


We are making great strides in working out the genetic and environmental factors that influence the development, function and appearance of physical features in many organisms. Malfunctioning genes as well are starting to yield their secrets about how they subvert normal physiological processes, while we continue to progress in our understanding of how genes interact with environmental factors to control normal human anatomy, physiology, metabolism and behaviour.
The complexity of genetic and biological systems allows them to modulate human behaviour at exquisite levels of subtlety and nuance. That same complexity, however, allows dysfunction to occur at a large number of sites and (depending on the nature of the deficit and its association to input or output neural pathways) to exert significant effects in areas distal to the source of the original insult. Thus, a relatively subtle biological deficit that may be difficult to detect (or easy to overlook), may occur in neural networks that are essential for normal function, and exert a profound effect on behaviour, the ultimate expression of central nervous system activity.
Because so many layers of regulation and organisation can intervene between a gene and its effects on behaviour, the issue of what is actually transmitted by genes in a complex disorder such as schizophrenia is a large and complicated topic. Yet that is precisely the issue we must address if we are to understand the origins of the disorder and, ultimately, its treatment and prevention. We approach this issue by first reviewing the genetic and environmental bases of schizophrenia. Evidence for genetic components in schizophrenia is extensive, and derives mainly from behavioural genetic and molecular biological studies. Evidence for environmental components derives from behavioural genetic studies as well, and from epidemiological analyses of risk factors. Following this discussion, we consider how these genetic and environmental components interact to produce either neurodevelopmental syndromes (‘schizotaxia’) or schizophrenia itself. Finally, treatment implications of the model are considered.
PARTITIONING THE GENETIC AND ENVIRONMENTAL CONTRIBUTIONS TO SCHIZOPHRENIA
That schizophrenia and schizophrenia spectrum disorders have a hereditary component is well established. While the general population life time prevalence is about 1%, relatives of schizophrenic probands have a higher risk of schizophrenia and related disorders. The risk of developing schizophrenia in family members increases with the degree of biological relatedness to the patient — greater risks are associated with higher levels of shared genes (Reference GottesmanGottesman, 1991). For example, third-degree relatives (e.g. first cousins) share about 12.5% of their genes, and show a risk of 2% for developing schizophrenia. Second-degree relatives (e.g. half-siblings) share about 25% of their genes and show a risk of 6%. Most first-degree relatives (e.g. siblings, dizygotic (DZ) twins) share about 50% of their genes and show a risk of about 9%. Monozygotic (MZ) twins share 100% of their genes, and show risks near 50%.
Observations of increased relative risk in patients' families are not sufficient to support the conclusion that genetic factors produce schizophrenia, because relatives share environments in addition to genes. Various types of behavioural genetic paradigms, however, including twin and adoption studies, provide converging evidence for both genetic and environmental aetiological components in schizophrenia. For example, when the biological offspring of a parent with schizophrenia are adopted away, they develop schizophrenia or related disorders at the elevated rates that would be expected of first-degree biological relatives, rather that at the lower population rates that are (usually) demonstrated by their adoptive families (e.g. Reference KetyKety, 1988; Reference Kety, Wender and JacobsenKety et al, 1994). MZ twins are genetically identical; if a phenotype is determined entirely by genetic factors, MZ twins should have a concordance of 100%. DZ twins, as well as other siblings and parents, share approximately 50% of their genes; the concordance should therefore be about half that of MZ twins. Estimates of concordance rates for schizophrenia, pooled from European studies over 30 years (Gottesman, Reference Gottesman1991; Reference Gottesman, Plomin and McClearn1993) clearly show higher mean rates for MZ than for DZ twins (48% v. 16%). Even allowing for the possibility that the relative contribution of genetic factors is inflated because MZ twins also share environmental factors such as prenatal and intra-uterine variables (Reference Reiss, Plomin and HetheringtonReiss et al, 1991), the pattern and consistency of the results underscores the imkportance of genetic components. Yet they also emphasise the importance of environmental factors: concordance rates are not 100% for MZ twins, and the concordance rate in the DZ twins is less than half the MZ rate.
