


Introduction
Psychiatric disorders are relatively common in terms of lifetime prevalence and are associated with considerable distress and functional impairment (Whiteford et al. Reference Whiteford, Degenhardt, Rehm, Baxter, Ferrari and Erskine2013). Understanding the etiology of these disorders is of critical importance to developing effective treatments and reducing suffering. There is strong evidence that these disorders are complex and partly genetic in origin, with twin study heritability estimates of 40–80% (Polderman et al. Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven and Visscher2015). Environmental factors also contribute and possibly moderate genetic risk. This review will consider two important related hypotheses: that psychiatric disorders share genetic risks with variation in relevant population traits (illustrated in Fig. 1a) and that there are shared genetic contributions across different psychiatric phenotypes (illustrated in Fig. 1b).
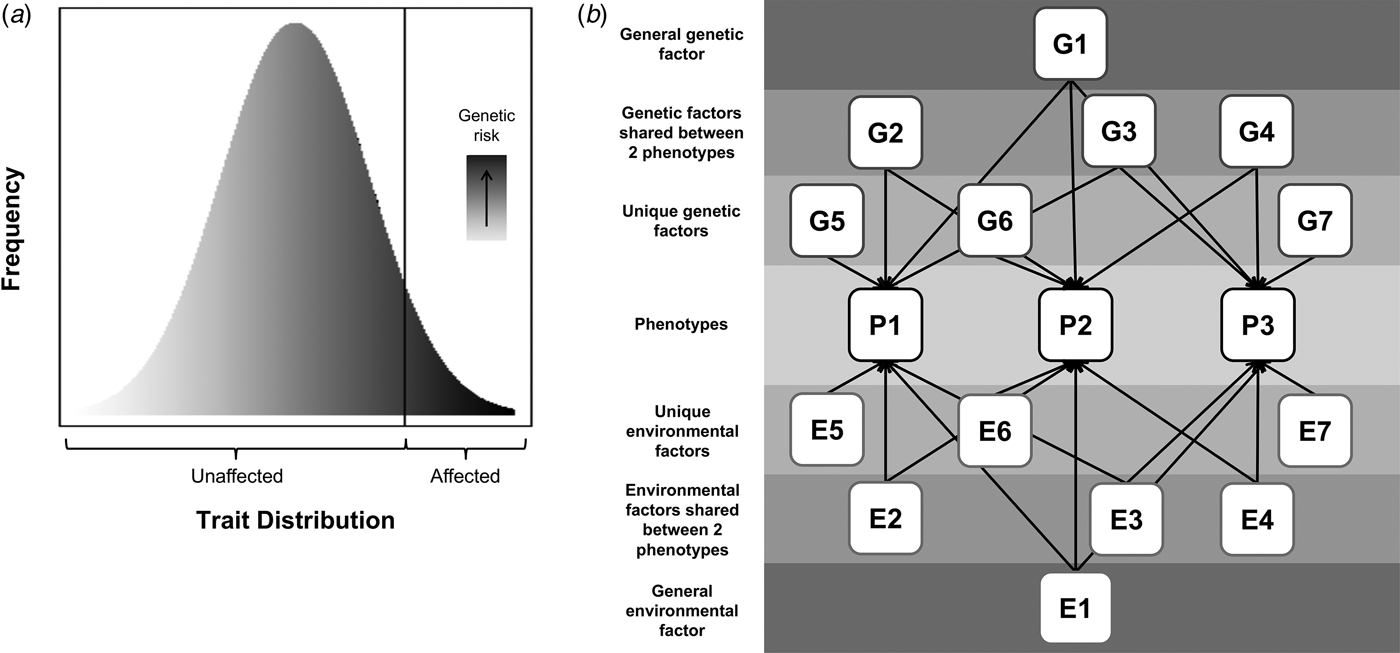
Fig. 1. Hypothesized models of: (a) shared genetic risks across disorder and population trait variation, where the extreme end of a continuous distribution of a trait is associated with a continuous underlying genetic liability, and (b) shared genetic risks across different disorders, where squares labeled ‘P’ represent phenotypes, and squares labeled ‘G’ and ‘E’, represent genetic or environmental contributions, respectively, that can be shared or unique (indicated by the number of arrows pointing to phenotypes). All G factors are uncorrelated with one another and thus the entire genetic contribution to a phenotype can be modelled as the sum of the genetic factors contributing to it (e.g. for P1 this would be G1 + G2 + G3 + G5). The same is true for environmental factors (i.e. environmental contribution to P1 is E1 + E2 + E3 + E5). As an illustrative example, if P1 were ADHD, P2 were ASD, and P3 were MDD, then G1 represents any genetic variants that are shared between ADHD, ASD, and MDD; G2–G4 represents genetic variants shared between only two of these disorders (e.g. G2 would be genetic risk for ADHD and ASD but not MDD); and G5–G7 represent unique genetic risks (e.g. G5 is genetic risk that is unique to ADHD and not shared with either ASD or MDD). N.B. The shapes are not indicative of whether a variable is latent or measured.
The hypothesis that psychiatric disorders are extreme manifestations of continuously distributed population traits is not new [e.g. for a theoretical review see (Plomin et al. Reference Plomin, Haworth and Davis2009)]. However, studies specifically testing whether categorical, clinical disorders share genetic risks with continuous variation in related sub-diagnostic traits in the population have been sparse until recently. A pressing matter that needs to be evaluated for specific psychiatric phenotypes, is the extent to which the current evidence supports this hypothesis. Recent years have also seen a dramatic increase in studies examining the related issue of shared genetic risks across different psychiatric disorders. Given the fast-growing body of research on this subject, the time is ripe to assess the strength of the evidence of shared risks for specific pairs of psychiatric phenotypes. In this review, we summarize and evaluate the evidence for the two hypotheses illustrated in Fig. 1, for a range of psychiatric phenotypes that have been extensively studied using both traditional twin and molecular genetic methods. We also discuss possible interpretations and implications for genetic research and clinical practice. Based on a non-exhaustive literature search, studies were included if they formally tested for shared genetic risks across a psychiatric disorder and traits related to the same disorder or across different psychiatric phenotypes (either defined as disorder or traits). Twin studies using DeFries-Fulker analysis were also included, although these studies do not directly test for genetic correlation; see additional discussion below.
