



INTRODUCTION
Carbon-14 (radiocarbon) is an important radionuclide in the inventory of radioactive waste produced in many European countries and, due to its long half-life (5730 yr), a significant dose-contributing radionuclide in safety assessments for deep geological repositories for radioactive waste (Nagra 2002; Yim and Carron Reference Yim and Caron2006; Johnson and Schwyn Reference Johnson and Schwyn2008; Nuclear Decommissioning Authority 2012). 14C has to be taken into account in safety assessments for various reasons: (1) 14C can be present both as dissolved and gaseous species in the disposal facility and the host rock; (2) gaseous and dissolved 14C-bearing organic compounds readily migrate in the near field and the geosphere due to weak interaction with mineral surfaces in alkaline to near neutral conditions; and (3) 14C can be incorporated in the human food chain. Thus, the chemical form of the 14C-containing species dictates the routes of 14C migration in the engineered barrier system of a deep geological repository and the surrounding host rock and therefore determines the long-term contribution of 14C to dose release from a repository for radioactive waste.
A compilation of the activity inventory in the already existing and future arising radioactive waste in Switzerland showed that the 14C inventory is mainly associated with irradiated steel while the 14C proportions from nuclear fuel components (e.g. Zircaloy) and from waste resulting from the treatment of reactor coolants are considerably smaller (Nagra 2014). In the light water reactors (LWR) operated in Switzerland, the formation of 14C is primarily caused by the activation of nitrogen impurities contained in nuclear fuel and metal components of the core structural materials based on the nuclear reaction 14N(n,p)14C. The main 14C source is its release during the corrosion of irradiated steel in the cementitious near field of an L/ILW repository.
Very limited information is available on the 14C speciation upon release from corroding irradiated steel (Swanton et al. Reference Swanton, Baston and Smart2015 and references therein; Wieland and Hummel Reference Wieland and Hummel2015). Current safety assessments are still based on specific, simplifying assumptions regarding 14C speciation. In previous experimental work both oxidized and reduced hydrocarbons have been observed in iron-water systems in anoxic, near neutral to alkaline conditions (Wieland and Hummel Reference Wieland and Hummel2015). The formation of reduced hydrocarbons, such as methane and other volatile compounds, is expected in view of the reducing conditions prevailing at the iron-water interface and as a result of the hydrolysis of carbide species on the iron surface. Nevertheless, the formation of oxidized hydrocarbons, such as carboxylic acids, alcohols, and aldehydes, has been observed in iron-water systems. In general, mostly organic compounds with a low molecular weight and with a low number of carbon atoms (in general between C1 and C5) were identified (Wieland and Hummel Reference Wieland and Hummel2015).
The present study was carried out with the aim to set up a well-controlled corrosion experiment with irradiated steel in anoxic alkaline conditions that can be sampled over a long time scale (i.e. up to few years). The development of a very sensitive analytical method for the detection of 14C-containing compounds on the basis of compound-specific 14C analysis (CSRA) using accelerator mass spectrometry (AMS) was required because (1) only small samples of irradiated steel (1–2 g) could be used in the laboratory setup for the corrosion experiment due to the high dose rate of irradiated steel (~30 mSv/h per gram irradiated steel); (2) the 14C inventory of irradiated steel was found to be relatively low (~18 kBq 14C/g); (3) the corrosion rate of stainless steel in alkaline conditions is extremely low (i.e. few nm/a) (Wieland et al. Reference Wieland, Cvetković, Kunz, Salazar and Szidat2017a, Reference Wieland, Cvetković, Kunz, Salazar and Szidat2018). AMS has a much lower detection limit than radioanalytical techniques for 14C detection, e.g. liquid scintillation counting, because AMS does not depend on the disintegration of the radionuclide. Furthermore, the method requires only a small sample volume and a single analysis is fast.
This study describes the setup of the corrosion experiment with irradiated steel and reports results from speciation measurements during the corrosion experiment including identification and quantification of the dissolved 14C-containing organic compounds by AMS and stable carbon bearing compounds by standard analytical techniques. Analysis of the latter compounds enabled us to assess the content of stable carbon to be added during sample preparation for CSRA and further allows a comparison of the stable carbon and 14C speciation.
MATERIALS AND METHODS
Irradiated Steel
The corrosion experiment was carried out with irradiated steel from the nuclear power plant (NPP) Gösgen (Switzerland). In the course of the annual maintenance work in NPP Gösgen in 2012 five guide-tube nuts (~5 g each) were retrieved from the storage pool and transferred to the PSI Hot Laboratory. The nuts made of stainless steel had been mounted at the top and bottom end of fuel rods and were exposed to irradiation in the nuclear reactor core for about two years. The activation cycle was terminated in June 2011. Two nuts were processed to prepare small specimens for laboratory experiments with a dose rate of ~30 mSv/h per gram steel (Wieland et al. Reference Wieland, Cvetković, Kunz, Salazar and Szidat2017a, Reference Wieland, Cvetković, Kunz, Salazar and Szidat2018).
The 14C content of the irradiated stainless steel nuts was determined experimentally by using a wet chemistry digestion technique in oxidative media (Schumann et al. Reference Schumann, Stowasser, Volmert, Guenther-Leopold, Linder and Wieland2014). 14C was released from irradiated steel as 14CO2, which was trapped in NaOH solution. Liquid scintillation counting was employed for 14C activity measurements. The 14C activity was determined to be 17.8 ± 2.5 kBq per gram steel, which corresponds to a 14C content of 0.107 ± 0.015 µg/g. Details of the experimental method and a comparison with theoretical predictions of the 14C content using a Monte Carlo reactor model for NPP Gösgen were reported by Schumann et al. (Reference Schumann, Stowasser, Volmert, Guenther-Leopold, Linder and Wieland2014). In contrast, the content of stable carbon in stainless steel is significantly higher than the 14C content. While the type of the steel used to fabricate the guide-tube nuts is not exactly known, there is evidence to suggest that the nuts were produced from one of the following alloys: X6CrNiTi18-10, X6CrNiNb18-10 or X6CrNiMoTi17-12-2, respectively. The stable carbon content of the latter steels is≤0.08 weight (wt.) % according to DIN 17440 specifications. Consequently, the lower limit of the intrinsic 14C/stable carbon ratio of the steel is 1.34·10–4.