To what extent do differences in concordance rates reflect the effects of heredity? Estimates of the heritability of schizophrenia vary across samples and methods of ascertainment. Kendler & Diehl, for example, reported the results of twin studies with an average heritability of around 70% (Reference Kendler and DiehlKendler & Diehl, 1993), and recent studies using DSM-III (American Psychiatric Association, 1980), DSM-III-R (American Psychiatric Association, 1987) or DSM-IV (American Psychiatric Association, 1994) diagnostic criteria also showed heritability estimates between 80% and 86% (Reference Farmer, McGuffin and GottesmanFarmer et al, 1987; Reference Onstad, Skre and TorgersenOnstad et al, 1991; Reference Cannon, Kaprio and LonnqvistCannon et al, 1998). Although the value obtained in a specific study might be accurate, estimates vary because heritability is a function of the population studied (including its size), the environment, the phenotypic criteria used for diagnosis, the statistical model used, the effect sizes of relevant variables and many other factors. Although the heritability of schizophrenia is substantial, its estimates do vary, and its contribution to the schizophrenia phenotype cannot be understood without taking account of the importance of environmental factors.
IDENTIFYING THE CAUSES OF SCHIZOPHRENIA
We have long suspected that schizophrenia is a heterogeneous disorder, both clinically and genetically. Individuals with schizophrenia or with the related conditions of schizoaffective disorder display a wide range of symptoms. Although dissimilar phenotypes (clinical heterogeneity) could suggest aetiological heterogeneity, such inferences must be conservative because homogeneous aetiologies can manifest with variable expressions in the presence of moderating variables, such as environmental factors (Reference Tsuang, Faraone and LyonsTsuang et al, 1993). Pleiotropy is a concept that describes a situation in which even a single genetic alteration can result in multiple physiological effects and phenotypic expressions, some of which might involve disease (Reference Vogel and MotulskyVogel & Motulsky, 1986). Conversely, patients with different diagnoses (e.g. other disorders with psychotic symptoms) or subtypes may share similar symptoms. Are these disorders different manifestations with the same cause, or is schizophrenia a disease with multiple causes? The link between phenotypic heterogeneity and aetiologic heterogeneity is tenuous, so attempts to use purely phenotypic data to infer aetiologic heterogeneity must be viewed cautiously. Phenotypic data are most useful when collected in the context of some aetiological theory that factors in one or more hypothetically causative agent(s) (for example, genes, viruses or obstetric complications).
GENES THAT CONTRIBUTE TO SCHIZOPHRENIA
Many theories have been offered to explain the genetic mechanisms that produce schizophrenia. One hypothesis is that schizophrenia has a homogeneous pathogenic genotype with pleiotropic effects. In a classic example of pleiotropy, Marfan's syndrome is a connective tissue disorder with at least 12 identifiable abnormalities. The most common of these occur in 88% of patients and the least common only in 12% (Reference LevitanLevitan, 1998). Similarly, individuals with schizophrenia may present with a variety of symptoms. Yet the preponderance of evidence argues against the possibility that most cases of schizophrenia are caused by a common gene (Reference GottesmanGottesman, 1991; Reference Tsuang, Stone and FaraoneTsuang et al, 1999a ). In particular, single major gene models do not explain familial patterns of illness accurately, in either families or twins; multi-factorial polygenic models explain such data better. In a model version of this view, the schizophrenia phenotype results from the additive effect of multiple genes and environmental factors. Each factor contributes effects until a critical threshold level is reached and the critical symptoms are manifested. Note that in this type of model, common genes of small effect may be involved, rather than rare genes of much larger effect, as is more likely in single major gene models. As much of the genetic effect is additive (although multiplicative or epistatic effects may occur as well), different combinations and/or numbers of genes may contribute to schizophrenia, in different families. Although models like these account best for family patterns of transmission, we will not know their accuracy with certainty until we identify the actual genes that are involved in schizophrenia.
Nevertheless, an important implication of multi-factorial polygenic models is that genetic heterogeneity at least partly accounts for phenotypic heterogeneity. In fact, such a state of affairs would be consistent with heterogeneous genetic aetiologies evident in a variety of complex disorders. Phenylketonuria (PKU), for example, is associated with up to 200 alleles of the phenylalanine hydroxylase gene, which produces varying degrees of mental illness in the absence of treatment (Reference Eisensmith, Martinez and KuzminEisensmith et al, 1996). Similarly, Gaucher's disease, a fatal neurological syndrome, can be produced by three different genotypes (Reference LevitanLevitan, 1998). The notion of genetic heterogeneity extends even further. While most cases of schizophrenia are accounted for best by polygenic models, some may indeed result from the effects of one or several genes of moderate to large effects. In some cases, for example, family transmission patterns are predicted about as well by oligogenic models (a type of polygenic model that posits a relatively small number of aetiological genes that have moderate or large effects) as they are by multi-factorial polygenic ones (Reference Tsuang, Stone and FaraoneTsuang et al, 1999a ).
