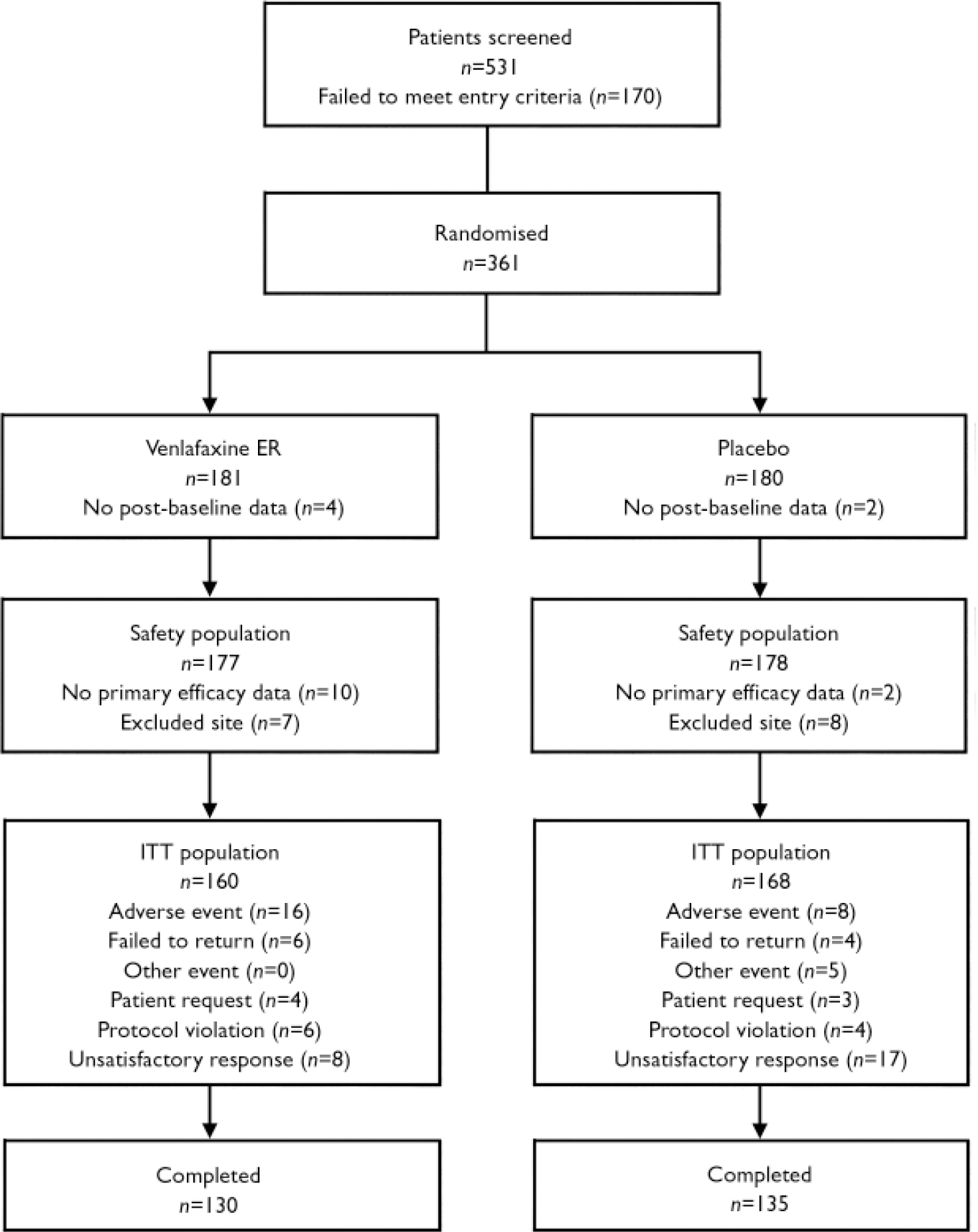
Fig. 1 Study flow chart. Data from one site (n=15) were excluded from the intent-to-treat (ITT) population because of poor compliance with good clinical practices. Inclusion of these15 individuals in the efficacy analyses, however, did not change the results obtained using the modified ITT population of 328 reported here. Of note, 49 participants had major protocol violations identified by the sponsor before unmasking. Efficacy analyses performed on the per-protocol population, excluding those with protocol violations that might affect the study results, did not differ significantly from the analyses for the modified ITT population. Therefore, only the results of the efficacy analyses with the modified ITT population described above are reported. Completers were defined as those in the ITT population whose last Panic and Anticipatory Anxiety Scale evaluation was at least 9 weeks after the first.
Participant characteristics and baseline severity
Most baseline and demographic characteristics were similar between the venlafaxine ER and placebo groups (Table 1), although there was a higher mean frequency of full-symptom panic attacks at baseline for the venlafaxine ER group compared with the placebo group (12.5 v. 9.5, respectively; P=0.078).
