


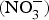
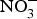
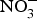
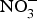
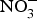
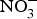
CVD are the leading cause of death worldwide and a major cause of morbidity and disability(Reference Townsend, Wickramasinghe and Bhatnagar1). In the UK, cardiovascular mortality accounts for 19 and 28 % of premature deaths among women and men, respectively(Reference Bhatnagar, Wickramasinghe and Williams2). CVD are characterised by a multifactorial pathogenesis including genetic, diet and lifestyle factors(Reference Frayn and Stanner3). The clinical outcomes of CVD, such as heart failure, atrial fibrillation and cerebrovascular disease, are largely attributed to a reduction in the blood supply to associated organs and tissues. This occurs secondary to thickening of the walls of the blood vessels and formation of obstructive atherosclerotic plaques(Reference Gimbrone and Garcia-Cardena4). Endothelial dysfunction (ED) appears to be a critical step in the initiation of the atherosclerotic process(Reference Gimbrone and Garcia-Cardena4).
The endothelium
The endothelium is a monolayer of cells separating the vascular lumen from the rest of the blood vessel. It is now recognised that the endothelium has vital paracrine, endocrine and autocrine functions(Reference Sena, Pereira and Seica5). Therefore, in addition to helping maintain blood flow, the main function of the endothelium is to serve as an endocrine organ(Reference Michiels6). The endothelium generates several extracellular messengers that mediate multiple functions including preserving haemostatic balance(Reference Rajendran, Rengarajan and Thangavel7). In addition to insulating the thrombogenic sub-endothelial layers, the endothelium secretes molecules that inhibit the inappropriate formation of thrombus including nitric oxide (NO), prostacyclin I2, tissue plasminogen activator and protein C/protein S(Reference Rajendran, Rengarajan and Thangavel7). However, in cases of vessel damage and exposure to certain pro-inflammatory substances, the balance is shifted towards a procoagulant/prothrombotic state(Reference Sena, Pereira and Seica5). This stimulates the endothelium to secrete agents that help with platelet aggregation and clot formation including platelet activating factor, von Willebrand factor and thromboxane A2(Reference Sena, Pereira and Seica5, Reference Sumpio, Riley and Dardik8).
Normal vascular endothelium has anti-proliferative and anti-apoptotic properties that are mediated through the activity of NO, prostacyclin I2 and C-type natriuretic peptide. Moreover, endothelial cells secrete factors that promote proliferation of smooth muscle cells and the formation of new blood vessels, e.g. vascular endothelial growth factor, angiopoietins and adropins(Reference Mensah9). Further, NO secreted by the normal endothelium prevents inflammatory response in the vascular wall secondary to local injury. Dysfunction in the endothelium is characterised by disturbed vasodilator and anticoagulant function, increased adhesiveness of the vessel wall for platelets and leucocytes (inflammation), reduced fibrinolytic activity and breakdown of barrier function causing leakage and oedema formation(Reference Vanhoutte, Shimokawa and Tang10).
Nitric oxide and endothelial function
NO is a free radical gas molecule that is involved in the regulation of multiple physiological processes such as blood pressure (BP), glucose metabolism, inflammation and coagulation(Reference Siervo, Capuano and Colantuoni11). Reduced availability of NO contributes to pathological conditions including hypertension, diabetes, chronic heart failure or kidney failure(Reference Yetik-Anacak and Catravas12). NO is regarded as one of the most important molecules secreted by the endothelium. It is a highly diffusible molecule with a very short half-life (<1 s)(Reference Channon13). The production of NO is catalysed by the nitric oxide synthase enzyme (NOS). There are three isoforms of this enzyme including: endothelial (eNOS), neuronal and inducible(Reference Lei, Vodovotz and Tzeng14). The eNOS is a homodimeric enzyme expressed constitutively in the endothelial cells that facilitate the conversion of the amino acid l-arginine into l-citrulline and NO(Reference Michiels6). This process requires molecular oxygen and reduced NADPH as co-substrates, and the following cofactors: FAD, FMN, tetrahydrobiopterin, haeme and Ca2±–calmodulin(Reference Forstermann and Sessa15) (see Fig. 1).

Fig. 1. Exogenous and endogenous sources of nitric oxide (NO). NO is produced by a family of enzymes known as NO synthases (NOS) which utilise the substrate l-arginine. The 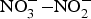 ${\rm NO}_3^ - {\rm -} {\rm NO}_2^ - $–NO pathway has been proposed as an alternative pathway for NO generation. ADMA, asymmetric dimethylarginine; BH4, tetrahydrobiopterin; cGMP, cyclic GMP; ONOO−, peroxynitrite; sGC, soluble guanylate cyclase.