In contrast to the high genetic ‘loading’ apparent in many familial cases of schizophrenia, sporadic (or non-familial) cases of schizophrenia may be phenocopies that result primarily from factors other than schizophrenia genes. Psychosocial causes, amphetamine misuse, schizophrenia-like psychosis of epilepsy and other brain trauma, brain disorders, infections in utero and/or complications of pregnancy are among the variables likely to contribute to the development of such cases. Finally, some isolated cases may be owing to gross chromosomal abnormalities (Reference Tsuang and FaraoneTsuang & Faraone, 1995).
In the light of the high heritability estimates for schizophrenia, the discovery of genes that cause the disorder has been eagerly anticipated since the 1980s, spurred in part by the development of polymorphic DNA markers that can detect multiple forms of genes (alleles) in chromosomal locations (Reference Tsuang, Stone and FaraoneTsuang et al, 1999a ).
While several forms of molecular genetic analyses are in use, linkage analyses provide a particularly versatile procedure that is helping to explain the familial basis of schizophrenia. Briefly, linkage analysis makes use of events that occur during meiosis, when chromosomes cross over and exchange segments of DNA. Genetic loci that are closer to each other have a higher probability of being inherited together than do loci that are further away. The probability of identifying a disease gene is increased if it co-segregates (i.e. is ‘linked’) with an allele whose chromosomal location can be identified among members of a family. Linkage analysis assesses the probability that a marker gene and a disease gene do, in fact, co-segregate. Results from linkage studies depend on a variety of factors, including the presumption of the mode of inheritance, the involvement of genes whose effects are large enough to detect, and/or the extent to which family members are diagnosed accurately as either affected or unaffected with a disorder in the schizophrenia spectrum. Since all of these factors (and others, such as the importance of statistical power) are in some way problematic, it is not surprising that linkage studies have thus far been less than conclusive, and have yet to identify the genes that cause schizophrenia.
Hopes were high in the late 1980s when two groups reported linkage to a gene on chromosome 5 (Reference Bassett, McGillivray and JonesBassett et al, 1988; Reference Sherrington, Brynjolfsson and PeturssonSherrington et al, 1988). Unfortunately, numerous attempts at replication failed, and the findings came to be regarded as false positives (cf. Reference Tsuang, Stone and FaraoneTsuang et al, 1999a ). This became a familiar pattern in the early 1990s, with initial reports of linkage raising hopes that were then lowered by subsequent non-replications. Yet progress was made. New generations of more precise DNA markers were developed, and the need for greater statistical power was met (partly) through the formation of large international collaborative efforts. In the past few years, evidence for linkage has been strengthened by replications of positive findings. Although non-replications have also been reported at the same sites, the first of these replications were reported to sites in chromosomes 6p and 8p in 1995 (e.g. Reference Moises, Yang and KristbjarnarsonMoises et al, 1995a ; Reference Pulver, Lasseter and KaschPulver et al, 1995; Reference Schwab, Albus and HallmayerSchwab et al, 1995; Reference Wang, Sun and WalczakWang et al, 1995; Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 6, 1996), and replications have now been reported at several additional sites including chromosomes 10p (Reference Faraone, Matise and SvrakicFaraone et al, 1998; Reference Schwab, Hallmayer and AlbusSchwab et al, 1998; Straub et al, 1998), 13q (Reference Blouin, Dombroski and NathBlouin et al, 1998), 15q (Reference Freedman, Coon and Myles-WorsleyFreedman et al, 1997; Reference Kaufmann, Suarez and MalaspinaKaufmann et al, 1998) and 22q (e.g. Reference Polymeropoulos, Coon and ByerleyPolymeropoulos et al, 1994; Reference Pulver, Karayiorgou and WolyneicPulver et al, 1994; Reference Moises, Yang and HavsteenMoises et al, 1995b ; Schizophrenia Collaborative Linkage Group (Chromosome 22), 1996).