Table 1 Baseline and demographic characteristics1,2
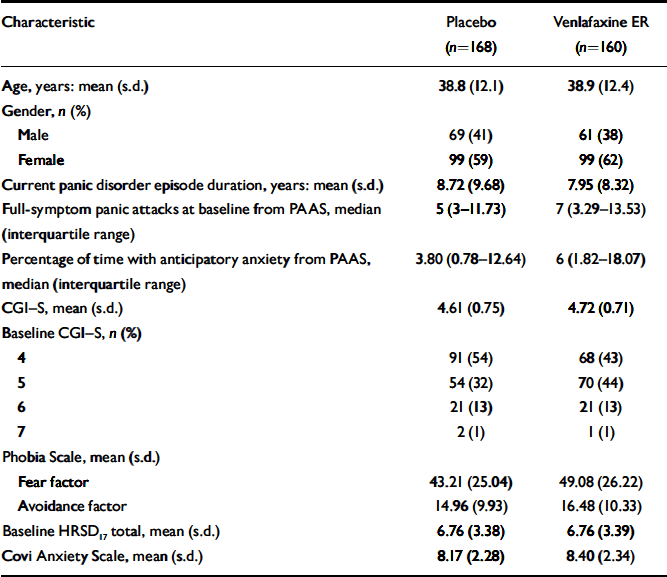
| Characteristic | Placebo (n=168) | Venlafaxine ER (n=160) |
|---|---|---|
| Age, years: mean (s.d.) | 38.8 (12.1) | 38.9 (12.4) |
| Gender, n (%) | ||
| Male | 69 (41) | 61 (38) |
| Female | 99 (59) | 99 (62) |
| Current panic disorder episode duration, years: mean (s.d.) | 8.72 (9.68) | 7.95 (8.32) |
| Full-symptom panic attacks at baseline from PAAS, median (interquartile range) | 5 (3-11.73) | 7 (3.29-13.53) |
| Percentage of time with anticipatory anxiety from PAAS, median (interquartile range) | 3.80 (0.78-12.64) | 6 (1.82-18.07) |
| CGI—S, mean (s.d.) | 4.61 (0.75) | 4.72 (0.71) |
| Baseline CGI—S, n (%) | ||
| 4 | 91 (54) | 68 (43) |
| 5 | 54 (32) | 70 (44) |
| 6 | 21 (13) | 21 (13) |
| 7 | 2 (1) | 1 (1) |
| Phobia Scale, mean (s.d.) | ||
| Fear factor | 43.21 (25.04) | 49.08 (26.22) |
| Avoidance factor | 14.96 (9.93) | 16.48 (10.33) |
| Baseline HRSD17 total, mean (s.d.) | 6.76 (3.38) | 6.76 (3.39) |
| Covi Anxiety Scale, mean (s.d.) | 8.17 (2.28) | 8.40 (2.34) |
Efficacy
At the final on-therapy evaluation, the primary end point, 55.0% of venlafaxine ER- and 52.4% of placebo-treated participants were free from full-symptom panic attacks, a statistically non-significant difference (Fig. 2). The median reduction from baseline in full-symptom panic frequency was significantly greater for the venlafaxine ER group (Fig. 3). A significantly higher proportion of the venlafaxine ER group responded to treatment (CGI–I score of 1 or 2), beginning at week 3 (Fig. 4) and experienced CGI–I remission, beginning at week 6 (Fig. 5). Improvement in fear and avoidance factors of the Phobia Scale was significantly greater for the venlafaxine ER group than for the placebo group, beginning at week 6 and continuing through the remainder of the study, with the exception of week 8 for the avoidance factor (Fig. 6). Venlafaxine ER was associated with significantly greater improvement than placebo in observed scores for the ‘work’ and ‘social life/leisure activities’ domains of the Sheehan Disability Scale, but not ‘family life and home responsibilities’ (Fig. 7). A significantly greater proportion of venlafaxine ER-treated participants were free from limited-symptom panic attacks at the final on-therapy evaluation compared with those who received placebo, and the venlafaxine ER group showed significantly greater improvement in adjusted mean CGI–S scores, CGI–I total scores and Covi Anxiety Scale scores, median time spent experiencing anticipatory anxiety, and Q–LES–Q ‘overall life satisfaction’ (Table 2).
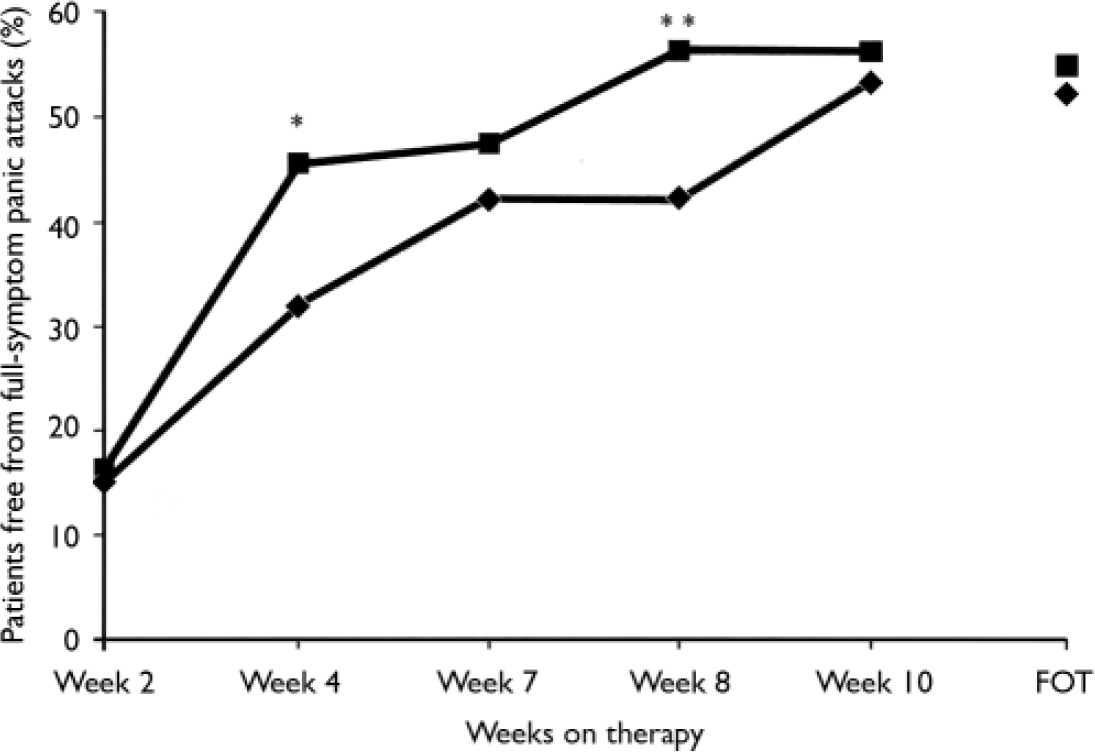
Fig. 2 Percentage of patients free from full-symptom panic attacks (intent-to-treat population, last-observation-carried-forward analysis) measured by the Panic and Anticipatory Anxiety Scale (during each preceding 2-week period). χ2 P values obtained from logistic regression model logit (response)=treatment+centre; *P<0.05, **P<0.01 venlafaxine ER v. placebo; FOT, final on-therapy evaluation; –♦–, placebo (n=168); –▪–, venlafaxine ER (n=160).