${\rm NO}_3^ - {\rm -} {\rm NO}_2^ - $–NO pathway has been proposed as an alternative pathway for NO generation. ADMA, asymmetric dimethylarginine; BH4, tetrahydrobiopterin; cGMP, cyclic GMP; ONOO−, peroxynitrite; sGC, soluble guanylate cyclase.
The triggers for NO synthesis and release are either mechanical stretching of the vessel wall or release of receptor-mediated agonists such as bradykinin, acetylcholine or histamine(Reference Khazaei, Moien-Afshari and Laher16). These signals lead to an increase in intracellular calcium concentration. Intracellular Ca2+ binds to calmodulin to form Ca2+–calmodulin complex that mobilises eNOS from its binding to caveolin, thereby allowing the activated eNOS to catalyse the synthesis of NO from l-arginine(Reference Khazaei, Moien-Afshari and Laher16). Because of the gaseous nature of NO, it diffuses from where it is synthesised in the endothelium to the vascular smooth muscle where it activates soluble guanylate cyclase leading to increasing intracellular cyclic guanosine monophosphate. The cyclic guanosine monophosphate causes smooth muscle relaxation and, eventually, arterial dilation(Reference Forstermann17).
In addition to arterial dilation, NO has many other vital protective functions in blood vessels including decreasing: (1) smooth muscle proliferation; (2) platelet aggregation; (3) endothelin production; (4) monocytes and platelets adhesion; (5) expression of adhesion molecules; and (6) oxidation of LDL(Reference Vanhoutte, Shimokawa and Tang10). Because of the vital role of NO, researchers have suggested that reduced NO availability is the major cause of ED. This deficiency activates atherogenic processes in the vessel wall, which include vasoconstriction, monocyte activation and adherence to vascular endothelium, proliferation of smooth muscle cells, thrombosis and impaired coagulation and, eventually, atherosclerosis(Reference Versari, Daghini and Virdis18).
Many factors modulate NO synthesis and degradation, and therefore, affect endothelial function (EF). Asymmetric dimethyl l-arginine is a product of protein metabolism formed secondarily to methylation of l-arginine(Reference Giles19). Asymmetric dimethyl l-arginine decreases the synthesis of NO by reducing the expression and/or activity of eNOS. Asymmetric dimethyl l-arginine is increased in many pathological conditions such as hypercholesterolaemia, atherosclerosis, hypertension, chronic heart failure, diabetes mellitus and chronic renal failure(Reference North and Sinclair20). Further, uncoupling of eNOS as a result of the oxidation of tetrahydrobiopterin or depletion of l-arginine and the accumulation of endogenous methylarginines may lead to reduced formation of NO, i.e. the eNOS enzyme is converted from NO-producing enzyme to O2−-producing enzyme(Reference Schmidt and Alp21). Overproduction of reactive oxygen species (ROS) is the major cause of reduced NO availability in CVD. NO reacts with superoxide anion with high affinity forming the harmful free radical peroxynitrite (ONOO−)(Reference Sena, Pereira and Seica5). Lipid peroxyl radicals and oxidised LDL react with endothelial NO before it reaches the vascular smooth muscle cells and, therefore, inhibit NO from dilating blood vessels(Reference Sena, Pereira and Seica5).
The ability of the endothelium to maintain the integrity of the vessel wall can be affected by both the biochemical and pathophysiological states of the rest of the body. For example, chronic smoking deteriorates EF by decreasing NO production and enhancing its degradation via the generation of oxygen-free radicals(Reference Toda and Toda22). Further, hypercholesterolaemia and high homocysteinaemia may reduce the availability of NO secondary to oxidative stress(Reference Sibal, Agarwal and Home23, Reference Dayal and Lentz24).
Endothelial dysfunction and hypertension
ED has been demonstrated both in the resistance and conduit arteries of several animal models of hypertension(Reference Tang and Vanhoutte25). In human subjects, reduced forearm blood flow responses to endothelium-dependent vasodilator agonists, such as acetylcholine and bradykinin(Reference Linder, Kiowski and Buhler26, Reference Panza, Quyyumi and Brush27), and increased vasoconstrictor responses to locally administered NOS inhibitors(Reference Panza, Casino and Kilcoyne28) have been observed in hypertensive patients. The cause of ED associated with hypertension is speculated to be a reduction in NO bioavailability (increased degradation by oxidative stress, reduced production by eNOS inactivation) and abundance of vasoconstrictor agents in the circulation such as angiotensin II and prostaglandins(Reference Tang and Vanhoutte25).