Solution
Throughout this study, the solutions were prepared using Fluka or Merck analytical-grade (“pro analysis”) chemicals. Ultrapure water (Milli-Q® water; 18.2 MΩ·cm) used for the preparation of solutions and standards was generated by a Milli-Q Gradient A10 purification system (Millipore, Bedford, MA, USA).
The corrosion experiment was performed in an artificial cement pore water (ACW) which had concentrations of the major cation (Ca(II)) and anion (OH–) in equilibrium with cement paste degraded to the second stage of cement degradation, that is a pore solution buffered by portlandite (e.g. Berner Reference Berner1992). The solution was prepared as follows: CaO was prepared by heating CaCO3 at 1000°C until the weight remained constant. 2 g CaO was added to 1000 mL degassed ultrapure water, which was prepared by boiling ultrapure water for about 2 hr under a continuous N2 purge to remove dissolved oxygen and carbon dioxide. The suspension was equilibrated for 2 days by using an end-over-end shaker inside a glove box with N2 atmosphere, left standing for 14 hr, filtered under N2 atmosphere through 100 nm polyethersulfone membrane filters (Criticap-M™, Gelman Science, USA) and stored in inert atmosphere conditions until further use. The pore water had a pH=12.5 and Ca was the main cation ([Ca]=2.0·10-2 M).
Analytical Methods
The solutions sampled from the corrosions reactor were analyzed to quantify the dissolved and gaseous stable carbon compounds by high performance ion exchange chromatography (HPIEC) and gas chromatography (GC), respectively, both with mass spectrometry (MS) detection. Furthermore, the total dissolved organic carbon content of stable carbon (TOC) and 14C (TO14C), as well as the dissolved 14C-containing organic compounds by CSRA AMS were determined.
Radioanalysis
Liquid scintillation counting (LSC) was used to check whether the 14C activity was below the limit of detection (LOD). Thus, the LOD of LSC indicated the upper limit of the 14C activity in the sample solutions. LSC was carried out as follows: A 1.5 mL aliquot of the sample was mixed with 3.5 mL ultrapure water and 15 mL scintillator (Ultima Gold XR, Packard Bioscience S.A., Meriden, USA). Beta counting was carried out on a Canberra Packard Tricarb 2250 CA LSC in the energy window between 4 and 65 keV. Standards for radio assay were prepared in the same manner by adding appropriate volumes of a 14C-labeled acetate tracer solution to 1.5 mL acidified ACW and 15 mL scintillator. The counting (2σ) statistics was typically≤2% or better. A count rate of ∼25 cpm was determined for 5 mL ACW blanks.
A series of calibrations were performed on samples containing both 60Co and 14C with the aim to determine the contribution of 60Co to the beta spectrum of 14C. Note that 60Co was found to be the main activation product in irradiated steel (Schumann et al. Reference Schumann, Stowasser, Volmert, Guenther-Leopold, Linder and Wieland2014). To this end, 60Co standards were analyzed both by gamma (energy window between 1050–1400 keV) and beta counting and a conversion factor accounting for the activity contribution of 60Co to the 14C spectrum in the energy window between 4 and 65 keV was assigned.
Ion Chromatography with Mass Spectrometry Analysis
HPIEC was carried out by using an ICS-5000 ion chromatography (IC) system (Dionex/Thermo Fisher, Sunnyvale, CA, USA) consisting of an AS 50 auto sampler, a 250 mm × 2 mm i.d. IonPac AS11-HC column in combination with the corresponding guard column, an EG 40 eluent generator, a 2 mm AERS–500 suppressor operated in the external water mode, a CD 25 conductivity detector (CD) and a coupled MSQ™ Plus MS (Thermo Fisher, Sunnyvale, CA, USA). Data acquisition, processing and IC operation are controlled by the Chromeleon 6.80 (SR 11d) software while the Xcalibur Finnigan Surveyor MSQ 1.1 ELMO is the MS control software. A fraction collector (Foxy Jr. Fraction collector, Teledyne Isco, Lincoln, NE, USA) was connected to the exit of the CD in order to sample fractions based on their previously determined retention times. Parameters and conditions used to operate the equipment (e.g. eluent, temperature, etc.) and a description of the analytical method (preparation of standards, calibration, dynamic range, repeatability and data analysis) are reported in detail elsewhere (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). The following carboxylic acids were analyzed by HPIEC-MS: formic (C1), acetic (C2), oxalic (C2), malonic (C3), glycolic (C2), and lactic (C3) acids, which had been identified as the main oxygenated hydrocarbons in corrosion experiments with non-irradiated iron powders (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). Note that the concentration of the corresponding 14C-bearing compounds was extremely low and therefore could not be quantified by HPIEC-MS.
Gas Chromatography with Mass Spectrometry Analysis
A GC-MS system consisting of a TRACE™ GC Ultra gas chromatograph (Thermo Fisher Scientific Inc., Waltham, MA, USA) equipped with an ISQ mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) in the electron ionization (EI) mode was used. The system is operated with the Thermo Xcalibur 3.0.63 (Thermo Fisher Scientific Inc., Waltham, MA, USA) and the PAL Sample Control (PAL3 System software, CTC Analytics AG, Switzerland) software for data acquisition and processing. Hydrocarbons were analyzed by using a headspace method and a Restek Rt®-Msieve 5A column (30 m × 0.32 mm with 0.025-mm film). Parameters and conditions used to operate the GC-MS (carrier gas, temperature, etc.) and the analytical method (e.g. preparation of standards, calibration) are reported elsewhere (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). The main compounds that were analyzed are volatile hydrocarbons, such as methane, ethane, and ethene, which had been identified as important corrosion products in experiments with non-irradiated iron powders (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). As for the dissolved phase, the concentration of the corresponding 14C-bearing hydrocarbons was too low in the gas phase and could not be quantified by standard GC-MS.