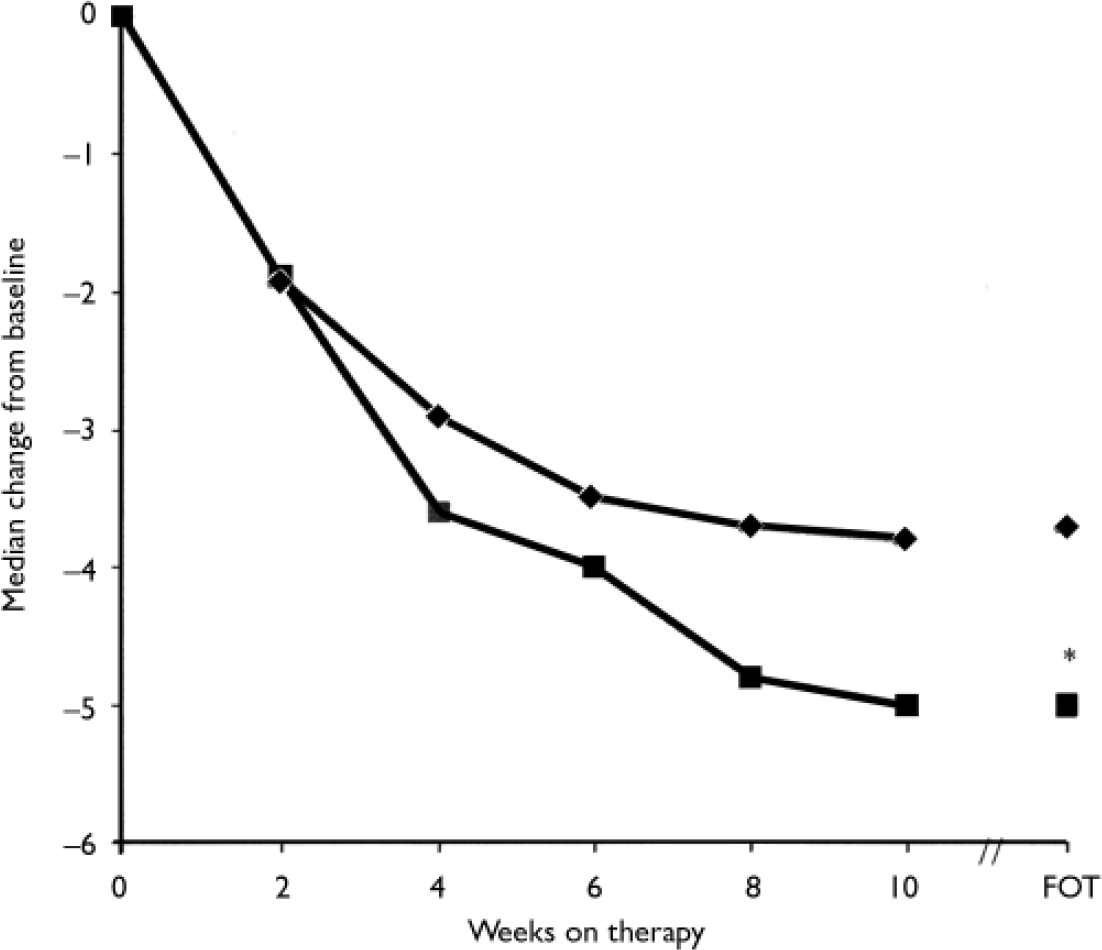
Fig. 3 Panic attack frequency (intent-to-treat population, last-observation-carried-forward analysis) measured by the Panic and Anticipatory Anxiety Scale (PAAS) (during each preceding 2-week period). Wilcoxon rank sum test on change from baseline for PAAS full-symptom panic attacks. P obtained from Z approximation to Wilcoxon statistics on difference from baseline; *P<0.05 venlafaxine ER v. placebo; FOT, final on-therapy evaluation; –♦–, placebo (n=168); –▪–, venlafaxine ER (n=160).