Vascular ageing
The ageing process is characterised by a progressive decline of cellular integrity and function resulting from the structural modification of macromolecules including formation of oxidised lipid species, advanced glycated products, nitrosylated proteins and DNA mutations(Reference Dobrovic and Kristensen29, Reference Vijg and Suh30). The accumulation of modified molecules and their incorporation into cellular components are responsible for the structural and functional deterioration of tissues and organs with time(Reference Ashor, Siervo, Mathers and Mocchegiani31).
Whilst the complexity of the biological mechanisms contributing to the ageing process is still poorly understood, a comprehensive summary of some of these mechanisms has been proposed recently by Lopez-Otin et al. (Reference Lopez-Otin, Blasco and Partridge32). The authors proposed the following set of hallmarks of ageing: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication(Reference Lopez-Otin, Blasco and Partridge32). Many factors contribute to the age-related molecular damage, but it seems likely that much damage is due to three common stressors including oxidative stress/redox changes, inflammation and metabolic stress(Reference Mathers33) (see Fig. 2).

Fig. 2. Factors associated with vascular ageing and atherosclerosis. The triad of oxidative stress, inflammation and endothelial cell senescence contribute to reduced nitric oxide (NO) availability, endothelial dysfunction and the subsequent atherosclerosis.
Ageing and CVD risk
Age-specific mortality rates from heart disease and stroke increase exponentially with age and account for more than 40 % of all deaths worldwide among individuals aged 65–74 years and almost 60 % at age 85 years and older(Reference Ungvari, Kaley and de Cabo34). In the UK, although death rates from CVD have been declining over the past four decades, IHD is ranked as the number one for years of life lost due to premature mortality(Reference Bhatnagar, Wickramasinghe and Williams2). Importantly, key cardiovascular risk factors including lifestyle factors such as smoking, poor diet and lack of physical activity are the major causes of morbidity measured by disability-adjusted life years(Reference Murray, Richards and Newton35).
Ageing is associated with complex structural and functional changes in all tissues including the vascular system, and these changes increase CVD risk independent of other risk factors such as hypertension, diabetes or hypercholesterolaemia(Reference Jani and Rajkumar36). These functional changes include widespread ED, dilation of the central arteries and increased arterial stiffness(Reference Mitchell, Parise and Benjamin37, Reference Taddei, Virdis and Ghiadoni38). Development of strategies to attenuate ageing of the vascular system could make a substantial contribution to lowering CVD risk and improving the quality of life of older people(Reference El Assar, Angulo and Vallejo39).
Factors that impair endothelial function with ageing
Oxidative stress
ROS encompass a large family of oxidant molecules such as superoxide (O2−), hydrogen peroxide (H2O2), hydroxyl radical (OH.) and ONOO−. The accumulation of ROS and the resulting oxidative modification of cellular macromolecules (lipids, proteins and nucleic acids/DNA) have been suggested to contribute to ageing in all organisms(Reference Puca, Carrizzo and Villa40). Indeed, increased production of free radicals, secondary to mitochondrial dysfunction, causes oxidative damage to cells including vascular cells(Reference Fusco, Colloca and Lo Monaco41). ROS formation can also lead to a propagation of the activity where the effect of a single reactive molecule can be amplified due to a series of chain reactions causing further damage and the loss of cell homeostasis(Reference Zorov, Juhaszova and Sollott42). Beside their damaging effect, ROS are also important secondary messengers in physiological process regulating enzymatic activity, gene expression and have a key role in response to pathogens infections(Reference Bedard and Krause43). For this reason, ROS production is tightly regulated by key antioxidant enzymes such as superoxide dismutase, glutathione peroxidase and catalase that when not jeopardised are able to keep the balance between the production and elimination of these oxidant species(Reference Birben, Sahiner and Sackesen44).