Non-Purgeable Organic Carbon Content
A 2.5-mL cartridge-treated solution (Figure 1) was used to determine the concentration of non-purgeable organic carbon (NPOC) by using a Shimadzu TOC-WP® analyzer. The samples were acidified with 20% H3PO4 prior to analysis. Determination of NPOC is based on UV-promoted persulfate wet oxidation and detection of CO2 by infrared (IR) detection (Glaus and Van Loon Reference Glaus and Van Loon2003). The samples were diluted with ultrapure water to conduct the NPOC measurements in the dynamic range of the method. The samples were analyzed along with 6 standard solutions with carbon concentrations ranging between 1 mg/L and 30 mg/L. The detection limit was ~0.5 mg/L carbon.
Total Organic Radiocarbon
0.8 mL of the aqueous sample collected after cartridge treatment (Figure 1) was transferred into a vial and spiked with 1.25 M 14C-free acetate solution to give a final 12C concentration of 2 µg/µL. The vial was tightly closed and directly used for AMS analysis as described in the Accelerator Mass Spectrometry section. The removal of inorganic 14C by cartridge treatment was determined to be >97% with the help of a 14CO3 2- radiotracer indicating that the AMS analysis accounts for total organic radiocarbon.

Figure 1 Scheme of the analytical protocol of the aqueous samples (dashed: stable carbon analytics; plain: AMS analytics; dotted: radioanalysis).
Compound-Specific Radiocarbon Analysis
Compound-specific radiocarbon analysis (CSRA) with AMS involved two analytical steps: Firstly, chromatographic fractionation of the individual 14C-containing compounds and secondly, 14C detection by AMS. Fractionation of the individual compounds was achieved by using the HPIEC system described previously herein and the analytical procedure reported by Cvetković et al. (Reference Cvetković, Salazar, Kunz, Szidat and Wieland2018b).
The concentrations of the 14C-containing organic compounds in the collected IC fractions were below the detection limit of 14C AMS in the first samplings. As a consequence, a pre-concentration step was developed on the basis of a freeze-drying procedure as follows: 3 or 4 replicates of each compound were collected in pre-cleaned and weighed vials after IC separation (500 µL fraction volume) and 1 µL 0.1 M NaOH was added to adjust pH ~11. At this pH, loss of carboxylates by evaporation was negligible. All samples were immediately frozen in liquid nitrogen, transferred into the freeze dryer (Alpha 1-2 LDplus, Christ, Osterode, Germany), dried for 3 hr and weighed to determine the pre-concentration factor. Then, the replicates were combined to one single sample and subjected to an additional freeze-drying step for about 3 hr. After the second freeze-drying step, the samples were weighed to determine the overall pre-concentration factor. The samples were thawed and the pH was re-adjusted to ~5–6 by adding the appropriate volume of 0.1 M HCl.
In addition to the IC fractions, ultrapure water blank samples were prepared and treated in the same manner to account for possible contaminations during the pre-concentration step. Eventually, the samples were spiked with fossil 12C carrier (acetate) to give a final carbon concentration of 2 µg/µL 12C in the AMS measurement.
Accelerator Mass Spectrometry
Carbon-14 AMS measurements were carried out on the MIni CArbon DAting System (MICADAS) (Synal et al. Reference Synal, Stocker and Suter2007) at the Laboratory for Radiocarbon Analysis (LARA) at the University of Bern (Szidat et al. Reference Szidat, Salazar, Vogel, Battaglia, Wacker, Synal and Türler2014). The samples were analyzed with an elemental analyzer (EA) coupled online with the MICADAS based on a gas interface, which is reported elsewhere (Ruff et al. Reference Ruff, Fahrni, Gäggeler, Hajdas, Suter, Synal, Szidat and Wacker2010; Salazar et al. Reference Salazar, Zhang, Agrios and Szidat2015). Briefly, aqueous samples or standards (both 10 µL) were tightly packed in tin foil for flash combustion, loaded into the oxidation oven of the EA (ELEMENTAR, Hanau, Germany) at 850°C by an autosampler and combusted with a pulse of oxygen. The EA directs the gases through a water trap containing Sicapent (Merck, Germany) and through a zeolite trap, which is heated up stepwise to consecutively release N2, CO2 and residual gases. The outlet of the EA is connected to a gas interface system (GIS) through a 1/16′′ O.D. tubing (10 m long), which directs the flow (80 mL/min) to a second zeolite trap (zeolite X13, Sigma–Aldrich, Germany) located at the GIS. The GIS trap is gradually heated to 450°C and releases the CO2 into a syringe of known volume. A manometer is used to determine the amount of carbon. Helium was added to make a mixture of 10% CO2 at ~0.16 MPa, and finally the mix is transferred into the gas ion source of the MICADAS at a flow rate of ~40 µL/min. The EA-GIS system can be flushed (ca. 4 min) at the same time that the sample is being analyzed. This analytical procedure includes running a blank through the whole process in between each sample. This blank not only cleans the EA from residues of the previous sample, but it also cleans the heated GIS trap at the high helium flow provided by the EA. The procedure is automated and controlled by a LabView program based on an earlier version reported by Wacker et al. (Reference Wacker, Fahrni, Hajdas, Molnar, Synal, Szidat and Zhang2013). The results can be corrected for cross contamination thanks to the fact that the carry-over effect is relatively constant for the given working range of 14C/12C concentrations (Cvetković et al. Reference Cvetković, Salazar, Kunz, Szidat and Wieland2018b).
The standards used to calibrate the AMS were solid crystals of sodium acetate (fossil; p.a., Merck, Germany), C5, C6 and C7 from IAEA and Oxalic Acid II from NIST (SRM 4990C) with a fraction modern (F14C) of 0.2305±0.0002, 1.5061±0.0011, 0.4935±0.0012 and 1.3407±0.0005, respectively. Each sample has to contain a defined 12C content by adding 14C-free (fossil) carrier to allow for measurement of the 14C/12C by AMS. To this end, a sodium acetate solution (30 µg 12C/µL) was prepared to spike the samples with 12C. The final 12C concentration of the samples was 2 µg 12C/µL. After addition of the 12C carrier, the samples were immediately frozen and stored at −20°C prior to the AMS measurements.