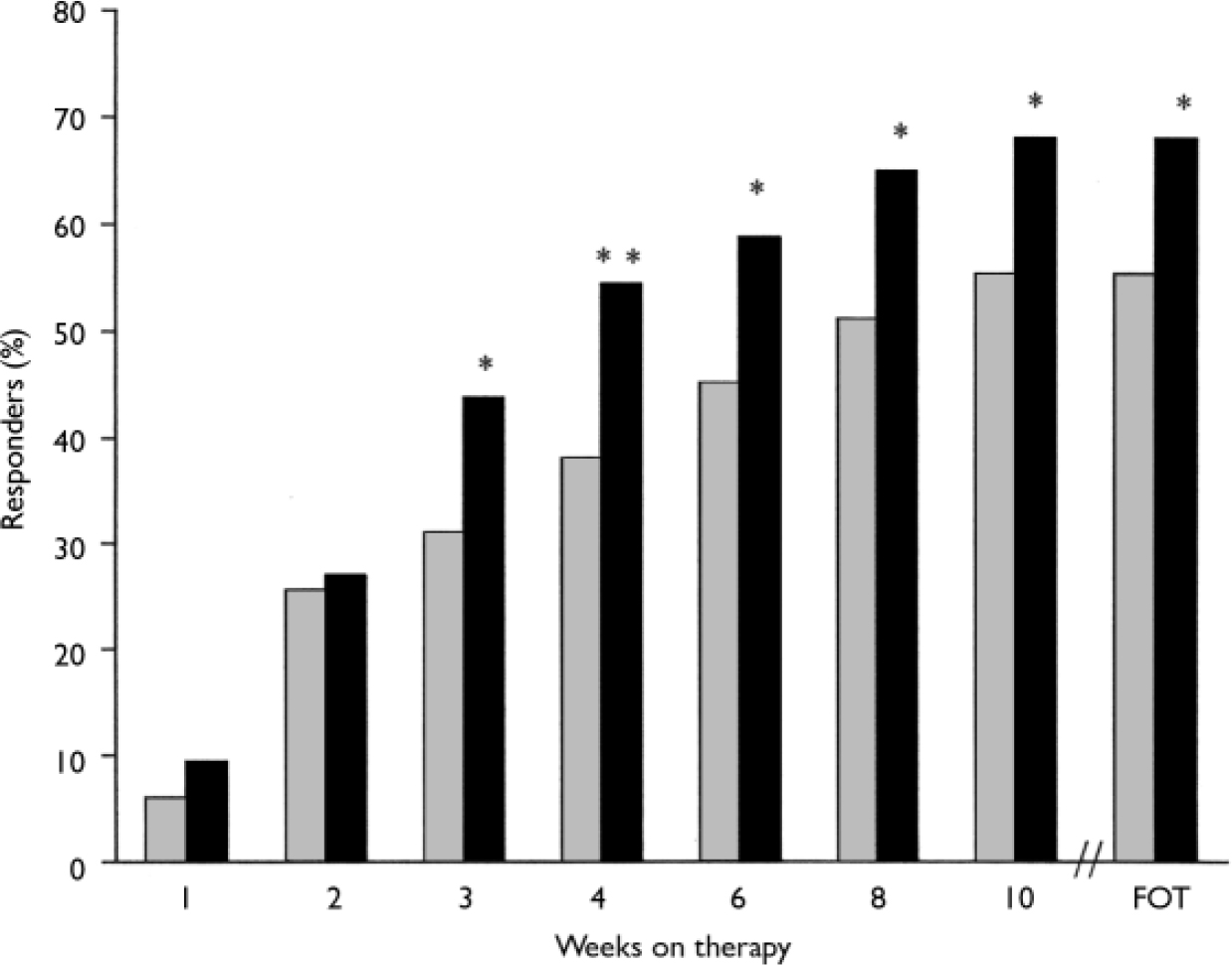
Fig. 4 Response rates (Clinical Global Impression – Improvement=1 or 2; intent-to-treat population, last-observation-carried-forward analysis). P obtained from Fisher's exact test; *P<0.05, **P<0.01 venlafaxine ER v. placebo; FOT, final on-therapy evaluation; ![]() , placebo (n=168);▪, venlafaxine ER (n=160).
, placebo (n=168);▪, venlafaxine ER (n=160).