The major sources of ROS in CVD are represented by NADPH(Reference Drummond and Sobey45, Reference Turgeon, Haddad and Dussault46), mitochondrial respiration(Reference Kornfeld, Hwang and Disatnik47), xanthine oxidase(Reference Panth, Paudel and Parajuli48), lipoxygenase and uncoupled NOS(Reference Kuroda, Ago and Matsushima49). The putative mechanism by which the dysregulated enzymatic functions are linked to CVD are thought to be connected to the excessive O2− generation that may act as a NO scavenger causing both a reduction of NO bioavailability in the vascular tissue and the production of the highly reactive ONOO−, which in turn can negatively modulate protein functions through nitrosylation of tyrosine residues. Mitochondria also represent an important source of ROS (mtROS) that have been associated with CVD pathogenesis(Reference Dromparis and Michelakis50), and the role of nitrate 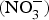 ${\rm \;} ({\rm NO}_3^ - )\; $ and nitrite
${\rm \;} ({\rm NO}_3^ - )\; $ and nitrite 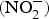 ${\rm \;} ({\rm NO}_2^ - )\; $ in the regulation of mitochondrial function and ROS generation is becoming an area of interest in the context of CVD prevention(Reference Shiva and Gladwin51).
${\rm \;} ({\rm NO}_2^ - )\; $ in the regulation of mitochondrial function and ROS generation is becoming an area of interest in the context of CVD prevention(Reference Shiva and Gladwin51).
Inflammation
Chronic inflammation is a driver of ageing and contributes to the pathology of many age-related diseases including atherosclerosis(Reference Chung, Cesari and Anton52). Observational and experimental studies have demonstrated the importance of inflammation as a determinant of an unhealthy ageing phenotype. For example, the Whitehall II study reported that a high level of IL-6 almost halved the odds of successful ageing after 10 years (OR 0·53) and increased the risk of cardiovascular events and non-cardiovascular mortality(Reference Akbaraly, Hamer and Ferrie53). Growing evidence suggests important cross-talk between oxidative stress, inflammatory processes and the onset of ED prior to atherosclerosis(Reference Ungvari, Kaley and de Cabo34). ROS induce pro-inflammatory changes in the vascular endothelium, described as endothelial activation, which involves secretion of autocrine/paracrine factors, leucocyte-endothelial interaction and the up-regulation of expression of cellular adhesion molecules(Reference Herrera, Mingorance and Rodriguez-Rodriguez54). Oxidative stress activates redox-sensitive transcription factors including the activator protein and NF-κB, increasing the expression of cytokines (TNF-α, IL-1 and IL-6), adhesion molecules (intercellular adhesion molecule and vascular cell adhesion molecule) and pro-inflammatory enzymes (inducible NOS and cyclooxygenase-2)(Reference El Assar, Angulo and Vallejo39).
Ageing is associated with higher circulating concentrations of cytokines, especially TNF-α, IL-1β and IL-6, which mediate the acute phase protein C-reactive protein(Reference El Assar, Angulo and Vallejo39). These factors contribute significantly to the pro-inflammatory microenvironment and facilitate the development of vascular dysfunction(Reference El Assar, Angulo and Vallejo39). Among middle-aged and older adults, the Framingham heart study showed that brachial flow-mediated dilation is inversely related to C-reactive protein, IL-6 and intercellular adhesion molecule inflammatory markers(Reference Vita, Keaney and Larson55). Further, inhibition of NF-κB signalling improved EF significantly in middle-aged and older adults(Reference Pierce, Lesniewski and Lawson56).
Senescence
Cellular senescence is characterised by telomere shortening and permanent loss of mitotic capability, which are associated with morphological and functional changes and impaired cellular homeostasis(Reference Erusalimsky57). Risk factors for atherosclerosis including oxidative stress, inflammation, smoking, diabetes and hypertension have all been associated with accelerated telomere shortening(Reference Fyhrquist and Saijonmaa58). Telomere length in endothelial cells is inversely proportional to patient age(Reference Aviv, Khan and Skurnick59), and this shortening is exacerbated in older aged patients with coronary artery disease(Reference Ogami, Ikura and Ohsawa60). Cross-sectional studies demonstrate that those with increased arterial stiffness, an indicator of vascular ageing, have shorter telomeres(Reference Nawrot, Staessen and Holvoet61). Hypertensive patients have shorter telomeres than their normotensive peers and hypertensives with shorter telomeres are more likely to develop atherosclerosis over 5 years follow-up(Reference El Assar, Angulo and Vallejo39). The afore-mentioned observations suggest that telomere length might be a potential candidate marker for cardiovascular ageing. Vascular endothelial cell senescence in vivo has also been observed(Reference Minamino, Miyauchi and Yoshida62). The development of more senescent endothelial cells has been linked to a shift from an anti-atherosclerotic phenotype (characterised by decreased levels of NO, eNOS activity and shear stress-induced NO production) to a pro-atherosclerotic phenotype (indicated by increased ROS, thromboxane A2 and endothelin-1). These observations implicate endothelial cell senescence in the initiation and progression of atherosclerosis(Reference Minamino and Komuro63). NO increases telomerase activity and promotes mobilisation of endothelial progenitors cells, which have the potential to delay endothelial cell ageing by replacing damaged endothelial cells to maintain physical and functional integrity of the endothelium(Reference Farsetti, Grasselli and Bacchetti64).