The results reported for 14C measurements in this study are presented in terms of molarity (mol 14C/L) of the sampled solutions. The latter unit depends on the experimental conditions, in particular volume of sample solution and addition of carrier. For the current experimental setup, a conversion factor can be applied to interconvert the different units (Cvetković et al. Reference Cvetković, Salazar, Kunz, Szidat and Wieland2018b):
 $${\rm 1}\,{\rm F}^{{{\rm 14}}} {\rm C}\,\equiv\,{\rm 2}{\rm .74\cdot10}^{{{\rm -12}}} \,{\rm g}^{{{\rm 14}}} \,{\rm C\,/\,L {\equals} 195}{\rm .7}\,{\rm fmol}^{{{\rm 14}}} {\rm C\,/\,L {\equals} 0}{\rm .452 Bq\,/\,L}$$
$${\rm 1}\,{\rm F}^{{{\rm 14}}} {\rm C}\,\equiv\,{\rm 2}{\rm .74\cdot10}^{{{\rm -12}}} \,{\rm g}^{{{\rm 14}}} \,{\rm C\,/\,L {\equals} 195}{\rm .7}\,{\rm fmol}^{{{\rm 14}}} {\rm C\,/\,L {\equals} 0}{\rm .452 Bq\,/\,L}$$
14C-free sodium acetate solution was used to prepare blank samples (2 µg 12C/µL). The blanks were run to identify possible contaminations and were subjected to the same treatments as the samples. Furthermore, blank samples were run during the HPIEC analysis and prepared for each set of injections. The blank samples were either prepared in ultrapure water or ACW solution that had been passed through the OnGuard® cartridge. The blanks were treated and injected into the IC system in the same conditions as the samples and the blank fractions were collected at the retention time of the targeted sample fractions and analyzed by AMS along with the sample fractions.
Experimental Protocol
Setup of Reactor
The experimental setup for the corrosion experiment with irradiated steel consists of a custom-made gas-tight overpressure reactor (stirred autoclave type “Versoclave Type 3”, Büchi AG, Uster, Switzerland) placed inside an in-house made cage with a 10-cm-thick lead shielding (Wieland et al. Reference Wieland, Cvetković, Kunz, Salazar and Szidat2017a, Reference Wieland, Cvetković, Kunz, Salazar and Szidat2018). Leak tests had confirmed gas-tightness of the reactor. An in-house liquid and gas sampling system was constructed to allow regular sampling without opening the reactor (see Sampling section). The reactor has a PEEK inlet and is equipped with a digital pressure transmitter, a temperature sensor and a sensor to detect dissolved oxygen (Visiferm DO Arc, Hamilton, USA). Pressure, temperature, and the concentration of dissolved O2 were recorded during the samplings and checked between the samplings (Wieland et al. Reference Wieland, Cvetković, Kunz and Tits2017b). Temperature was found to vary between 22.7 and 24.4 °C. Pressure varied only slightly between the samplings. Small pressure changes might be due to temperature variation. The O2 concentration ranged from ~39 to ~59 ppb, which is considered to be close to the detection limit of the sensor. The apparent increase in the O2 concentration could be caused by changes in the sensitivity of the sensor with time (e.g. also due to temperature changes).
The overpressure reactor was designed in such a way that all manipulations necessary for regular sampling can be carried out outside the lead shielding to minimize exposure to radiation (Wieland et al. Reference Wieland, Cvetković, Kunz, Salazar and Szidat2017a, Reference Wieland, Cvetković, Kunz, Salazar and Szidat2018).
Start of Experiment
The reactor vessel was pre-washed with ultrapure water and ACW. After closing the reactor and purging it with N2, 300 mL ACW was introduced. To this end, a gas-tight metallic sample container (500 mL) was evacuated, purged with N2 and again evacuated to transfer 300 mL ACW under the inert atmosphere conditions. The container was connected to the inlet and outlet of the sampling system for fluids. The solution was transferred into the reactor by applying a N2 gas overpressure. After closing the inlet valve, an absolute pressure of 5 bar was applied to the reactor. The solution was regularly sampled over a time period of 14 days and analyzed with the aim to determine background 14C activities and concentrations of the organic compounds in the aqueous and gaseous phase (total organic 14C by AMS, carboxylic acids by HPIEC, hydrocarbons by GC-MS, NPOC).
The corrosion experiment was started after completely removing the ACW solution used for the background tests. Two irradiated steel nut specimens were mounted to a sample holder which is connected to the stirring unit of the reactor (Wieland et al. Reference Wieland, Cvetković, Kunz, Salazar and Szidat2017a). 300 mL ACW was transferred into the reactor. An absolute pressure of 5 bar N2 was applied to the gas volume (200 mL). The system was equilibrated for 2 days. Then, the entire volume of 300 mL ACW was sampled and discarded. 300 mL fresh ACW solution was transferred into the reactor and the pressure was adjusted to 5 bar N2.
Sampling
7-mL sampling tubes made of stainless steel were used to store the aqueous samples withdrawn from the corrosion reactor. Each tube is equipped with two valves at the inlet and outlet. A sampling tube was connected to the liquid outlet valve of the corrosion reactor, evacuated and purged with N2 for at least three times to remove oxygen. Fluid was withdrawn from the reactor by opening the valves in-between the reactor and the evacuated sampling tube. The tube was closed, disconnected from the reactor and transferred into a glove box with N2 atmosphere. Each sampling tube was weighed before and after sampling to determine the exact sample volume.
In the glove box, the valves of the sampling tube were opened and the alkaline solution released into a pre-cleaned 20 mL vial. The sample was split: One portion was directly used for further analysis (2.5 mL; no pretreatment) and the second portion (4.5 mL) was subjected to a cartridge pre-treatment (OnGuard Ag/Ba/H-cartridge, ThermoFisher Scientific/Dionex, Sunnyvale, CA, USA) to remove interfering anions (e.g. Cl–) and to adjust pH ~6 (Figure 1).
Gas samples were collected by using a 50-mL gas collecting tube, which was connected to the gas outlet of the corrosion reactor. The gas collecting tube was evacuated and purged with nitrogen for at least three times prior to the sampling. The gas samples were collected instantaneously by opening the valves in-between reactor and the gas collecting tube due to the applied absolute pressure of 5 bar in the reactor.