Overview of twin and molecular genetic methods
Most evidence concerning shared genetic risks within and across phenotypic constructs comes from twin studies and common variant genome-wide analyses. The twin design, which relies on comparison of identical (monozygotic) and non-identical (dizygotic) individuals, is commonly used to estimate the heritability of individual traits. Of particular relevance is the DeFries-Fulker analytic method, which estimates group heritability. Group heritability indicates the degree to which the mean difference between a proband group and the rest of a given sample is influenced by genetic factors. Significant group heritability indicates similar etiology for milder variation in continuous traits and more severe manifestations. An extension of the twin design, the bivariate twin model, allows one to estimate the degree of genetic correlation (r g) between two phenotypes. Complementarily, molecular genetic methods directly test for shared genetic risks across phenotypes. One method is the estimation of genetic correlation [e.g. using LDSC or GREML-GCTA (Yang et al. Reference Yang, Lee, Goddard and Visscher2011; Bulik-Sullivan et al. Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day and Loh2015a)] from millions of common variants (single nucleotide polymorphisms; SNPs), for example using a case-control sample of one psychiatric disorder and another sample assessed for a relevant continuous trait or a different disorder. Such methods provide correlation estimates of the degree to which genetic risks are shared. However, practical limitations include a need for very large sample sizes and for some methods (e.g. GREML-GCTA), access to raw genotypes, limiting the application of these tools. A second approach uses a genome-wide association study (GWAS) ‘discovery’ sample to calculate polygenic risk scores (PRS) (Wray et al. Reference Wray, Lee, Mehta, Vinkhuyzen, Dudbridge and Middeldorp2014) for individuals in an independent ‘target’ sample. PRS for a phenotype of interest can be tested for association with another phenotype (e.g. another psychiatric disorder or trait variation) in the target sample, to establish whether there are shared genetic risks across phenotypes. Although studies using PRS methods can show direct evidence for shared genetic risks, typically modest effect sizes are observed (Wray et al. Reference Wray, Lee, Mehta, Vinkhuyzen, Dudbridge and Middeldorp2014), whereas molecular genetic studies that estimate genetic correlation provide a more precise assessment of the degree of shared genetic risks across phenotypes using different definitions.
It is important to note several differences in the meaning of results obtained from twin and molecular genetic analyses. For a more thorough review of different methods for estimation of univariate heritability and genetic correlation, please see Yang et al. (Reference Yang, Zeng, Goddard, Wray and Visscher2017). In brief, the correlation estimates from twin studies capture all inherited genetic variants shared by monozygotic twins. These estimates are likely to be different and higher than those from SNP-based studies as the latter is only based on additive common variant effects tagged by genotyping arrays. However, the source of any shared genetic effects cannot be discerned from twin studies; effects may be driven by or limited to specific types of variants (e.g. rare mutations) but not to other classes of variants (e.g. SNPs). Genetic studies assessing multiple classes of variants are needed to determine the source of genetic correlations estimated using twin studies. It is also worth noting that evidence from common and rare variant studies regarding shared genetic risks between two phenotypes might not be consistent.
Shared genetic risks across categorical disorders and population trait variation
Genetic studies have consistently demonstrated that thousands of common variants of small effect, as well as rare variants of larger effect, increase the risk for psychiatric disorders (Sullivan et al. Reference Sullivan, Daly and O'Donovan2012; Cross-Disorder Group of the PGC, 2013a; Davis et al. Reference Davis, Yu, Keenan, Gamazon, Konkashbaev, Derks and Keller2013; Robinson et al. Reference Robinson, Neale and Hyman2015). This complex polygenic architecture supports a model where a quantitatively distributed liability (influenced by numerous genes) is associated with one or more continuous phenotypes that underlie the diagnostic distinction between cases and controls. According to such a model (Fig. 1a), genetic risks that contribute to clinical diagnoses will also influence variation in related quantitative traits in the general population. See Table 1 for a summary of studies that have addressed this hypothesis for specific psychiatric disorders.
Table 1. Summary of studies investigating shared genetic risks across disorders and trait variation

ASD, autism spectrum disorder; ADHD, attention-deficit hyperactivity disorder; ID, intellectual disability; OCD, obsessive-compulsive disorder; MDD, major depressive disorder; SCZ, schizophrenia
Group heritability (implemented in DeFries-Fulker analysis) (DeFries & Fulker, Reference DeFries and Fulker1985) refers to the degree to which genetic factors influence the mean difference between extreme groups and the rest of a sample; significant group heritability implies a genetic link between milder and more severe manifestations of a trait
Linkage disequilibrium score correlation (LDSC) (Bulik-Sullivan et al. Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day and Loh2015a, Reference Bulik-Sullivan, Loh, Finucane, Ripke, Yang and Pattersonb) estimates the contribution of all SNPs from genome-wide data and indexes this as an estimate of SNP-heritability; which is different to twin heritability (Wray et al. Reference Wray, Lee, Mehta, Vinkhuyzen, Dudbridge and Middeldorp2014). This method can be applied to examine shared genetic risks between disorders and population traits to give an estimate of genetic correlation. Genome-wide association studies (GWAS) directly assess the independent association of many millions of common genetic variants (single nucleotide polymorphisms; SNPs) with a phenotype. Polygenic risk score (PRS) analysis, uses a GWAS ‘discovery’ sample to calculate genetic risk scores for individuals in an independent ‘target’ sample with genetic data; scores are derived by calculating the number of risk alleles weighted by the discovery effect size for each SNP and then summing these values for the set of SNPs, for each target individual (The International Schizophrenia Consortium, 2009). Regression analyses are used to test whether PRS for the discovery phenotype (e.g. clinical disorder) are associated with phenotypes of interest in the independent target sample (e.g. symptom variation in the population)
Disorders with early onset
Twin studies have reported significant group heritability using several different definitions of ASD (Robinson et al. Reference Robinson, Koenen, McCormick, Munir, Hallett and Happé2011; Lundström et al. Reference Lundström, Chang, Råstam, Gillberg, Larsson and Anckarsäter2012). One study employed a novel twin model to estimate the genetic correlation between ASD diagnoses and traits (r g = 0.70) (Colvert et al. Reference Colvert, Tick, McEwen, Stewart, Curran and Woodhouse2015). PRS studies show mixed results, with association between clinical ASD PRS with social-communication problems at age 8 but not later ages (St Pourcain et al. Reference St Pourcain, Robinson, Anttila, Bulik Sullivan, Maller and Golding2017), with self-assessed autistic traits in adults (Bralten et al. Reference Bralten, van Hulzen, Martens, Galesloot, Arias Vasquez and Kiemeney2017) and null results in a third study (Krapohl et al. Reference Krapohl, Euesden, Zabaneh, Pingault, Rimfeld and von Stumm2016). Modest, genetic correlation (r g = 0.27–0.34) was estimated between clinical ASD and social-communication traits at age 8, with non-significant estimates at ages 11–17 years (Robinson et al. Reference Robinson, St Pourcain, Anttila, Kosmicki, Bulik-Sullivan and Grove2016; St Pourcain et al. Reference St Pourcain, Robinson, Anttila, Bulik Sullivan, Maller and Golding2017). The rate of rare de novo mutations was associated with autism-related behaviors not only in children with ASD but also in unaffected siblings (Robinson et al. Reference Robinson, St Pourcain, Anttila, Kosmicki, Bulik-Sullivan and Grove2016).