Despite this progress, there is a long way to go before we can identify the genes that cause schizophrenia. Both phenotypic and (probably) genetic heterogeneity make the classification of affected v. unaffected relatives difficult. As noted above, this is an important factor in determining the success or failure of linkage analyses. Part of the problem is that clinical symptoms — or their absence — often reflect a remote consequence of the events that gave rise to them. Between the actions of genes and the behaviours we interpret as clinically meaningful (symptoms) are numerous levels of biological and environmental modulation that can weaken the connection between genetic determinant and behavioural outcome. For this reason, it is important to examine phenotypes that are closer to their genetic or biological aetiologies than are clinical symptoms, in order to determine more specific consequences of schizophrenia genes.
Candidate measures include neuropsychological, physiological or anatomical measurements as surrogates for classical clinical criteria. For example, Arolt et al (Reference Arolt, Lencer and Achim1996), using deficits in eye-tracking as a phenotype, studied German multiplex pedigress and obtained evidence for linkage on chromosome 6p. A second paradigm is based on the difficulties demonstrated by many patients with schizophrenia in filtering sensory input from the environment (Reference Coon, Plaetke and HolikCoon et al, 1993). Unlike normal subjects, patients with schizophrenia cannot filter out or ‘gate’ the second of two sensory stimuli (e.g. Reference Waldo, Cawthra and AdlerWaldo et al, 1994). One way to measure sensory gating involves the measurement of P50 waves in auditory evoked potential paradigms. Deficits in P50 waves are both heritable (Reference Myles-Worsley, Coon and ByerleyMyles-Worsley et al, 1996) and evident at higher rates in relatives of patients with schizophrenia than they are in appropriate controls (Reference Siegel, Waldo and MiznerSiegel et al, 1984; Reference Waldo, Carey and Myles-WorsleyWaldo et al, 1991). It is relevant to the current discussion that the P50 deficit showed evidence for linkage to a locus near chromosome 15q13-14 in families with schizophrenia.
Both patients with schizophrenia and their relatives show a broad range of deficits or alterations in neuropsychological and neurobiological functions (Reference SeidmanSeidman, 1997) that are potentially useful phenotypes in genetic studies. More generally, some subset of these measures, in conjunction with selected clinical symptoms, may provide a more accurate picture of the liability produced by schizophrenia genes than is available currently. We have modified Meehl's use of the term ‘schizotaxia’ (Reference MeehlMeehl, 1962) to describe this liability, based on a combination of neuropsychological deficits and negative clinical symptoms (Reference Faraone, Green and SeidmanFaraone et al, 2001). We also assume that schizotaxia, which is an evolving concept, will come to incorporate biological and possibly psychosocial measures as well. The notion of schizotaxia may provide a basis for the development of treatment strategies in vulnerable, nonpsychotic individuals, which is a point we will return to after reviewing the role of the environment in schizophrenia.
IDENTIFYING ENVIRONMENTAL RISK FACTORS FOR SCHIZOPHRENIA
The degree of risk of schizophrenia in members of families with one or more patients with schizophrenia correlates with the degree of biological relatedness between the relative and the patient: the closer the relationship, the higher the level of risk. Yet even if an individual has an identical twin with schizophrenia, or two affected parents, the risk is nowhere near 100%. In the case of identical twins with one affected member, the genetic predisposition is present in both individuals, but is expressed only in the twin who has undergone certain environmental experiences as well. Consistent with this view, Gottesman & Bertelsen (Reference Gottesman and Bertelsen1989) showed that the offspring of identical twins who were discordant for schizophrenia showed similar (elevated) rates of developing the disorder, regardless of whether their parent was the affected or the unaffected twin.
Environmental components are either shared by individuals in the same household, (whether related or not) or unshared (that is, unique, even for MZ twins). Components of the environment include psychosocial, biological and physical factors experienced by the individual from the moment of conception, through development, birth and maturation. Monozygotic twins may experience different prenatal and perinatal factors such as adequacy of blood supply, position in the womb and birth complications. Later, they may experience different home and school environments, as well as different marital experiences, occupational events or surroundings (Reference Reiss, Plomin and HetheringtonReiss et al, 1991; Reference Pike and PlominPike & Plomin, 1996). Such differences in environment are likely to be meaningful, as non-shared environmental influences accounted for almost all of the variance in liability to schizophrenia attributable to environmental effects in several twin studies (Reference Kendler, Gruenberg and KinneyKendler et al, 1994; Reference Kety, Wender and JacobsenKety et al, 1994; Reference Cannon, Kaprio and LonnqvistCannon et al, 1998).