The gas sample was released from the gas collecting tube into a plastic gas-sampling bag (0.5 L gas bag, SKC Limited, Dorset, UK) to allow pressure balance to ambient conditions (1 bar). For the analysis a headspace vial was evacuated and a 10-mL sample withdrawn from the gas-sampling bag was injected through the septum by using a gas-tight syringe equipped with a valve (Hamilton AG, Bonaduz, Switzerland). A second gas-sampling bag filled with nitrogen was connected to the GC vial for 5 seconds to allow pressure compensation in the sample vial. The sample was injected into the GC-MS system using the headspace method. It should be noted that the concentrations reported for the gaseous species relate to pressure adjusted to ambient conditions (1 bar).
After sampling both the aqueous and gaseous phase 7 mL fresh ACW solution was transferred into the reactor via the sampling tube by applying N2 overpressure. The gas pressure in the reactor was then adjusted to its initial value (5 bar N2 absolute pressure).
RESULTS
Determination of Background Parameters
The background parameters of the reactor system (e.g. 14C activities, concentration of organic compounds etc.) were determined prior to starting the corrosion experiment with irradiated steel. The solution was regularly sampled over a period of 15 days (Table 1).
Table 1 Background concentrations of carboxylic acids, NPOC and TO14C.

* Analysis of carboxylic acids: FA: formic acid (LOD=3 µM, LOQ=5 µM); AA: acetic acid (LOD=3 µM, LOQ=5 µM); OA: oxalic acid (LOD=0.05 µM, LOQ=0.1 µM; MA: malonic acid (LOD=0.05 µM, LOQ=0.15 µM); GA: glycolic acid (LOD=0.2 µM, LOQ=0.5 µM); LA: lactic acid (LOD=0.2 µM, LOQ=0.5 µM). Presence of additional carboxylates was checked, e.g. butyric acid (LOD=0.2 µM, LOQ=0.5 µM), propionic acid (LOD=0.2 µM, LOQ=0.5 µM) and valeric acid (LOD=1 µM, LOQ=3 µM), which were found to be below the detection limit (LOD).
# An aliquot of the sampled solution was subjected to chromatographic separation and the concentration of 14C-bearing formate, acetate, oxalate, and malonate was determined in the respective HPIEC fractions by AMS. The following concentrations were determined: formate: 0.011 F14C; acetate: 0.020 F14C; oxalate; 0.071 F14C; malonate: 0.043 F14C.
The NPOC of ACW sampled from the reactor was ~650 µM. Hence, it increased by ~300 µM within 1 hr upon contact with the PEEK liner as the blank NPOC of ACW was determined to be a factor ~2 lower (342 µM) (Table 1). Note that both parameters, i.e. NPOC and the concentrations of the carboxylates were constant after 1 hr contact between the PEEK liner and ACW. Hence, it seems that the PEEK liner is a significant carbon source in the reactor. However, HPIEC-MS analysis of carboxylates, which are considered to be important corrosion products (Wieland and Hummel Reference Wieland and Hummel2015), showed that their concentrations were very low in these samples, i.e. slightly above the LOD in the case of formate and acetate and below the LOD in the case of oxalate, malonate, glycolate, and lactate. The above findings suggest that the high NPOC background is presumably caused by the presence of polymeric, still unidentified organic material rather than small organic molecules such as carboxylates.
The blank concentration of 14C was determined to be 0.095 F14C in ACW before injection into the reactor system (Table 1). The background could be caused by the presence of trace concentrations of 14CO2 in ACW. This value is a factor ~2 higher than the 14C blank obtained during chromatographic separation of the 14C-containing organic compounds by HPIEC (Cvetković et al. Reference Cvetković, Salazar, Kunz, Szidat and Wieland2018b). The 14C blank (TO14C) further increased to 0.36 F14C after contacting ACW with the PEEK liner in the closed reactor system. Chromatographic analysis of an aliquot showed, however, that the14C blank of the individual carboxylate fractions ranged between 0.011 F14C in the case of formate and 0.071 F14C in the case of oxalate (Table 1). These values are close to the background value determined earlier (0.06±0.02 F14C) (Cvetković et al. Reference Cvetković, Salazar, Kunz, Szidat and Wieland2018b) revealing that the concentration of 14C-containing carboxylates is negligible in the blank solutions.
The results from the background tests showed that other carbon sources in addition to the irradiated steel could be present in the reactor. Nevertheless, this material was not contained in any of the fractions of the individual carboxylates that were collected after chromatographic separation. Therefore, the carbon contaminants do not contribute to the 14C concentration of the individual 14C-bearing organic compounds that are expected to be formed during the corrosion of irradiated steel and that are sampled after chromatographic separation. Note further that the blank 14C activity was below the LOD of LSC in the ACW solution used to pre-clean and pre-condition the PEEK liner.
Corrosion Experiment with Irradiated Steel
The results obtained within the first 412 days reaction are summarized in Tables 2–5. The concentrations have been corrected for each dilution of both the aqueous and the gaseous phase caused by the injection of 7 mL fresh ACW and pressure adjustment after each sampling.
Table 2 NPOC and concentrations of the stable carbon carboxylic acids (formic acid (FA), acetic acid (AA), oxalic acid (OA), malonic acid (MA), glycolic acid (GA), lactic acid (LA) determined by HPIEC-MS.

* For LOD and LOQ see Table 1. n.d.=not determined.
Table 3 GC-MS measurements of gaseous stable carbon compounds in samples at ambient pressure.

* LOD and LOQ of hydrocarbons are as follows: methane (LOD=0.02 µM, LOQ=0.06 µM) and ethene (LOD=0.01 µM, LOQ=0.03 µM). Presence of additional hydrocarbons was checked, e.g. ethane (LOD=0.07 µM, LOQ=0.2 µM), acetylene (LOD=0.005 µM, LOQ=0.016 µM), propane (LOD=0.05 µM, LOQ=0.15 µM), propene (LOD=0.01 µM, LOQ=0.02 µM) and butane (LOD=0.03 µM, LOQ=0.09 µM) which were found to be below the LODs.
Table 4 Total 14C activity determined by LSC and AMS (TO14C) and 60Co activity.

* LOD of LSC: 25 cpm in 5 mL ~100 Bq/L. LOQ of LSC: 35 cpm in 5 mL ~ 140 Bq/L.
# The data are corrected for the background value (0.59 F14C or 0.27 Bq/L, respectively). Conversion factor as given in Equation (1).