Twin studies of attention-deficit hyperactivity disorder (ADHD) traits have also revealed substantial group heritability for extreme scores on ADHD traits (Levy et al. Reference Levy, Hay, McStephen, Wood and Waldman1997; Larsson et al. Reference Larsson, Anckarsater, Råstam, Chang and Lichtenstein2011), albeit extremely low ADHD scores are a potential exception (Greven et al. Reference Greven, Merwood, van der Meer, Haworth, Rommelse and Buitelaar2016). Multiple PRS analyses have demonstrated that genetic risk for clinically-diagnosed ADHD is shared with ADHD traits assessed between ages 3 and 17 years (Groen-Blokhuis et al. Reference Groen-Blokhuis, Middeldorp, Kan, Abdellaoui, van Beijsterveldt and Ehli2014; Martin et al. Reference Martin, Hamshere, Stergiakouli, O'Donovan and Thapar2014a; Stergiakouli et al. Reference Stergiakouli, Martin, Hamshere, Langley, Evans and St Pourcain2015, Reference Stergiakouli, Davey Smith, Martin, Skuse, Viechtbauer and Ring2017; Riglin et al. Reference Riglin, Collishaw, Thapar, Dalsgaard, Langley and Smith2016; Brikell et al. Reference Brikell, Larsson, Lu, Pettersson, Chen and Kuja-Halkola2017; Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017). Estimates of genetic correlation between ADHD diagnosis and traits are very high (r g = 0.94–0.96) (Middeldorp et al. Reference Middeldorp, Hammerschlag, Ouwens, Groen-Blokhuis, St. Pourcain and Greven2016; Demontis et al. Reference Demontis, Walters, Martin, Mattheisen, Als and Agerbo2017), with a moderate genetic correlation (r g = 0.30) between ADHD diagnosis and extraversion traits in the population (Lo et al. Reference Lo, Hinds, Tung, Franz, Fan and Wang2016).
Cognitive abilities display a similar pattern of significant group heritability in studies of mild intellectual disability (ID) (Spinath et al. Reference Spinath, Harlaar, Ronald and Plomin2004), different quantiles of reading assessments (Logan et al. Reference Logan, Petrill, Hart, Schatschneider, Thompson and Deater-Deckard2012), and high levels of intelligence (Shakeshaft et al. Reference Shakeshaft, Trzaskowski, McMillan, Krapohl, Simpson and Reichenberg2015). However, severe ID appears to be an exception to this pattern (Reichenberg et al. Reference Reichenberg, Cederlöf, McMillan, Trzaskowski, Kapara and Fruchter2016). Molecular genetic studies of ID have focused on very rare mutations (Girirajan et al. Reference Girirajan, Brkanac, Coe, Baker, Vives and Vu2011; The Deciphering Developmental Disorders Study, 2014) and there is some evidence that rare, likely pathogenic copy number variants (CNVs) are associated with poor performance on cognitive tasks in the population (Männik et al. Reference Männik, Mägi, Macé, Cole, Guyatt and Shihab2015; Kendall et al. Reference Kendall, Rees, Escott-Price, Einon, Thomas and Hewitt2016). Studies assessing the degree of shared common variants between ID and cognition in the population are lacking.
Converging evidence from twin and molecular genetic methods so far shows reasonably strong support for certain child-onset neurodevelopmental disorders (i.e. ADHD, ASD, and mild ID) as the extreme ends of continuous distributions of population traits.
Disorders with onset in adolescence and adulthood
There is a lack of studies testing for shared genetic risks across disorder and traits for anxiety disorders and obsessive-compulsive disorder (OCD). Although twin studies have established the heritability of anxiety traits, only two studies reported significant group heritability for anxiety disorders (Stevenson et al. Reference Stevenson, Batten and Cherner1992; Goldsmith & Lemery, Reference Goldsmith and Lemery2000). Twin studies of OCD indicate that traits characteristic of OCD are heritable and present throughout the population (van Grootheest et al. Reference van Grootheest, Cath, Beekman and Boomsma2005), although no twin studies have tested whether extreme OCD traits share genetic risks with milder traits. One recent study found associations between OCD PRS and continuously-distributed obsessive-compulsive traits in the population (den Braber et al. Reference den Braber, Zilhão, Fedko, Hottenga, Pool and Smit2016).
Twin studies of group heritability for depressive traits have found mixed results (Rende et al. Reference Rende, Plomin, Reiss and Hetherington1993; Eley, Reference Eley1997). Shared genetic influences across major depressive disorder (MDD) and depressive traits have been reported in an elderly population using PRS analysis (Demirkan et al. Reference Demirkan, Penninx, Hek, Wray, Amin and Aulchenko2011) but not in a childhood sample assessing internalizing traits at ages 3–10 years (Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017). Recent common variant analyses showed very high genetic correlation (r g = 0.91–1.00) between MDD and depressive symptoms (Direk et al. Reference Direk, Williams, Smith, Ripke, Air and Amare2016; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Major Depressive Disorder Working Group of the PGC et al. Reference Wray and Sullivan2017) and moderate correlation between MDD and personality measures, notably neuroticism (r g = 0.56–0.74), in the general population (Lo et al. Reference Lo, Hinds, Tung, Franz, Fan and Wang2016; Major Depressive Disorder Working Group of the PGC et al. Reference Wray and Sullivan2017).