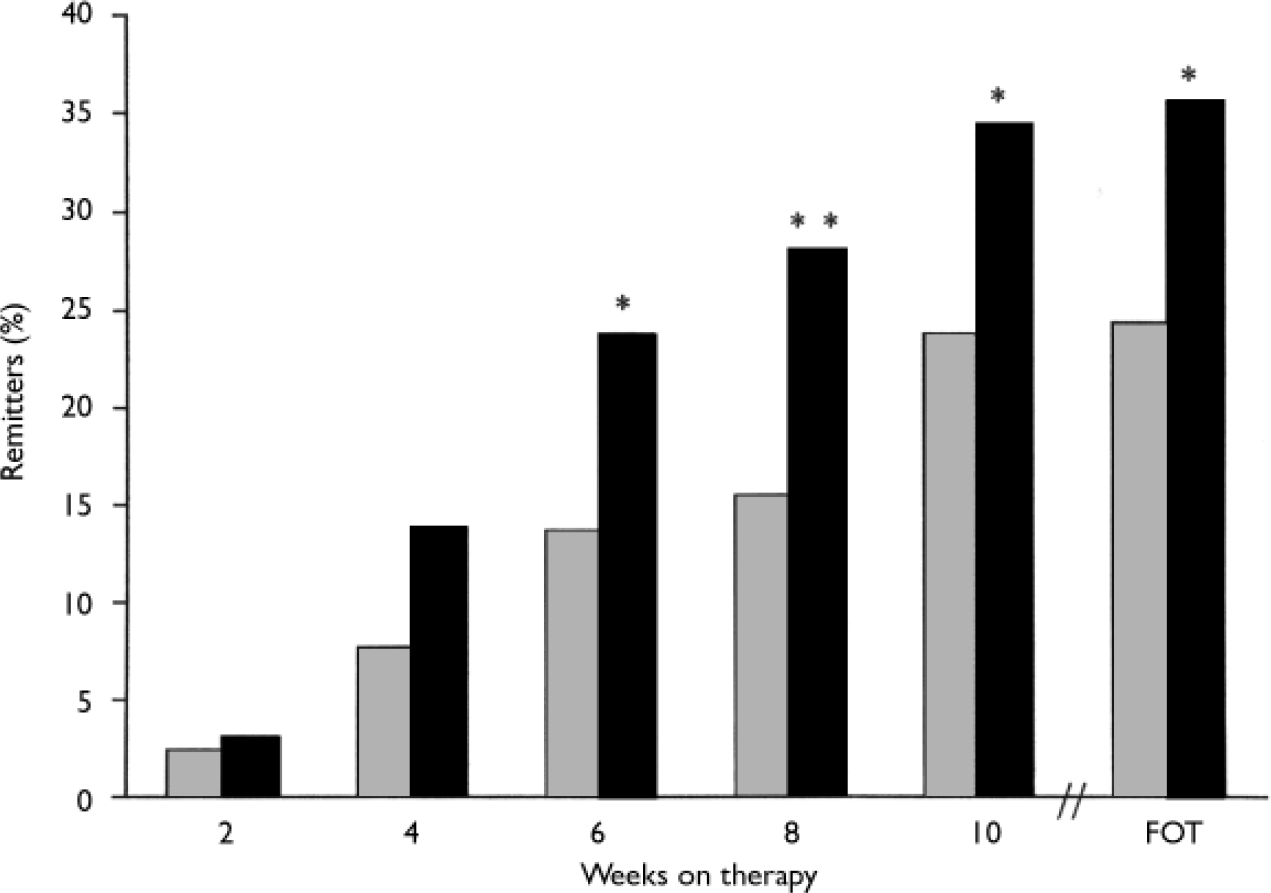
Fig. 5 Remission rates (Clinical Global Impression – Improvement=1 and panic free; intent-to-treat population, last-observation-carried-forward analysis); full-symptom panic attacks measured by the Panic and Anticipatory Anxiety Scale. P obtained from Fisher's exact test; *P<0.05, **P<0.01 venlafaxine ER v. placebo; FOT, final on-therapy evaluation; ![]() , placebo (n=168); ▪, venlafaxine ER (n=160).
, placebo (n=168); ▪, venlafaxine ER (n=160).
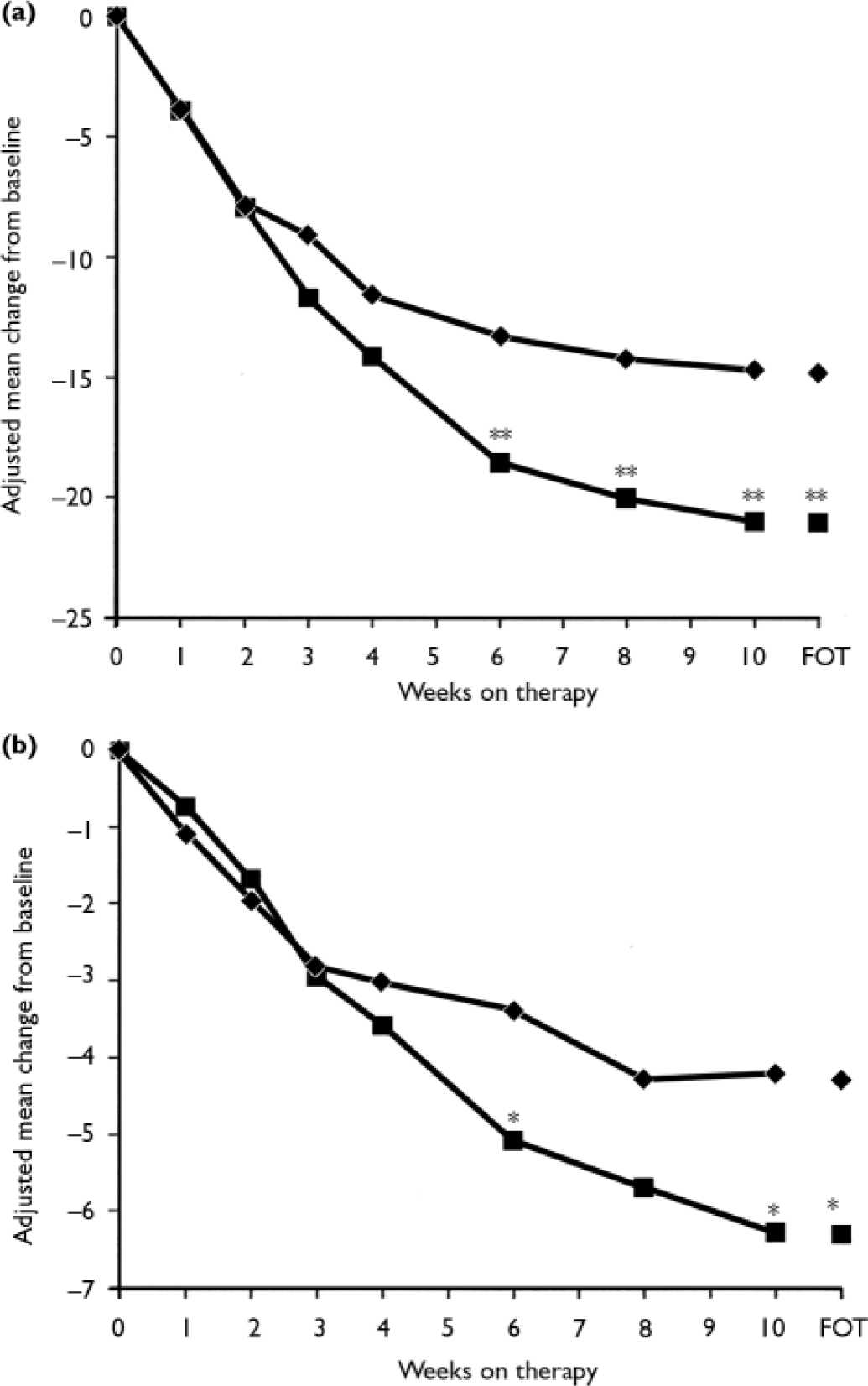
Fig. 6 Phobia scale fear (a) and avoidance (b) factors (intent-to-treat population, last-observation-carried-forward analysis). P obtained from ANCOVA model: change from baseline=baseline+treatment+centre; *P<0.05, **P<0.01 venlafaxine ER v. placebo; FOT, final on-therapy evaluation; –♦–, placebo (n=167); –▪–, venlafaxine ER (n=160).