+n.d.=not determined. LOD of gamma counting: 20 cpm in 5 mL ~300 Bq/L. LOQ of gamma counting: 30 cpm in 5 mL ~450 Bq/L.
Table 5 14C-bearing formate, acetate and lactate analyzed by AMS after 412 days reaction by applying the pre-concentration step. Note that malonate and oxalate fractions were not analyzed. The concentrations of the individual 14C-bearing carboxylic acids were below the LOD of the 14C AMS for a direct measurement without pre-concentration.

* LOD of AMS method with pre-concentration: 0.007 F14C
# Measured concentrations were corrected using the dilution factor resulting from chromatographic separation by HPIEC.
NPOC increased with progressive reaction from initially ~200 µM to ~550 µM after 412 days (Table 2). Note that the NPOC almost doubled between the early phase of the experiment (t<30 days; 204 µM) and after 93 days reaction (390 µM). The concentration of formate increased with time, i.e. from 2 µM after 29 days to 15 µM after 412 days reaction. In the same time interval the acetate concentration increased from<3 µM to 10 µM. The lactate concentration slightly increased over the same time interval while its concentration was substantially lower than those of formate and acetate. In contrast, the concentrations of the other carboxylates, i.e. oxalate, malonate, and glycolate, were found to be almost constant within the uncertainty of the analytical method while their values were still close to the LOD.
Methane and ethene were the only hydrocarbons detected in the gas phase (Table 3). Note that the concentration of ethene is close to the LOD while methane is the only gaseous carbon species showing a pronounced increase in concentration with time. Other hydrocarbons that had previously been observed in iron-water systems (Wieland and Hummel Reference Wieland and Hummel2015) were below the LOD of GC-MS. At the time being, however, we are not able to identify and quantify the 14C-bearing hydrocarbons in the gas phase. The required analytical procedure based on compound-specific 14C AMS is still under development.
The 14C activity determined by LSC corresponds to the total 14C activity in an aliquot of the sample collected from the reactor, i.e. prior to cartridge pretreatment (Figure 1). Thus, the 14C activity results from the presence of all 14C-containing compounds irrespective of their speciation. The 14C activity measurements have large uncertainties (estimated at>50%) due to presence of 60Co, which significantly contributes to the count rate in LSC in the energy range between 4 and 65 keV (>80% of counts). Therefore, the 14C activity determined by LSC is considered to be a non-specific measure of the 14C activity poorly constrained by carbon speciation. In the present study, the 14C activity determined by LSC was only used as guidance in conjunction with the preparation of the 14C AMS samples.
The TO14C accounts for the 14C activity associated with all 14C-containing organic compounds produced during the corrosion of irradiated steel. The results from AMS measurements (F14C) were corrected by subtracting the background determined immediately after starting the corrosion experiment (0.59 F14C). TO14C increased up to 93 days reaction, thus indicating progressive corrosion of irradiated steel (Table 4). The TO14C content of the solution sampled at 412 days reaction, however, was significantly lower.
Identification and quantification of the individual 14C-containing organic compounds by CSRA was not possible for the samplings up to 286 days, because the concentration of these compounds was below the LOD. The pre-concentration step based on freeze-drying (section Compound-Specific Radiocarbon Analysis) allowed the 14C concentration of the separated carboxylate fractions to be increased above the LOD of CSRA. The activities of the acetate and formate fractions were determined to be 0.007 Bq/L (0.015 F14C) in the sample collected after 412 days reaction (Table 5), which demonstrates the need of a pre-concentration step for detecting the very low concentrations of 14C-bearing organic compounds in this experiment with irradiated steel. Lactate was also detected slightly above the LOD (0.007 F14C).
The TO14C content was calculated to be 0.75 Bq/L (1.66 F14C) by adding up the activities of the three individual fractions. Note that this estimate of TO14C was based on the assumption that the three carboxylates carry only one 14C (single labelled). The calculated TO14C agrees well with the result from the direct measurement of TO14C (1.46 F14C, Table 4). This comparison shows that the entire TO14C content can be attributed to the three fractions formate, acetate, and lactate, which account for 112 ± 25 % of the directly measured TO14C listed in Table 4. This finding further reveals that the cartridge treatment removed possible 14C background contributions from solution, in particular 14C inorganic carbon.
DISCUSSION
This study shows that the concentrations of the 14C-bearing organic compounds are extremely low (fmol to pmol 14C/L) for the given setup of a corrosion experiment with small irradiated steel nut specimens. Nevertheless, CSRA by AMS allows quantification of these compounds at the given very low concentrations. Formate and acetate were identified as the most important dissolved 14C-bearing compounds formed during the anoxic corrosion of irradiated steel. Lactic acid was also present, however, at concentrations close to the LOD. Formate and acetate have previously been identified as the most important dissolved carbon species in corrosion studies with non-irradiated iron powders (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). In general, it was observed that, firstly, only a limited number of small organic compounds with a carbon chain length between C1 and C5 are formed during anoxic corrosion of iron/steel, and secondly, both oxidized (carboxylic acids, probably also alcohols and aldehydes) and reduced (methane etc.) carbons species are present in anoxic iron-water systems (Wieland and Hummel Reference Wieland and Hummel2015; Swanton et al. Reference Swanton, Baston and Smart2015). The first results from the corrosion experiment with irradiated steel thus show that similar carboxylic acids are formed during the anoxic corrosion of non-irradiated and irradiated iron/steel.
After 412 days reaction, the TO14C content was directly determined in the sample solution subjected to a cartridge treatment (TO14C=(2.86±0.31) · 10–13 M). The TO14C content can further be calculated from the sum of the concentrations of the individual 14C-bearing carboxylates determined in the same sample (TO14C=(3.21±0.41) · 10–13 M). Hence, both values agree well within the given uncertainty. This finding shows that formate, acetate and lactate belong to the main 14C-bearing species that contribute to the TO14C of the solution.