The genetic evidence for a continuous spectrum of psychosis in the population is more complex. Psychotic experiences (e.g. paranoia and hallucinations) show low-to-moderate heritability (15–59%), with significant group heritability implying a genetic link between mild and severe psychotic experiences (Zavos et al. Reference Zavos, Freeman, Haworth, McGuire, Plomin and Cardno2014). However, it is unclear from twin studies whether psychotic experiences are related to schizophrenia. Findings from PRS studies are mixed, with several studies finding no association of schizophrenia or bipolar disorder (BD) PRS with adolescent psychotic experiences (Sieradzka et al. Reference Sieradzka, Power, Freeman, Cardno, McGuire, Plomin and Potash2014; Krapohl et al. Reference Krapohl, Euesden, Zabaneh, Pingault, Rimfeld and von Stumm2016), others reporting an association in the opposite direction to that expected (Zammit et al. Reference Zammit, Hamshere, Dwyer, Georgiva, Timpson and Moskvina2013) and others finding associations between schizophrenia PRS and adolescent negative symptoms (e.g. apathy or lack of energy) related to schizophrenia (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016) and ‘thought problems’ at age 10 (Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017). Schizophrenia PRS are higher in unaffected relatives of schizophrenia probands compared with controls (Bigdeli et al. Reference Bigdeli, Bacanu, Webb, Walsh, O'Neill and Fanous2014) and in individuals with more strictly defined schizophrenia, in terms of chronicity or severity of disorder (Meier et al. Reference Meier, Agerbo, Maier, Pedersen, Lang and Grove2016).
Evidence for shared genetic risks across disorders and traits is limited for adolescent- and adult-onset psychiatric disorders. Preliminary supporting evidence is seen for OCD and MDD. The picture is quite complex for schizophrenia and there is insufficient evidence to conclude whether anxiety disorders share genetic risks with related population traits.
Limitations and interpretation
There are several limitations of existing studies and important issues that have not been sufficiently addressed. First, many twin studies use percentile-based cut-offs to identify probands, rather than using clinical diagnoses. Second, twin studies have largely employed DeFries-Fulker analysis, which does not directly estimate genetic correlation between psychiatric disorders and related traits; rather, significant group heritability suggests a link between extreme values of a trait and variation in the trait. Direct estimation of the genetic correlation, as done for ASD (Colvert et al. Reference Colvert, Tick, McEwen, Stewart, Curran and Woodhouse2015), would likely be informative in future twin research.
Although analyses of population traits do not include many individuals who have psychiatric diagnoses, it is important to determine whether associations persist when such individuals are excluded. If not, this might suggest that any association signal is driven by extreme cases and not continuous variation in the trait of interest. Another important issue is the strength of any observed genetic correlations. It is entirely likely that even if there is some degree of shared genetic risk between a disorder and related traits, this will be partial and unique genetic effects will also contribute [e.g. as may be the case with ASD and social-communication traits, given somewhat modest genetic correlations (Robinson et al. Reference Robinson, St Pourcain, Anttila, Kosmicki, Bulik-Sullivan and Grove2016)].
Given that most psychiatric disorders consist of multiple domains, another challenge is identifying whether relevant population traits show different degrees of shared genetic risk with a given psychiatric disorder, as seems to be the case for schizophrenia genetic risk in relation to psychotic experiences and negative symptoms in the population (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016). Another difficulty with analyzing continuously distributed psychiatric traits is capturing the full spectrum of a relevant behavior, as most measurement instruments are optimized for detecting difficulties not abilities, thereby resulting in highly zero-inflated and skewed distributions that often violate modeling assumptions. It is unknown whether normalizing such scores through transformations or by regressing out covariates and rank-transforming a variable is an optimal solution and such methods may introduce technical artifacts (Pain et al. Reference Pain, Dudbridge and Ronald2017). Skewed variables need to be analysed using models that appropriately account for non-normal distributions of data. Ideally, measures that better capture the full variability of behavioral phenotypes are also needed.
We suggest that the assessment of the degree to which a heritable disorder can be considered as an extreme manifestation of population traits should include the following investigations: estimation of the heritability of relevant population traits, estimation of genetic correlation between the disorder and traits, and sensitivity analyses to determine whether any correlation is explained entirely by inclusion of individuals scoring at the extreme end of the trait distribution.
Shared genetic risks across different psychiatric phenotypes
Whilst the degree to which many specific psychiatric disorders share genetic risk with related population traits is yet to be determined, there is much more evidence regarding shared genetic risks across different disorders. Below we consider the strength of the evidence examining this hypothesis, as illustrated in Fig. 1b. See Table 2 for a summary. It is important to note that many studies have examined shared genetic risk between one psychiatric disorder and population traits related to another phenotype, thereby providing additional, albeit indirect, evidence for sharing of genetic risks across psychiatric disorders and continuous traits.
Table 2. Summary of studies investigating shared genetic risks across disorders

ADHD, attention-deficit hyperactivity disorder; AN&ED, anorexia nervosa and other eating disorders; ASD, autism spectrum disorder; AXD, anxiety disorders; BD, bipolar disorder; ID, intellectual disability; MDD, major depressive disorder; OCD, obsessive-compulsive disorder; SCZ, schizophrenia; TS, Tourette's syndrome and other tic disorders; SNP, single nucleotide polymorphism; CNV, copy number variant; PRS, polygenic risk score analysis; ns, non-significant estimates based on published studies.
Twin rg is the correlation between the additive genetic variance components from twin studies. Note that the ‘twin rg’ in Lichtenstein et al. (Reference Lichtenstein, Yip, Björk, Pawitan, Cannon and Sullivan2009) & Song et al. (Reference Song, Bergen, Kuja-Halkola, Larsson, Landén and Lichtenstein2015) are estimated from family studies but with a similar approach as in twin studies. SNP rg: is the estimated genetic correlation from genome-wide association studies using LDSC (linkage disequilibrium score correlation) or GCTA (genome-wide complex trait analysis). Only results estimated to be nominally significantly different from zero (p < 0.05) are presented. For a more detailed explanation of the methods, please refer to the caption of Table 1. The GREML-GCTA method (genetic relatedness estimation through maximum likelihood using the GCTA software) (Yang et al. Reference Yang, Lee, Goddard and Visscher2011; Lee et al. Reference Lee, Yang, Goddard, Visscher and Wray2012) is conceptually similar to LDSC; it is used to estimate the contribution of all SNPs from genome-wide data (SNP-heritability) and can be applied to examine shared genetic risks between disorders and population traits to give an estimate of genetic correlation.