The importance of environmental factors is made more apparent by the nature of their interactions with genetic determinants. Genes and environmental factors were formerly thought to be mainly additive, with the outcome reflecting the sum of their influence; in fact, they are interactive as well (Reference KendlerKendler, 1995). In this newer view, environmental factors may have varied effects on individuals with different genotypes; genotype-environment interactions result from genetically mediated differences in sensitivity to environmental factors, and/or from environmentally mediated genetic effects. In some ways this is obvious, as genes and environments always interact; yet the concept deserves attention, because it emphasises different ways in which to consider the importance of both genetic and environmental variables. For example, it is consistent with the notion that individuals with particular genotypes seek out or construct specific environments that are consistent with their genetically based predilections (Reference Scarr and McCartneyScarr & McCartney, 1983). Consideration of genetic-environmental influences may also help us to understand the nature of at least some environmental risk factors. Just as geneticists search the entire genome for all of the many genes that affect susceptibility to schizophrenia, so must environmental researchers search the entire ‘envirome’ for all environmental risk factors that affect the disorder. When we understand the sum and interaction of all effects from the genome and from the envirome, we will have solved the puzzle of schizophrenia. To date, there are at least two features of the envirome that are candidate risk factors for schizophrenia: psychosocial factors and delivery/birth complications.
Psychosocial factors
Several adoption studies provide evidence of genetic-environmental interactions. Kinney et al (Reference Kinney, Holzman and Jacobsen1997), using the Danish sample, found elevations of the Thought Disorder Index (TDI) (Reference Johnston and HolzmanJohnston & Holzman, 1979) in biological relatives of patients with schizophrenia, compared with normal controls. In contrast, elevations of the TDI were not evident in the adoptive relatives of either subjects suffering from schizophrenia or control subjects. Findings from the Finnish adoption studies (Reference TienariTienari, 1991; Reference Tienari, Wynne and MoringTienari et al, 1994; Reference Wahlberg, Wynne and OjaWahlberg et al, 1997) were consistent with those reported by Kinney and colleagues. Wahlberg et al, for example, showed that young adult offspring of mothers with schizophrenia were more likely to show symptoms of thought disorder when they were raised by adoptive mothers who themselves showed elevated levels of ‘communication deviance’. In contrast, adoptees who were raised by adoptive parents with low communication deviance were less likely to show thought disorder. This pattern was not evident in control adoptees, who showed no discernible relationship between thought disorder in the adoptees and communication deviance in the adoptive parents. In other words, these findings did not detect the presence of a ‘schizophrenogenic environment’ for individuals who did not demonstrate a preexisting genetic liability. These examples support the view that genetic factors alone do not explain the development of schizophrenia, and that interactions with the environment provide important mediating variables.
Pregnancy and delivery complications
The examination of developmental abnormalities such as pregnancy and delivery complications, especially in conjunction with genetic risk factors, has provided useful information on precursor states for schizophrenia (Reference Buka, Goldstein and SeidmanBuka et al, 1999). For example, patients with schizophrenia have experienced a greater number of labour and delivery complications at birth than have normal controls (Reference Lewis and MurrayLewis & Murray, 1987; Reference Geddes and LawrieGeddes & Lawrie, 1995; Reference JablenskyJablensky, 1995; Reference Tsuang and FaraoneTsuang & Faraone, 1995). Among these complications is pre-eclampsia, which results in foetal hypoxia, and leads to a nine-fold increase in the risk for subsequent schizophrenia (Reference Kendell, Juszczak and ColeKendell et al, 1996). Cannon (Reference Cannon1996) found a dose-dependent relationship between risk of schizophrenia and severity of perinatal hypoxia. Interestingly, the same researchers (Cannon et al, Reference Cannon, Mednick and Parnas1993, Reference Cannon, Zorrilla and Shtasel1994) found that among the offspring of mothers with schizophrenia, rates of birth complications were higher in the group who eventually developed schizophrenia than in the group who did not, or who developed schizotypal personality disorder. Moreover, birth complications were unrelated to the development of schizophrenia in a control, low-risk group whose parents did not have schizophrenia.