The time-dependent release of organic stable carbon- and 14C-containing compounds in the reactor (in moles) was tentatively calculated as a function of corrosion rate as follows:
 $${\rm m}_{{{\rm C}{\minus}12\,/\,13}} ({\rm t}){\equals}\mathop{\int}\limits_0^t {{{{\rm R}_{{\rm c}} {\rm A\rho }v_{{{\rm C}{\minus}12\,/\,13}} } \over {{\rm M}_{{\rm w}} }}} {\rm dt}\quad {\rm m}_{{{\rm C}{\minus}14}} ({\rm t}){\equals}\mathop{\int}\limits_0^t {{{{\rm R}_{{\rm c}} {\rm A\rho }v_{{{\rm C}{\minus}14}} } \over {{\rm M}_{{\rm w}} }}} {\rm dt}\quad {\rm [mol]}$$
$${\rm m}_{{{\rm C}{\minus}12\,/\,13}} ({\rm t}){\equals}\mathop{\int}\limits_0^t {{{{\rm R}_{{\rm c}} {\rm A\rho }v_{{{\rm C}{\minus}12\,/\,13}} } \over {{\rm M}_{{\rm w}} }}} {\rm dt}\quad {\rm m}_{{{\rm C}{\minus}14}} ({\rm t}){\equals}\mathop{\int}\limits_0^t {{{{\rm R}_{{\rm c}} {\rm A\rho }v_{{{\rm C}{\minus}14}} } \over {{\rm M}_{{\rm w}} }}} {\rm dt}\quad {\rm [mol]}$$
Rc: Corrosion rate [m/a]
A: Surface area of specimens in reactor (A = mFe · Sm) [m2]
mFe: Mass of the two specimens (= 1.718·10–3 kg)
Sm: Mass-related specific surface area (= 0.649 m2/kg)
Mw: Molecular weight of iron (0.055845 kg/mol)
ρ: Density of iron (7855 kg/m3)
υ: Stoichiometric factor of 14C (= 4.27 · 10–7) and stable carbon (12/13C) (= 3.72 · 10–3) expressed in terms of mol 14C/mol Fe or mol stable carbon/mol Fe of irradiated steel (see Irradiated Steel section).
Equation (2) is based on the assumption that (1) irradiated steel is the only source of 14C and stable carbon in the reactor, (2) 14C and stable carbon released from irradiated steel was converted either completely into gaseous or aqueous carbon species, respectively, and (3) no other sources and sinks for 14C and stable carbon exist. The release of 14C and stable carbon concentrations to the aqueous and gaseous phases, respectively, was calculated with Equation (2) and normalized to the surface area of irradiated steel. In case of the gaseous components the calculated concentration in the gas phase may be underestimated as methane and ethene are partially soluble in the aqueous phase. However, scoping calculations showed that the error is negligibly small as only 1.0±0.3% of the gases are dissolved in the aqueous phase based on Henry’s law solubility constants of methane and ethene corrected for the “salting out” effect according to the ionic strength of the pore solution (Burkholder et al. Reference Burkholder, Sander, Abbatt, Barker, Huie, Kolb, Kurylo, Orkin, Wilmouth and Wine2015; Sander Reference Sander2015).
It should be noted that the above assumption only allows a simplified prediction of the concentrations with time as it has been shown that both gaseous and aqueous carbon species are formed simultaneously during the course of the corrosion process of irradiated steel (De Visser-Tynová et al. Reference De Visser-Tynová, Stijkel, Swanton, Otlet and Walker2017). However, the ratio of gaseous to aqueous 14C-bearing species can presently not be quantified from this study, as the CSRA AMS method for the gaseous 14C species is not yet fully developed. It should further be noted that 14C is considered to be the relevant tracer to study the corrosion process rather than stable carbon, because only the source of 14C in the system can clearly be assigned, that is irradiated steel.
The release of the total 14C to solution (TO14C) can be modeled on the assumption that fast corrosion occurred in the early phase (20 nm/a) followed by significantly slower corrosion in the long term (2 nm/a) (Figure 2a). As previously noted, the TO14C may not account for all 14C species present in the reactor because gaseous 14C species and 14C-containing inorganic species may also be formed during corrosion of irradiated steel. Therefore, the rates of 2 and 20 nm/a correspond to lower limits as they are based on the assumption that the total amount of 14C released from irradiated steel is present in the dissolved organic chemical form (TO14C).
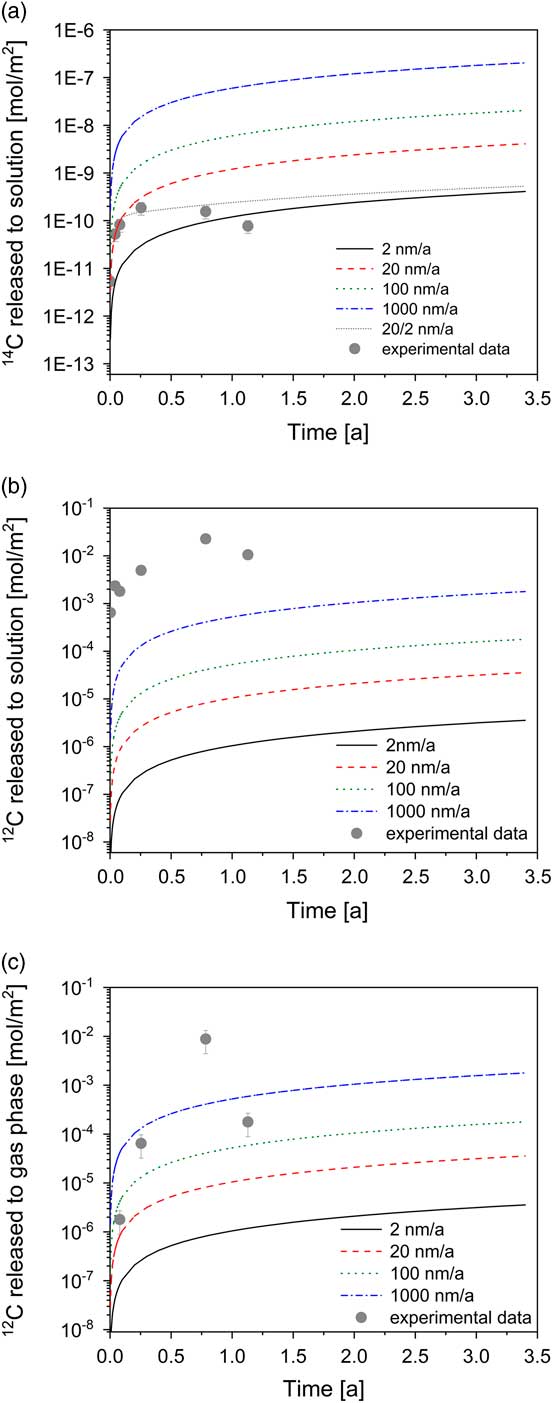
Figure 2 Modeling of the total 14C and stable carbon concentrations in the aqueous and gas phase as function of the corrosion rate (lines) for the experimental setup and comparison with experimental data. Conservative modeling assumption: total amount of 14C and stable carbon released from steel is present either completely in the aqueous or completely in the gas phase (expressed in terms of moles).