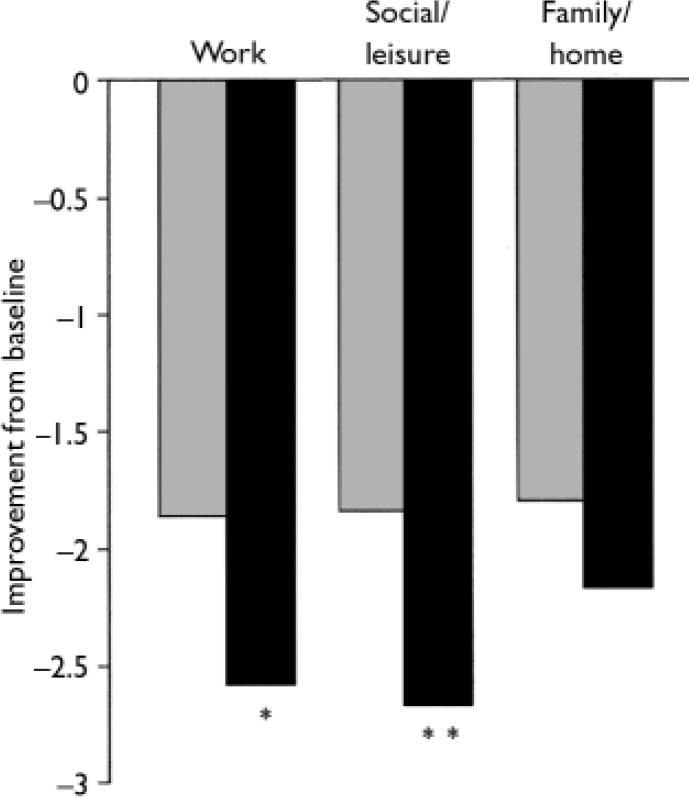
Fig. 7 Sheehan Disability Scale (intent-to-treat population, observed cases). P obtained from ANCOVA model: change from baseline=baseline +treatment+centre; *P<0.05, **P <0.01 v. venlafaxine ER; ![]() , placebo (n=168); ▪, venlafaxine ER (n=160).
, placebo (n=168); ▪, venlafaxine ER (n=160).
Table 2 Final on-therapy values for selected outcomes
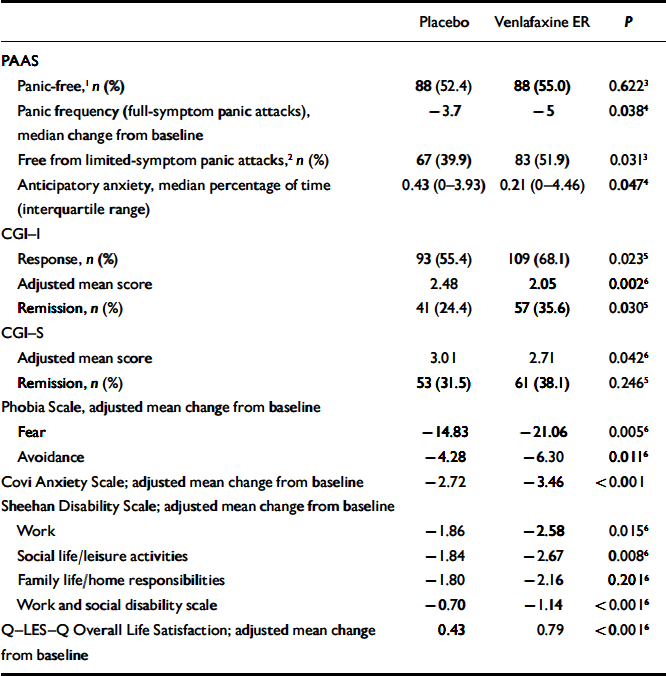
| Placebo | Venlafaxine ER | P | |
|---|---|---|---|
| PAAS | |||
| Panic-free,1 n (%) | 88 (52.4) | 88 (55.0) | 0.6223 |
| Panic frequency (full-symptom panic attacks), median change from baseline | -3.7 | -5 | 0.0384 |
| Free from limited-symptom panic attacks,2 n (%) | 67 (39.9) | 83 (51.9) | 0.0313 |
| Anticipatory anxiety, median percentage of time (interquartile range) | 0.43 (0-3.93) | 0.21 (0-4.46) | 0.0474 |
| CGI—I | |||
| Response, n (%) | 93 (55.4) | 109 (68.1) | 0.0235 |
| Adjusted mean score | 2.48 | 2.05 | 0.0026 |
| Remission, n (%) | 41 (24.4) | 57 (35.6) | 0.0305 |
| CGI—S | |||
| Adjusted mean score | 3.01 | 2.71 | 0.0426 |
| Remission, n (%) | 53 (31.5) | 61 (38.1) | 0.2465 |
| Phobia Scale, adjusted mean change from baseline | |||
| Fear | -14.83 | -21.06 | 0.0056 |
| Avoidance | -4.28 | -6.30 | 0.0116 |
| Covi Anxiety Scale; adjusted mean change from baseline | -2.72 | -3.46 | <0.001 |
| Sheehan Disability Scale; adjusted mean change from baseline | |||
| Work | -1.86 | -2.58 | 0.0156 |
| Social life/leisure activities | -1.84 | -2.67 | 0.0086 |
| Family life/home responsibilities | -1.80 | -2.16 | 0.2016 |
| Work and social disability scale | -0.70 | -1.14 | <0.0016 |
| Q—LES—Q Overall Life Satisfaction; adjusted mean change from baseline | 0.43 | 0.79 | <0.0016 |
Significant differences in favour of venlafaxine on the Q–LES–Q were also demonstrated in ‘subjective feelings of well-being’ (0.63 v 0.36, respectively; P<0.001), ‘general activities’ (0.58 v. 0.36, respectively; P=0.002), ‘satisfaction with medication’ (0.85v. 0.45, respectively; P=0.001), ‘physical health/activities’ (0.65 v. 0.45, respectively; P=0.021), ‘work’ (0.39 v. 0.18, respectively; P=0.041), and ‘social relations’ (0.46 v. 0.31; P=0.026). No significant differences were observed between the venlafaxine ER group and the placebo group for the remaining three domains (‘household duties’, ‘school/coursework’ and ‘leisure time activities’). No significant between-group differences in healthcare utilisation were observed.
Safety and tolerability
Overall, treatment-emergent adverse events were reported by 138 (78%) of the placebo-treated group and 152 (86%) of the venlafaxine ER-treated group (Table 3). Eleven individuals had events that precipitated withdrawal from the trial: 5 in the placebo group (1 each with unintended pregnancy, infection, vascular purpura, myocardial infarction and anxiety) and 6 in the venlafaxine ER group (1 each with accidental overdose, unintended pregnancy, deep thrombophlebitis, colitis, panic attack and metrorrhagia). The treatment-emergent adverse events that most frequently caused discontinuation of treatment in the venlafaxine ER group (reported by ≥2%) were anorexia, nausea, insomnia and sweating.