Findings from the Philadelphia cohort of the National Collaborative Perinatal Project (NCPP) indicated that the risk of developing schizophrenia among children of parents with schizophrenia increased as a function of the number of hypoxiarelated birth complications (Reference CannonCannon, 1996). Hypoxia-related birth complications, but not pregnancy and other delivery complications, predicted subsequent schizophrenia among children of patients with schizophrenia.
Zornberg et al (Reference Zornberg, Buka and Tsuang2001) stressed the importance of distinguishing between the relative risks of different types of obstetric complications, because grouping them produces inconsistent results. This point was underscored in a recent study using the New Zealand cohort of the NCPP, in which three measures of obstetric complications were assessed in relationship to deficits in neuropsychological performance at age 7 years (Reference Seidman, Buka and GoldsteinSeidman et al, 2000). Low birth-weight had the strongest association with neuropsychological impairments, followed by an index of inferred hypoxic insults, and then by maternal conditions suggesting chronic hypoxia. Future analyses with this sample will determine how interactions between individual obstetrical complications and family history (i.e. genetic risk status) affect the liability for schizophrenia.
INTEGRATION OF GENETIC AND ENVIRONMENTAL RISK: A NEURODEVELOPMENTAL VIEW
In addition to the examples described above, a variety of other environmental risk factors for schizophrenia have been identified (Reference Jones and CannonJones & Cannon, 1998), including viral infections (e.g. Reference Torrey, Kaufmann, Nasrallah and WeinbergerTorrey & Kaufmann, 1986; Reference Mednick, Huttunen and MachonMednick et al, 1994; Reference Tsuang and FaraoneTsuang & Faraone, 1995). How do genetic and environmental risk factors fit together? Whether they are mainly additive or more interactive is one level of analysis. Another involves a neuro-developmental perspective. It is significant that many genetic and environmental risk factors are apparent either prior to or around the time of birth (Reference Jones and CannonJones & Cannon, 1998), and interact well before the onset of psychosis. For this reason, neurodevelopmental models of schizophrenia are particularly useful for understanding the course of their influence. These models have refined the diathesis-stress theory by proposing that a combination of genetic and non-genetic errors leads to maldevelopment of the brain (Reference SeidmanSeidman, 1990; Weinberger, Reference Weinberger, Hirsch and Weinberger1994, Reference Weinberger, Bloom and Kupfer1995; Reference Goldman-RakicGoldman-Rakic, 1995).
The importance of neurodevelopmental factors in schizophrenia is indicated by the presence of neuropsychological abnormalities in children of patients with schizophrenia, by the high frequency of obstetric complications in schizophrenia, and by direct observations of the brain. For example, some brain alterations found at autopsy are consistent with a neurodevelopmental hypothesis. Among these are abnormalities of cell formation in the hippocampus, cingulate gyrus and prefrontal cortex, and temporal lobe sulcal-gyral patterns, which suggests either second- or third-trimester pregnancy abnormalities (Reference BogertsBogerts, 1993). Moreover, some patients with schizophrenia exhibit a cavum septi pellucidi, representing the failure of two laminar membranes to fuse, a process that usually occurs in the third trimester of gestation (Reference Shenton, Wible, McCarley, Krishnan and DoraiswamyShenton et al, 1997). These data thus suggest that well before the onset of the illness, the brains of people who develop schizophrenia are not entirely normal.
One of the neurodevelopmental models of schizophrenia attributes the usual postpubertal onset of psychosis to the continuing development of the brain (especially the frontal cortex) throughout adolescence (Reference Weinberger, Bloom and KupferWeinberger, 1995). This model postulates that patients with schizophrenia express psychosis when the frontal cortex comes ‘on-line’ in a deficient fashion, as a result of long-standing neurologic abnormalities that may interact with environmental stressors. Essentially, the neurodevelopmental model assumes that the substrate for schizophrenia is formed when development goes awry and key neural networks do not develop properly. Thus, both the diathesis-stress and neurodevelopmental theories predict that the children of patients with schizophrenia will show neurobiologic abnormalities linked to the predisposition to schizophrenia.
In endorsing a neurodevelopmental model, we are not discounting the possibility that neurodegeneration occurs after the onset of illness. For example, the glutamate hypothesis of schizophrenia suggests that hypoactivity of glutamate transmission through the N-methyl-D-aspartate (NMDA) receptor could lead to increased activity at non-NMDA glutamate receptors, which then produces signs of neurodegeneration (Reference CoyleCoyle, 1996; Reference WoodsWoods, 1998).