Disorders with early onset
Twin studies of neuropsychiatric diagnoses and childhood traits consistently show significant genetic correlations. Associations have been seen between ADHD inattentive symptoms and difficulties in reading and mathematics (Greven et al. Reference Greven, Harlaar, Dale and Plomin2011, Reference Greven, Kovas, Willcutt, Petrill and Plomin2014; Wadsworth et al. Reference Wadsworth, DeFries, Willcutt, Pennington and Olson2015), categorically and continuously defined ADHD and ASD (Reiersen et al. Reference Reiersen, Constantino, Grimmer, Martin and Todd2008; Ronald et al. Reference Ronald, Simonoff, Kuntsi, Asherson and Plomin2008, Reference Ronald, Larsson, Anckarsäter and Lichtenstein2014a; Lichtenstein et al. Reference Lichtenstein, Carlström, Råstam, Gillberg and Anckarsäter2010; Taylor et al. Reference Taylor, Charman, Robinson, Plomin, Happé and Asherson2012), and ASD with learning difficulties and tics (which are associated with Tourette's syndrome) (Lichtenstein et al. Reference Lichtenstein, Carlström, Råstam, Gillberg and Anckarsäter2010). However, two other twin studies of ASD and intellectual ability have reported low genetic correlations, although this might have been related to measurement differences (Hoekstra et al. Reference Hoekstra, Happé, Baron-Cohen and Ronald2009, Reference Hoekstra, Happé, Baron-Cohen, Ronald, Baron-Cohen and Ronald2010).
Analyses of common genetic variants so far have not confirmed the genetic correlation between ADHD and ASD observed in twin studies (Cross-Disorder Group of the PGC, 2013a, b; Bulik-Sullivan et al. Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day and Loh2015a; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017). Clinical ADHD shares some genetic risk with social-communication traits (Martin et al. Reference Martin, Hamshere, Stergiakouli, O'Donovan and Thapar2014a) and other neurodevelopmental and externalizing traits that make up a general factor of childhood psychopathology (Brikell et al. Reference Brikell, Larsson, Lu, Pettersson, Chen and Kuja-Halkola2017). Clinical ADHD shares genetic risk with lower cognitive abilities in children and adults in the general population (Martin et al. Reference Martin, Hamshere, Stergiakouli, O'Donovan and Thapar2014b; Clarke et al. Reference Clarke, Lupton, Fernandez-Pujals, Starr, Davies and Cox2016; Stergiakouli et al. Reference Stergiakouli, Martin, Hamshere, Heron, St Pourcain and Timpson2016; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Demontis et al. Reference Demontis, Walters, Martin, Mattheisen, Als and Agerbo2017; Riglin et al. Reference Riglin, Collishaw, Richards, Thapar, Maughan and O'Donovan2017; Sniekers et al. Reference Sniekers, Stringer, Watanabe, Jansen, Coleman and Krapohl2017). In ASD, there is a positive genetic correlation with common variants associated with cognitive ability, suggesting that these variants operate differently to common risk variants for other psychiatric phenotypes and to rare variants in the context of ASD (Clarke et al. Reference Clarke, Lupton, Fernandez-Pujals, Starr, Davies and Cox2016; Robinson et al. Reference Robinson, St Pourcain, Anttila, Kosmicki, Bulik-Sullivan and Grove2016; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Sniekers et al. Reference Sniekers, Stringer, Watanabe, Jansen, Coleman and Krapohl2017; Weiner et al. Reference Weiner, Wigdor, Ripke, Walters, Kosmicki and Grove2017). With regard to rare variants, studies of CNVs have implicated the same genomic regions in multiple disorders, including ASD, ID, and ADHD (Guilmatre et al. Reference Guilmatre, Dubourg, Mosca, Legallic, Goldenberg and Drouin-Garraud2009; Sebat et al. Reference Sebat, Levy and McCarthy2009; Pinto et al. Reference Pinto, Pagnamenta, Klei, Anney, Merico and Regan2010; Williams et al. Reference Williams, Zaharieva, Martin, Langley, Mantripragada and Fossdal2010, Reference Williams, Franke, Mick, Anney, Freitag and Gill2012; Cooper et al. Reference Cooper, Coe, Girirajan, Rosenfeld, Vu and Baker2011; Lionel et al. Reference Lionel, Crosbie, Barbosa, Goodale, Thiruvahindrapuram and Rickaby2011; Sanders et al. Reference Sanders, Ercan-Sencicek, Hus, Luo, Murtha and Moreno-De-Luca2011; Pescosolido & Gamsiz, Reference Pescosolido and Gamsiz2013). Recent large exome sequencing studies have identified the first robust rare de novo protein-truncating mutations (variants which disrupt protein formation and are likely highly deleterious) associated with ASD, with many of the same genes found to harbor de novo mutations linked to ID (De Rubeis et al. Reference De Rubeis, He, Goldberg, Poultney, Samocha and Ercument Cicek2014; Iossifov et al. Reference Iossifov, O'Roak, Sanders, Ronemus, Krumm and Levy2014; Samocha et al. Reference Samocha, Robinson, Sanders, Stevens, Sabo and McGrath2014; The Deciphering Developmental Disorders Study, 2014).
Twin and molecular studies have yielded some consistent findings, but larger genetic studies are needed to further understand the degree and source of shared genetic risks in these early-onset disorders. The association between ASD and ID is particularly complex, with shared risk for these phenotypes seen at the level of rare risk variants but a positive association seen for common variants; indeed these mixed genetic results may partly explain the low genetic correlations between these phenotypes in twin studies (Hoekstra et al. Reference Hoekstra, Happé, Baron-Cohen and Ronald2009, Reference Hoekstra, Happé, Baron-Cohen, Ronald, Baron-Cohen and Ronald2010).