Table 3 ‘Treatment-emergent’ adverse events reported by ≥5% of the venlafaxine ER-treated individuals and at a frequency equal to or greater than twice the rate for placebo-treated participants

| Event, n (%) | Placebo (n=178) | Venlafaxine ER (n=177) |
|---|---|---|
| Any adverse event data | 138 (78) | 152 (86) |
| Insomnia | 8 (4) | 33 (19) |
| Dry mouth | 15 (8) | 31 (18) |
| Sweating | 5 (3) | 28 (16) |
| Abnormal ejaculation/orgasm1 | 0 (0) | 9 (13) |
| Constipation | 4 (2) | 17 (10) |
| Libido decreased | 3 (2) | 16 (9) |
| Somnolence | 8 (4) | 14 (8) |
| Anorexia | 6 (3) | 14 (8) |
| Impotence1 | 0 (0) | 4 (6) |
| Palpitation | 2 (1) | 9 (5) |
| Vasodilatation | 1 (<1) | 9 (5) |
Venlafaxine ER treatment was associated with few clinically important changes in laboratory tests, vital sign results or ECG assessments. Mean changes in laboratory values at final on-therapy evaluation included significant increases from baseline in total cholesterol (0.18 mmol/l, P<0.01) and low-density lipoprotein (LDL) cholesterol (0.12 mmol/l, P<0.05) in the venlafaxine ER group, and nonsignificant decreases in the placebo group (–0.05 mmol/l and –0.07 mmol/l, respectively). Between-group comparisons were significant for both variables (total cholesterol, P=0.032; LDL, P=0.017).
Two participants, both in the venlafaxine ER group, experienced clinically important changes in vital signs (one with tachycardia and one with increased blood pressure). The venlafaxine ER and placebo groups differed significantly in mean changes from baseline in supine pulse rate (2.18 beats/min v. 0.12 beats/min, respectively; P=0.007) and weight (–0.72 kg v. 0.21 kg, respectively; P=0.002).
Individual clinically important ECG results were observed for four participants, all in the placebo group. No participant had a QTc interval greater than 500 ms. Mean changes from baseline in ECG variables at the final on-therapy evaluation included, for the venlafaxine ER group, a decrease in PR interval (–4.58 ms, P≤0.01) and increases in QTc interval (4.02 ms, P=NS) and ECG-measured heart rate (3.38 beats/min, P<0.001), and for the placebo group, a non-significant increase in PR interval (1.72 ms) and non-significant decreases in QTc interval (-4.32 ms) and ECG- measured heart rate (-1.45 beats/min). Between-group comparisons were significant for all three variables (PR interval, P=0.01; QTc interval, P= 0.035; ECG-measured heart rate, P<0.001). None of the participants had any clinically important findings on physical examination, and no other statistically significant changes from baseline or differences between groups were deemed clinically important by the medical monitor.