Additional evidence for a neurodegenerative component to schizophrenia comes from clinical studies indicating that patients treated with antipsychotic medicine during their first or second hospitalisation had a better outcome than patients who had not been treated early in their course of illness (Reference WyattWyatt, 1995). This finding has led to the hypothesis that psychotic episodes are neurotoxic, having adverse effects on the brain through ‘kindling’ or some other as yet unknown process (Reference WyattWyatt, 1995; Reference McGlashan and JohannessenMcGlashan & Johannessen, 1996).
The suggested developmental pathways are summarised in the heuristic model presented in Fig. 1. As the upper part of the figure suggests, schizotaxia (introduced earlier as a condition reflecting the genetic liability for schizophrenia) emerges from the effects of an early environmental insult in conjunction with a genetic predisposition to schizophrenia. This view differs somewhat from Meehl's original formulation (Reference MeehlMeehl, 1962). Meehl defined schizotaxia as reflecting the genetic predisposition, and schizotypy as its necessary clinical manifestation. We view schizotaxia as the predisposition to schizophrenia (which has both genetic and environmental components), and schizotypy as only one of its possible outcomes. This minor reformulation of schizotaxia reflects our theoretical premise that the neurobiological substrate for schizophrenia is formed by the joint effect of genes and adverse environmental events.
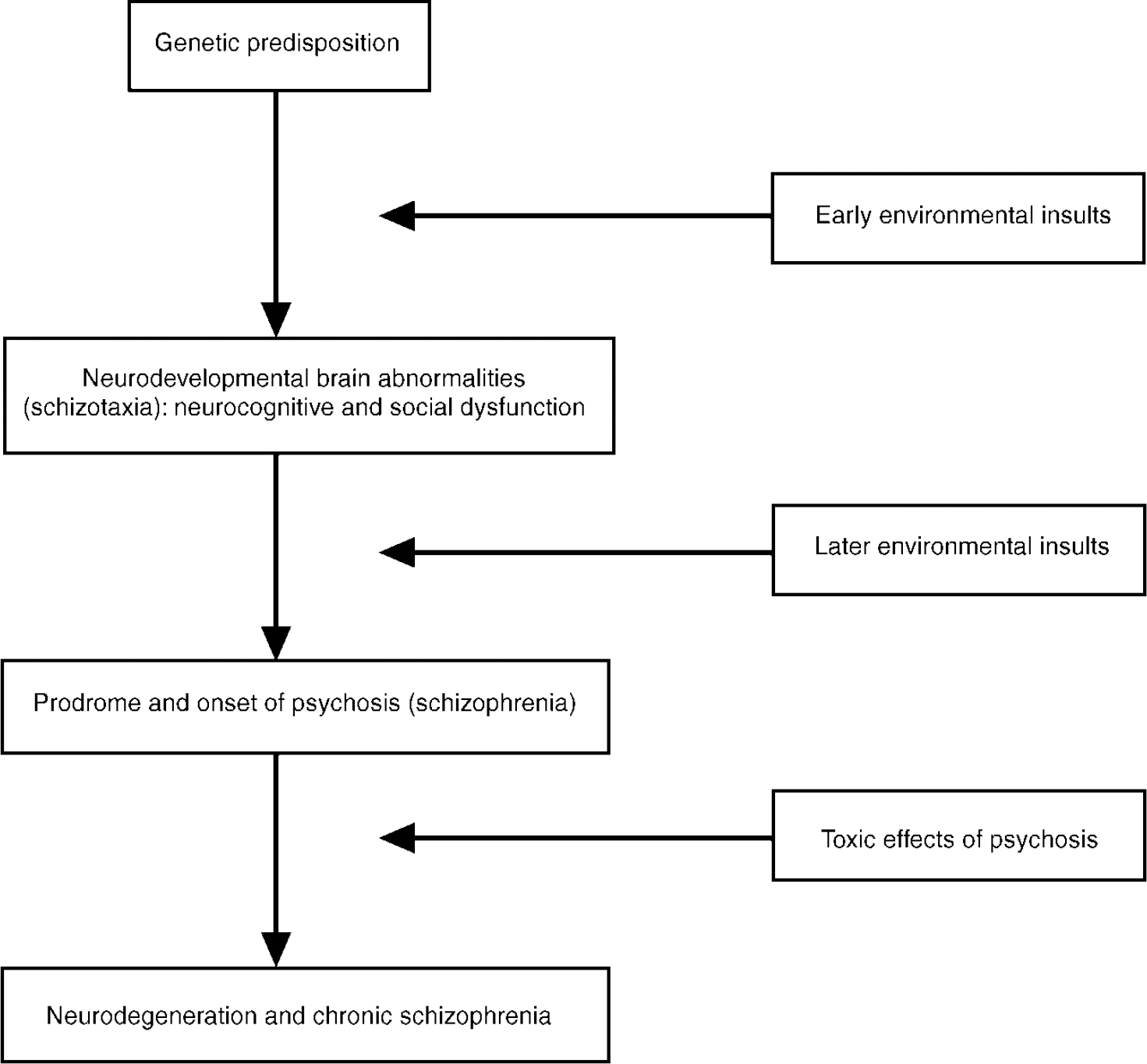
Fig. 1 Hypothetical developmental pathways to schizotaxia and schizophrenia.