Disorders with onset in adolescence and adulthood
Twin studies have found substantial evidence of genetic correlations across schizophrenia and BD (Cardno et al. Reference Cardno, Rijsdijk, Sham, Murray and McGuffin2002; Lichtenstein et al. Reference Lichtenstein, Yip, Björk, Pawitan, Cannon and Sullivan2009), BD and MDD (Song et al. Reference Song, Bergen, Kuja-Halkola, Larsson, Landén and Lichtenstein2015), anxiety disorder subtypes (Mosing et al. Reference Mosing, Gordon, Medland, Statham, Nelson and Heath2009), specific anxiety disorders and MDD (Roy et al. Reference Roy, Neale, Pedersen, Mathé and Kendler1995; Kendler et al. Reference Kendler, Gardner, Gatz and Pedersen2007; Mosing et al. Reference Mosing, Gordon, Medland, Statham, Nelson and Heath2009), traits of anxiety and depressive symptoms (Thapar & McGuffin, Reference Thapar and McGuffin1997), MDD and psychotic experiences in adolescence (Zavos et al. Reference Zavos, Eley, McGuire, Plomin, Cardno and Freeman2016), depressive symptoms and disordered eating scores (Slane et al. Reference Slane, Burt and Klump2011), OCD and MDD (Bolhuis et al. Reference Bolhuis, McAdams, Monzani, Gregory, Mataix-Cols and Stringaris2014), and OCD with anxiety-related behaviors and anorexia nervosa (AN) (Cederlöf et al. Reference Cederlöf, Thornton, Baker, Lichtenstein, Larsson and Rück2015; López-Solà et al. Reference López-Solà, Fontenelle, Bui, Hopper, Pantelis and Yücel2016).
GWAS of adult psychiatric disorders have confirmed that common genetic variants associated with one disorder also play an important role in other disorders. Recent analyses using multiple genome-wide methods report shared genetic risks across schizophrenia, BD, MDD, and OCD, across schizophrenia, AN and OCD, and between MDD with anxiety disorders and AN (Cross-Disorder Group of the PGC, 2013a, b; Bulik-Sullivan et al. Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day and Loh2015a; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Major Depressive Disorder Working Group of the PGC et al. Reference Wray and Sullivan2017). Shared genetic risks are seen across different anxiety disorders (generalized anxiety disorder, panic disorder and phobias) and with MDD, though not with BD or schizophrenia (Otowa et al. Reference Otowa, Hek, Lee, Byrne, Mirza and Nivard2016). General population studies of schizophrenia PRS report associations with anxiety symptoms, with mixed evidence for association with depressive symptoms between ages 7 and 15 (Jones et al. Reference Jones, Stergiakouli, Tansey, Hubbard, Heron and Cannon2016; Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017; Nivard et al. Reference Nivard, Gage, Hottenga, van Beijsterveldt, Abdellaoui and Bartels2017). MDD PRS were also associated with anxiety symptoms in an elderly population sample (Demirkan et al. Reference Demirkan, Penninx, Hek, Wray, Amin and Aulchenko2011). Thus, there is evidence that a considerable degree of genetic influences are shared across multiple phenotypes, assessed categorically or continuously.
Shared genetic risks across child- and adult-onset disorders
Childhood-onset disorders and disorders with an onset typically in adolescence or adulthood also share genetic risks. For example, twin studies find that early-onset-neurodevelopmental disorders share genetic risk with anxiety (Hallett et al. Reference Hallett, Ronald, Rijsdijk and Happé2010; Michelini et al. Reference Michelini, Eley, Gregory and McAdams2015; Chen et al. Reference Chen, Ji, Wang, Lichtenstein, Larsson and Chang2016), MDD (Cole et al. Reference Cole, Ball, Martin, Scourfield and Mcguffin2009; Lundström et al. Reference Lundström, Chang, Kerekes, Gumpert, Råstam and Gillberg2011), affective problems (Rydell et al. Reference Rydell, Taylor and Larsson2017), and OCD (Pinto et al. Reference Pinto, Monzani, Leckman, Rück, Serlachius and Lichtenstein2016). In a study of specific intellectual domains, problems with communication shared a modest degree of genetic risk with adolescent hallucinations and mania (Cederlöf et al. Reference Cederlöf, Ostberg, Pettersson, Anckarsäter, Gumpert and Lundström2014b). Molecular genetic studies have reported genetic correlations between both ADHD and ASD with MDD, schizophrenia and BD (Cross-Disorder Group of the PGC, 2013a, b; Bulik-Sullivan et al. Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day and Loh2015a; van Hulzen et al. Reference van Hulzen, Scholz, Franke, Ripke, Klein and McQuillin2016; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Demontis et al. Reference Demontis, Walters, Martin, Mattheisen, Als and Agerbo2017; Major Depressive Disorder Working Group of the PGC et al. Reference Wray and Sullivan2017; The ASD Working Group of The PGC, 2017). Tourette's syndrome shares genetic risks with OCD and MDD (Davis et al. Reference Davis, Yu, Keenan, Gamazon, Konkashbaev, Derks and Keller2013; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017). Genetic risk for schizophrenia is associated with numerous traits assessed across ages 3–15 years, including ADHD, aggression, irritability, language, and social abilities (Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017; Nivard et al. Reference Nivard, Gage, Hottenga, van Beijsterveldt, Abdellaoui and Bartels2017; Riglin et al. Reference Riglin, Collishaw, Richards, Thapar, Maughan and O'Donovan2017). BD and MDD PRS were not found to be associated with early life (age 3–10 years) internalizing and externalizing problems (Jansen et al. Reference Jansen, Polderman, Bolhuis, van der Ende, Jaddoe and Verhulst2017).
CNV loci implicated in children with ADHD, ASD, and ID have also been associated with schizophrenia (The International Schizophrenia Consortium, 2008; Guilmatre et al. Reference Guilmatre, Dubourg, Mosca, Legallic, Goldenberg and Drouin-Garraud2009; Sebat et al. Reference Sebat, Levy and McCarthy2009; Williams et al. Reference Williams, Zaharieva, Martin, Langley, Mantripragada and Fossdal2010, Reference Williams, Franke, Mick, Anney, Freitag and Gill2012; Lionel et al. Reference Lionel, Crosbie, Barbosa, Goodale, Thiruvahindrapuram and Rickaby2011; Pescosolido & Gamsiz, Reference Pescosolido and Gamsiz2013). Schizophrenia shares genetic risks with cognitive measures throughout the lifespan (McIntosh et al. Reference McIntosh, Gow and Luciano2013; Lencz et al. Reference Lencz, Knowles, Davies, Guha, Liewald and Starr2014; Hagenaars et al. Reference Hagenaars, Harris, Davies, Hill, Liewald and Ritchie2016; Hubbard et al. Reference Hubbard, Tansey, Rai, Jones, Ripke and Chambert2016; Krapohl et al. Reference Krapohl, Euesden, Zabaneh, Pingault, Rimfeld and von Stumm2016). General cognitive ability shows negative genetic correlations with schizophrenia and depressive symptoms, though not with BD, anxiety disorder, MDD, OCD or AN (Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Sniekers et al. Reference Sniekers, Stringer, Watanabe, Jansen, Coleman and Krapohl2017). Genetic correlations across several psychiatric disorders and personality measures have also been reported (Lo et al. Reference Lo, Hinds, Tung, Franz, Fan and Wang2016; Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017). Psychiatric phenotypes also more broadly share genetic contributions with other human complex traits, for example genetic risk for ADHD is shared with behavioral traits (e.g. smoking), brain- (e.g. migraine) and non-brain-based diseases (e.g. type-2-diabetes) and traits (e.g. body mass index) (Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017; Demontis et al. Reference Demontis, Walters, Martin, Mattheisen, Als and Agerbo2017). A wider review is beyond the scope of this paper.