Ageing-associated nitric oxide insufficiency
Vascular NO insufficiency in older people is mediated in part by decreased NO production by eNOS(Reference Cau, Carneiro and Tostes65). There is evidence that eNOS activity is reduced with age because of post-translational modification such as acylation, nitrosylation, glycation or phosphorylation(Reference Cau, Carneiro and Tostes65). Additionally, this reduction in eNOS activity might be secondary to the deficiency of cofactors required in the process of NO production (e.g. tetrahydrobiopterin)(Reference Seals, Kaplon and Gioscia-Ryan66). The age-associated increase in arginase activity may compete with eNOS for the critical substrate required in NO production, l-arginine(Reference Seals, Kaplon and Gioscia-Ryan66). Further, excessive O2− production with ageing may contribute to NO insufficiency. The interaction of O2− with NO produces the highly reactive ONOO−(Reference Sindler, Devan and Fleenor67). Due to its ability to restore reduced NO bioavailability, inorganic 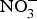 ${\rm NO}_3^ - $ represents a potential therapeutic strategy to treat age-associated vascular dysfunction(Reference Sindler, Devan and Fleenor67).
${\rm NO}_3^ - $ represents a potential therapeutic strategy to treat age-associated vascular dysfunction(Reference Sindler, Devan and Fleenor67).
The nitrate–nitrite−nitric oxide pathway
Epidemiological studies have consistently shown a protective effect of higher intake of fruit and vegetables and reduced risk of CVD(Reference Zhan, Liu and Cai68). Whilst the exact mechanism/s through which a fruit- and vegetable-rich diet reduces CVD risk remains to be fully elucidated, an increase in NO bioavailability is likely to be important. Eighty-five per cent of the dietary 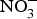 ${\rm NO}_3^ - $ is derived from vegetables and the remaining is mostly from drinking-water. Dietary intake of
${\rm NO}_3^ - $ is derived from vegetables and the remaining is mostly from drinking-water. Dietary intake of 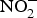 ${\rm NO}_2^ - $ principally comes from cured meat, to which
${\rm NO}_2^ - $ principally comes from cured meat, to which  ${\rm NO}_2^ - $ salts are added to prevent the development of botulinum toxin and to maintain product taste and colour(Reference Hord, Tang and Bryan69). Vegetables can be categorised according to their
${\rm NO}_2^ - $ salts are added to prevent the development of botulinum toxin and to maintain product taste and colour(Reference Hord, Tang and Bryan69). Vegetables can be categorised according to their 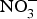 ${\rm NO}_3^ - $ contents into three categories: (1) high
${\rm NO}_3^ - $ contents into three categories: (1) high 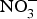 ${\rm NO}_3^ - $ contents: e.g. rocket, spinach, lettuce and beetroot (>1000 mg/kg); (2) medium
${\rm NO}_3^ - $ contents: e.g. rocket, spinach, lettuce and beetroot (>1000 mg/kg); (2) medium 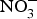 ${\rm NO}_3^ - $ contents: e.g. turnip, cabbage, green beans, cucumber and carrot (100–1000 mg/kg); and (3) low
${\rm NO}_3^ - $ contents: e.g. turnip, cabbage, green beans, cucumber and carrot (100–1000 mg/kg); and (3) low 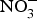 ${\rm NO}_3^ - $ contents: e.g. onion and tomato (<100 mg/kg)(Reference Lidder and Webb70). The concentration of
${\rm NO}_3^ - $ contents: e.g. onion and tomato (<100 mg/kg)(Reference Lidder and Webb70). The concentration of 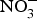 ${\rm NO}_3^ - $ in drinking-water varies according to the geographical location and regional rules regarding safe levels of
${\rm NO}_3^ - $ in drinking-water varies according to the geographical location and regional rules regarding safe levels of 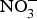 ${\rm NO}_3^ - $ in tap or bottled water(Reference Bryan and Ivy71).