Nevertheless, the results show that the release rates of 14C are consistent with corrosion rates reported for stainless steel in strongly alkaline conditions (e.g. Smart et al. Reference Smart, Blackwood, Marsh, Naish, O’Brien, Rance and Thomas2004; Diomidis Reference Diomidis2014; Swanton et al. Reference Swanton, Baston and Smart2015). In the long term, the TO14C seems to remain constant or even slightly decrease with time. A decrease would suggest decomposition of the dissolved compounds, e.g. by impurities of microorganisms, enzymes, or other catalytic agents. At present, however, it is not clear whether or not this decrease is statistically significant, i.e. outside the uncertainty range of the experiment. Further samplings will enable us to assess the trend.
Time evolution of the concentration of the stable carbon-containing compounds could provide additional information on the corrosion process if irradiated steel were the only source of stable carbon in the reactor. An increase in the formate, acetate and lactate concentrations within the first 286 days supports the idea that the production of the stable carbon-containing carboxylates results from the corrosion of irradiated steel (Table 2). In addition, the concentrations of formate and acetate decreased after 412 days in agreement with a decrease in the TO14C content (Figure 2a). Methane and ethene were found to be the dominant gaseous stable carbon species (Table 3). Note that both compounds had been identified as important gaseous carbon species formed during the corrosion of non-irradiated iron powders (Cvetković et al. Reference Cvetković, Rothardt, Büttler, Kunz, Schlotterbeck and Wieland2018a). The modeling results, however, indicate that the experimentally determined concentrations of both gaseous and dissolved stable carbon-bearing compounds are significantly higher than the modeled concentrations (Figures 2b, 2c). As it is unlikely that the carbon content of stainless steel was underestimated, which gives rise to underestimating the predicted stable carbon concentrations in the system, the discrepancy between the release rates of 14C and stable carbon suggests that either 14C and stable carbon are released by very different kinetics or that an additional source contributes to the production of dissolved (i.e. formate, acetate, lactate) and gaseous (methane, ethene) stable carbon-containing compounds with time. At the present time it is not possible to discriminate between the two scenarios while the high NPOC determined in solutions supports the idea of an additional stable carbon source in the reactor. Nevertheless, it is to be noted that the kinetics of stable carbon and 14C release are not necessarily identical as their chemical form in steel may be different, i.e. carbide carbon in the case of stable carbon and single 14C atoms produced by nitrogen activation
SUMMARY AND CONCLUSIONS
An experimental setup enabling corrosion experiments under reducing alkaline conditions in a gas-tight reactor with small sample specimens of irradiated steel was successfully developed. This reactor was placed behind a lead shielding to avoid excessive irradiation during sampling caused by the high dose rate of irradiated steel specimens. The setup allows continuous monitoring of important physico-chemical parameters (pressure, temperature, dissolved oxygen) and further allows aliquots of fluid and gas to be sampled from the reactor. It was estimated that, for the given experimental setup, the concentration of 14C-containing compounds in solution would be extremely low (fmol to pmol range), which prompted us to apply the very sensitive 14C detection by AMS. The AMS-based analytical methods used in this study enabled us to quantify TO14C and individual organic 14C-containing compounds by CSRA. In addition, the use of conventional techniques (HPIEC-MS, GC-MS) allowed the concentration of stable carbon-containing compounds in the aqueous and gaseous phase to be quantified.
The first results show that 14C AMS can be employed to determine the total concentration of 14C-bearing organic compounds (TO14C). The TO14C content was found to increase with time up to 92 days, thus indicating the production of aqueous 14C-bearing organic compounds during progressive corrosion of irradiated steel. The rate of release of dissolved 14C-containing compounds is ~20 nm/a in the early stage of the corrosion process while release decelerates in the long run (~2 nm/a). Both rates are considered to be lower limits while they are consistent with the corrosion rate of stainless steel in hyper-alkaline media.
Determination of individual 14C-bearing organic compounds was successful upon the development of a pre-concentration procedure in combination with CSRA AMS. The study clearly demonstrates that CSRA AMS is well suited to perform 14C speciation measurements in conjunction with the given setup of a corrosion experiment with irradiated steel.
The production of the stable carbon-bearing compounds is not yet understood. Discrepancy between experimental data and modeling suggest that either the release of stable carbon from irradiated steel is significantly enhanced compared to 14C or that an additional source of stable carbon exists in the reactor which gives rise to the high concentration of stable carbon carboxylates and stable carbon methane and ethane detected in the aqueous and gas phase.
Information on the 14C speciation during the corrosion of irradiated metallic waste materials (e.g. irradiated steel, Zircaloy) is required with the aim to support safety assessment and to allow long-term predictions of the 14C contribution to dose release from a repository for radioactive waste to be made. The recently started corrosion experiment with irradiated steel from NPP Gösgen, Switzerland, allows the required information on the 14C speciation to be acquired thanks to the development of a CSRA-based methodology for 14C quantification in the aqueous and gaseous phase.
ACKNOWLEDGMENTS
We thank NPP Gösgen for providing the irradiated steel nuts and Ines Günther-Leopold (PSI), Matthias Martin (PSI) and Robin Grabherr (PSI) for sample preparation. Partial funding for the project was provided by swissnuclear (Project title: “Investigation of the chemical speciation of 14C released from activated steel”) and by the National Cooperative for the Disposal of Radioactive Waste (Nagra), Switzerland. The project has further received funding from the European Union’s European Atomic Energy Community’s (Euratom) Seventh Framework Programme FP7/2007-2013 under grant agreement no. 604779, the CAST project.








 Open access
Open access