In summary, studies indicate that a considerable degree of genetic influences on particular disorders are shared with at least one other disorder, regardless of whether one focuses on childhood- or adulthood-onset conditions. It has been hypothesized that a single ‘general genetic factor’ underlies multiple psychiatric phenotypes (Lahey et al. Reference Lahey, Applegate, Hakes, Zald, Hariri and Rathouz2012; Caspi et al. Reference Caspi, Houts, Belsky, Goldman-Mellor, Harrington and Israel2014). Two twin studies supported this model, with a latent genetic factor accounting for 31% of variance in neurodevelopmental symptoms in a population-based sample (Pettersson et al. Reference Pettersson, Anckarsäter, Gillberg and Lichtenstein2013) and 10–36% of disorder liability across multiple clinical psychiatric diagnoses (Pettersson et al. Reference Pettersson, Larsson and Lichtenstein2015). A recent study further confirmed that common genetic risk variants contribute to this general factor, with an estimated SNP-heritability of approximately 0.38 (Neumann et al. Reference Neumann, Pappa, Lahey, Verhulst, Medina-Gomez and Jaddoe2016). As illustrated in Fig. 1b, the situation is likely to be even more complex, with not only a general genetic factor predisposing to multiple phenotypes but also disorder-specific genetic factors as well as genetic factors relevant only to specific pairs of disorders. Similarly, environmental factors could also be shared or unique and more complex effects, such as gene-environment interactions, could also exist.
Although several of the pairs of psychiatric disorders assessed using GWAS data do not show significant genetic correlations, some of the studies were relatively small and are likely to be underpowered. Notably, genetic correlations are present regardless of whether psychiatric phenotypes are conceptualized continuously or dichotomously, thus providing additional, albeit indirect, support for shared genetic risk across these disorders and related traits.
Interpreting the meaning of genetic correlations
The interpretation of what genetic correlations mean is complex, with a number of possibilities, some of which are not mutually exclusive. One possibility (Fig. 2a) is that of true biological pleiotropy, where the same risk variants (or variants within the same gene) are directly, causally impacting on multiple phenotypes, albeit possibly through separate biological pathways. Alternatively, the same genetic risk variants could be causally affecting a third, unmeasured phenotype which lies on the pathway between risk variants and measured phenotypes (Fig. 2b). A third possibility (Fig. 2c) is that observed genetic correlations are actually capturing different risk variants that are highly correlated but are acting through different mechanisms. For example, even though the same CNV loci have been implicated in multiple disorders (Guilmatre et al. Reference Guilmatre, Dubourg, Mosca, Legallic, Goldenberg and Drouin-Garraud2009; Lionel et al. Reference Lionel, Crosbie, Barbosa, Goodale, Thiruvahindrapuram and Rickaby2011; Williams et al. Reference Williams, Franke, Mick, Anney, Freitag and Gill2012; Pescosolido & Gamsiz, Reference Pescosolido and Gamsiz2013), different variants within these large loci might be associated with different phenotypes. Given that such large, rare variants are also shared by monozygotic twins, this could also influence estimates of genetic correlations based on twin studies. A fourth possibility (Fig. 2d) is that one phenotype mediates the association between genetic risk and a second phenotype and there is no direct causal relationship between the risk variant and this second phenotype. For example, it has been proposed that the genetic correlation between MDD and depressive symptoms in the population could be accounted for by shared genetic risk with low levels of subjective well-being (Direk et al. Reference Direk, Williams, Smith, Ripke, Air and Amare2016).

Fig. 2. Potential interpretations of genetic correlation across phenotypes: (a) true biological pleiotropy, where the same genetic risk variant is causally associated with two phenotypes; (b) unmeasured phenotype, where a third phenotype is on the causal pathway between genetic risk and the outcome phenotypes of interest; (c) correlated genetic risk, where different genetic risk variants that are highly correlated are causally associated with each phenotype; (d) mediation, where a genetic risk variant only acts on one of the phenotypes, which in turn influences a second phenotype; (e) Nosological issues, which blur the distinction between phenotypes, for example comorbidity, ascertainment bias, heterogeneity or diagnostic misclassification; (f) assortative mating, where individuals with the two phenotypes of interest are more likely to mate than expected at random, thereby leading to clustering of genetic risk for both phenotypes in the offspring. N.B. The shapes are not indicative of whether a variable is latent or measured.
Several nosological issues (Fig. 2e) may also explain genetic correlations to an extent. Comorbidity across disorders (e.g. anxiety and MDD) is frequently observed and certain symptom domains show similarities [e.g. manic (BD) or hyperactive (ADHD) symptoms]. Specific symptoms also overlap directly across disorders (e.g. concentration problems in ADHD, MDD or anxiety) and such overlap may largely account for comorbidity [e.g. anxiety and MDD (Cramer et al. Reference Cramer, Waldorp, van der Maas and Borsboom2010)]. Such phenotypic overlap could inflate genetic correlation estimates. Within-disorder heterogeneity could also induce an overall correlation across two phenotypes, when only a sub-group of individuals with one disorder (who may have a specific clinical profile) show genetic correlation with individuals with another phenotype. Another possibility is that of diagnostic misclassification or changes in meeting diagnostic criteria over time (e.g. individuals who are diagnosed with MDD but later develop manic features, leading to a diagnosis of BD). Given the similar diagnostic features across different disorders, accurate diagnosis is difficult. Fortunately, diagnostic changes over time can be taken into consideration using epidemiological family study designs (Song et al. Reference Song, Bergen, Kuja-Halkola, Larsson, Landén and Lichtenstein2015). Simulations show that a 10% rate in misclassification can inflate estimates of genetic correlation (Wray et al. Reference Wray, Lee and Kendler2012). However, very high degrees of such misclassification would be required to fully account for the observed genetic correlations across psychiatric phenotypes (Anttila et al. Reference Anttila, Bulik-Sullivan, Finucane, Bras, Duncan and Escott-Price2017). Such issues related to phenotype definition remain to be resolved as the underlying biology of psychiatric disorders is better understood. For now, careful ascertainment and better measurement of frequently co-occurring disorder-level and sub-threshold phenotypes is required.