DISCUSSION
The results of this study demonstrate that venlafaxine ER is safe, well-tolerated and efficacious in the short-term treatment of panic disorder. Baseline characteristics of the study population were consistent with the typical profile for panic disorder. Although the difference between groups in the percentage of those who achieved panic-free status, the primary outcome measure, was not statistically significant, venlafaxine ER-treated participants had a significantly greater reduction in the median number of full-symptom panic attacks compared with placebo at end point. The venlafaxine ER-treated group also had significantly greater improvement compared with placebo in most other secondary outcome measures.
Methodological issues
Assessment of panic attack frequency, and in particular panic-free status, in clinical trials is difficult and correlates poorly with global measures of efficacy (Reference Bandelow, Hajak and HolzrichterBandelow et al, 1995), as was the case here in the medication group (55% panic-free v. 68% responders). Panic attacks are known to be sporadic and highly variable in frequency and it is not uncommon for there to be a period of several weeks when no panic attacks occur (Reference Shear, Clark and FeskeShear et al, 1998). In fact, up to 50% of those receiving placebo in short-term clinical studies are ‘panic-free’ at end point (Reference den Boerden Boer, 1998), which is similar to the proportion in this study (i.e. 52%). A meta-analysis by Otto et al (Reference Otto, Tuby and Gould2001) found that more recently reported trials of selective serotonin reuptake inhibitors in panic disorder that included larger sample sizes were associated with smaller panic frequency effect sizes compared with earlier trials with small sample sizes. If both published and unpublished studies were available, it is likely that even higher panic-free rates for placebo-treated individuals would be observed (Reference Otto, Tuby and GouldOtto et al, 2001). Thus, failure to detect differences between active treatment and placebo groups in panic-free status is not uncommon, even with agents that are known to be highly effective in the treatment of panic disorder.
Multidimensional assessment
Multidimensional assessment of therapeutic interventions for panic disorder is therefore considered appropriate (Reference Ballenger, Davidson and LecrubierBallenger et al, 1998). In addition to panic attack frequency, four key domains have been identified as relevant for the assessment and treatment of panic disorder: anticipatory anxiety; panic-related phobias; well-being and overall severity of illness; and effect on work, social life, the family and quality of life (Reference Ballenger, Davidson and LecrubierBallenger et al, 1998). Global measures by clinicians are also clinically important in assessing how the individual has responded to treatment over time (Reference Pollack, Rapaport and FayyadPollack et al, 2002). Our study showed that the venlafaxine ER group had significant response (CGI–I of 1, very much improved, or 2, much improved) and remission (panic-free and CGI–I of 1) rates based on global measures of improvement compared with placebo, and significantly greater improvement in global severity and in symptoms of anticipatory anxiety and fear/avoidance. Finally, improvement in a majority of domains representing quality of life and functionality was observed.
Tolerability and safety
The increases in total plasma cholesterol and LDL levels observed in the venlafaxine ER group relative to the placebo group in this report are consistent with studies of healthy volunteers that show an increase in LDL after treatment with the selective serotonin reuptake inhibitor paroxetine (Reference Lara, Baker and ArcherLara et al, 2003) and an increase in total plasma cholesterol with mirtazapine, an agent that has both serotonergic and noradrenergic properties (Reference Lara, Baker and ArcherLara et al, 2003; Reference Nicholas, Ford and EspositoNicholas et al, 2003). Other studies have reported increased total plasma cholesterol levels in individuals with anxiety disorders (Reference Peter, Hand and HohagenPeter et al, 2002). The metabolic basis of such results and their clinical significance merits further investigation. Heart rate and QT interval variability in those with panic disorder relative to controls has been attributed to abnormal α2-adrenergic function (Reference Yeragani, Tancer and UhdeYeragani et al, 2003). Further investigation of the differences in heart rate and QTc interval changes from baseline between treatment and placebo groups, as in the present study, is desirable.
Summary
Venlafaxine ER was generally safe and well tolerated in this study. The type and frequency of adverse events and discontinuation rate attributable to adverse events in the venlafaxine ER group was similar to that seen in individuals with depression, generalised anxiety disorder and social anxiety disorder, indicating no additional risk of adverse events in those with panic disorder. Together with its superior efficacy relative to placebo across most outcome measures, the favourable safety and tolerability profile of venlafaxine ER suggests that it is a viable treatment option for panic disorder.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Venlafaxine extended-release (ER) is statistically significantly more effective than placebo across most outcome measures, including clinically important symptom domains, global scales and health outcomes.
-
▪ Venlafaxine ER is safe and well tolerated.
-
▪ The effective dose for venlafaxine ER in the treatment of panic disorder appears to be comparable to that used in the treatment of depression.
LIMITATIONS
-
▪ Venlafaxine ER was not associated with a significantly greater proportion of individuals who were free from full-symptom panic attacks at the final on-therapy evaluation.
-
▪ Those with comorbid depression or other clinically important psychiatric disorders were excluded from the study.
-
▪ Lack of an active comparator in this study limits the conclusions that can be drawn regarding the efficacy of venlafaxine ER compared with other antidepressants.











eLetters
No eLetters have been published for this article.