The model in Fig. 1 also allows for later environmental events (e.g. expressed emotion, life events and biological factors) to combine with schizotaxia to cause schizophrenia. The lower part of the figure indicates that psychosis may lead to neurodegeneration and chronic schizophrenia. This portion of the model underscores the significance of two areas of schizophrenia research: (a) facilitating the secondary prevention of schizophrenia deterioration; and (b) understanding the course of pathophysiology from its neurodevelopmental origins, through the onset of the illness, to its subsequent chronicity.
If the model is substantially accurate, then it follows that psychosis and the subsequent diagnosis (i.e. categorisation) of schizophrenia are events that occur well after the first manifestation of the genetic liability to schizophrenia. This view is consistent with the notion of an underlying continuum of genetic liability that has schizophrenia as only one of its possible outcomes.
SCHIZOTAXIA AND FUTURE RESEARCH
If our concept of schizotaxia is correct - the syndrome still requires validation - then the genetic liability to schizophrenia shows meaningful clinical manifestations that have several implications. First, schizotaxia may be a more specific expression of schizophrenia genes than is psychosis, which may be associated with additional and not entirely related pathophysiological manifestations. This may be particularly true if suggestions of neurodegeneration in psychosis (e.g. Reference CoyleCoyle, 1996) are correct, and have distinct aetiological bases.
Second, the treatment of schizotaxia in non-psychotic relatives could serve to attenuate clinically meaningful symptoms (Reference Faraone, Green and SeidmanFaraone et al, 2001). This course of action was tested in a pilot study of four relatives with schizotaxia (Reference Tsuang, Stone and SeidmanTsuang et al, 1999b ). The criteria for schizotaxia included moderate deficits in at least two neuropsychological domains, including long-term verbal memory, attention and executive functions (moderate deficits were defined as scoring at least 2 s.d. below normal in one domain, and at least 1 s.d. below normal in a second domain), and moderate levels of negative symptoms (defined as six or more scores on the Schedule for the Assessment of Negative Symptoms (Reference AndreasenAndreasen, 1983) rated 3 or higher). Individuals who met these criteria and agreed to treatment received low doses of risperidone (0.25-2.0 mg) for 6 weeks. Side-effects were temporary and mainly mild. All four individuals showed marked improvements in attention, and mild to moderate reductions in negative symptoms. We stress that these results are preliminary, and do not advocate this treatment until larger, more controlled trials have determined the reliability and validity of our findings.
Finally, if the above results are replicated, then treatment of schizotaxia could be conceptualised as a step towards the even more important goal of identifying people at high risk when they are young, and then providing them with treatments to prevent or attenuate schizophrenia. As we continue to attend to both the genetic and environmental components of schizotaxia, it may be hoped that the parallel goals of better treatment and prevention of schizophrenia will draw closer in the near future.
ACKNOWLEDGEMENTS
Preparation of this chapter was supported in part by the National Institute of Mental Health Grants 1 R01MH4187901, 5 UOI MH4631802 and 1 R37MH4351801 to Dr Ming T. Tsuang and the Veterans Administration's Medical Research, Health Services Research and Development and Cooperative Studies Programs.


eLetters
No eLetters have been published for this article.