Another possibility for interpreting observed genetic correlations between psychiatric disorders is that they arise through assortative mating (Fig. 2f). There are substantial effects of assortative mating both within and across multiple psychiatric disorders (Nordsletten et al. Reference Nordsletten, Larsson, Crowley, Almqvist, Lichtenstein and Mataix-Cols2016). Such assortative mating across disorders would likely increase genetic correlation estimates (Coop & Pickrell, Reference Coop and Pickrell2016). Finally, there are technical and methodological artifacts (e.g. overlapping or related individuals) that may induce spurious genetic correlations in molecular genetic studies, which need to be ruled out.
More research is needed to determine the extent to which comorbidity, ascertainment bias, heterogeneity, diagnostic misclassification, and assortative mating inflate genetic correlations across psychiatric disorders and how much of these estimates are due to true pleiotropy. Even so, the possible biological interpretations of genetic correlations described above are hard to distinguish using the methods described in this review, as genetic correlations do not pinpoint the source of shared genetic risks. Some clues might be gained by partitioning heritability based on SNP functional category, position or frequency (Finucane et al. Reference Finucane, Bulik-Sullivan, Gusev, Trynka, Reshef and Loh2015), to try to better identify the source of the genetic correlations. Large-scale GWAS meta-analyses and sequencing studies are needed to find robust risk variants associated with multiple disorders.
After identifying specific genetic risk variants that correlate across disorders and considering the above possibilities, well-phenotyped samples and new methods will be needed to interpret the meaning of genetic correlations. Several newly developed methods have the potential to help with interpretation. The method ‘pairwise-GWAS’ aims to determine whether the effect sizes of variants associated with one trait are correlated with effect sizes of those variants for another trait and vice versa (Pickrell et al. Reference Pickrell, Berisa, Liu, Ségurel, Tung and Hinds2016). Another method, BUHMBOX, aims to statistically differentiate between situations where there is sub-group heterogeneity (i.e. phenotype misclassification, different biological subtypes of a disorder, ascertainment bias or mediation) or whether there is true pleiotropy (Han et al. Reference Han, Pouget, Slowikowski, Stahl, Lee and Diogo2016).
Implications for research and clinical practice
Despite moderate to high degrees of genetic correlation between some pairs of phenotypes, unique genetic factors are also likely to be important, as illustrated in Fig. 1b. This unique genetic risk is associated with important clinical distinctions that exist between disorders and also between disorders and continuous traits. For example, certain medications are effective for one disorder (e.g. stimulants for ADHD), but do not impact the symptoms of other disorders (Thapar et al. Reference Thapar, Cooper and Rutter2017). Also, in the absence of severe impairment resulting from symptoms, the cost-benefit ratio of treatment needs to be considered. Since most genetic correlations are below 1, more insights into the meaning of these correlations are required before clinical practice can be advanced.
The assumption that there is some true sharing of genetic risks has already led to insights into the genetic architecture and biology of psychiatric disorders through combining phenotypes in joint analyses to boost statistical power. For example, a joint GWAS analysis of five psychiatric disorders led to a more powerful approach for identifying genetic variants associated with psychiatric disorders (Cross-Disorder Group of the PGC, 2013b). Similarly, using the results of a GWAS of multiple psychiatric disorders can substantially increase the accuracy of PRS analyses (Maier et al. Reference Maier, Moser, Chen, Ripke and Coryell2015). Also, a literature review of genetic sequencing studies of several childhood-onset neurodevelopmental disorders has shown the power of pooling information on multiple phenotypes to identify more robust genes implicated in neurodevelopmental disorders (Gonzalez-Mantilla et al. Reference Gonzalez-Mantilla, Moreno-De-Luca, Ledbetter and Martin2016). Gene discovery studies meta-analyzing GWAS of a clinical disorder with GWAS of population traits can benefit from substantially increased power to detect common variants, as can be seen for example for MDD and ADHD (Direk et al. Reference Direk, Williams, Smith, Ripke, Air and Amare2016; Demontis et al. Reference Demontis, Walters, Martin, Mattheisen, Als and Agerbo2017). Understanding the nature and degree of shared genetic risks across psychiatric phenotypes will be essential to most effectively using this observation for future research into the genetic architecture of these disorders.
One important limitation of existing molecular genetic studies is that for many psychiatric disorders, sample sizes are still relatively small and analyses are limited in statistical power. PRS studies, in particular, tend to find low effect sizes. As larger and more reliable genetic samples become available in the future, it will be possible to better determine the degree and source of shared genetic risks across psychiatric phenotypes.
Conclusion
Emerging evidence from twin and molecular genetic studies suggests that some genetic risk is shared between diagnosed disorders and variation in psychiatric traits in the population for certain disorders (e.g. ADHD) and across different psychiatric diagnoses (e.g. schizophrenia and BD). More research is needed to investigate the degree of genetic correlation across disorders and traits for other psychiatric phenotypes (e.g. anxiety or BD) and across pairs of different disorders (e.g. anorexia and OCD). Future research should then aim to identify specific genetic loci that are driving any genetic correlations and determine the nature of such correlations. However, recent insights into the genetic architectures of psychiatric disorders are already pointing towards new avenues for further research into the biology of these complex disorders.
Acknowledgements
This work was supported by the Wellcome Trust (grant 106047). Many thanks to Dr Elise Robinson and Prof Anita Thapar for helpful comments on an early draft of the manuscript. Prof Lichtenstein is funded by the Swedish Research Council for Health, Working Life and Welfare.
Declaration of interest
Drs Martin and Taylor report no biomedical financial interests or potential conflicts of interest. Dr Lichtenstein has served as a speaker for Medice.





 Open access
Open access