${\rm NO}_3^ - $ in tap or bottled water(Reference Bryan and Ivy71).
It is thought that the beneficial effect of NO disappears in a few seconds as this gasotransmitter is oxidised to 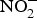 ${\rm NO}_2^ - $ and then to
${\rm NO}_2^ - $ and then to 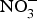 ${\rm NO}_3^ - $.
${\rm NO}_3^ - $. 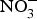 ${\rm NO}_3^ - $ is then excreted in urine as a cumulative by-product of NO metabolism and dietary
${\rm NO}_3^ - $ is then excreted in urine as a cumulative by-product of NO metabolism and dietary 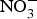 ${\rm NO}_3^ - \; $ intake(Reference Kelm72). Interestingly, in the past two decades, scientists discovered an alternative pathway for NO source other than the classical l-arginine–eNOS–NO pathway(Reference Zweier, Wang and Samouilov73). The other source of NO was found to be
${\rm NO}_3^ - \; $ intake(Reference Kelm72). Interestingly, in the past two decades, scientists discovered an alternative pathway for NO source other than the classical l-arginine–eNOS–NO pathway(Reference Zweier, Wang and Samouilov73). The other source of NO was found to be 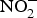 ${\rm NO}_2^ - $, which can be converted back to NO by the action of several enzymes and molecules including deoxyhaemoglobin, deoxymyoglobin, xanthine oxidoreductase, protons, polyphenols and ascorbic acid(Reference Lundberg, Weitzberg and Gladwin74). Of note, this pathway is more active and efficient in cases of hypoxia in which the level of both oxygen and NO are low(Reference Zweier, Samouilov and Kuppusamy75).
${\rm NO}_2^ - $, which can be converted back to NO by the action of several enzymes and molecules including deoxyhaemoglobin, deoxymyoglobin, xanthine oxidoreductase, protons, polyphenols and ascorbic acid(Reference Lundberg, Weitzberg and Gladwin74). Of note, this pathway is more active and efficient in cases of hypoxia in which the level of both oxygen and NO are low(Reference Zweier, Samouilov and Kuppusamy75).
Dietary 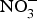 ${\rm NO}_3^ - $ is well absorbed in the upper gastrointestinal tract with approximately 100 % bioavailability and plasma concentration of
${\rm NO}_3^ - $ is well absorbed in the upper gastrointestinal tract with approximately 100 % bioavailability and plasma concentration of 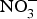 ${\rm NO}_3^ - $ peaking after 1 h(Reference Lidder and Webb70). About 25 % of the circulating pool of
${\rm NO}_3^ - $ peaking after 1 h(Reference Lidder and Webb70). About 25 % of the circulating pool of 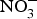 ${\rm NO}_3^ - $ is actively taken up from the blood via an anion exchange channel called sialin and secreted by the salivary glands into the saliva(Reference Bailey, Feelisch and Horowitz76). The salivary
${\rm NO}_3^ - $ is actively taken up from the blood via an anion exchange channel called sialin and secreted by the salivary glands into the saliva(Reference Bailey, Feelisch and Horowitz76). The salivary 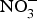 ${\rm NO}_3^ - $ is reduced to
${\rm NO}_3^ - $ is reduced to 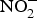 ${\rm NO}_2^ - $ by facultative anaerobic bacteria in the oral cavity, particularly those residing on the dorsal surface of the tongue(Reference Lundberg, Weitzberg and Gladwin74). This
${\rm NO}_2^ - $ by facultative anaerobic bacteria in the oral cavity, particularly those residing on the dorsal surface of the tongue(Reference Lundberg, Weitzberg and Gladwin74). This 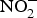 ${\rm NO}_2^ - $ and other inorganic
${\rm NO}_2^ - $ and other inorganic 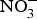 ${\rm NO}_3^ - $ travel to the stomach where they are converted to NO with the help of ascorbic acid. In this strong acidic environment of the stomach,
${\rm NO}_3^ - $ travel to the stomach where they are converted to NO with the help of ascorbic acid. In this strong acidic environment of the stomach, 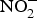 ${\rm NO}_2^ - $ is protonated to form nitrous acid (HNO2)(Reference Bailey, Feelisch and Horowitz76). Nitrous acid can spontaneously give rise to the generation of NO through the following sequence of reactions: 2HNO2 → H2O + N2O3 and N2O3 ↔ NO + NO2(Reference Butler and Feelisch77). The liberated NO has been found to be protective for the gastric mucosa, i.e. enhances blood supply(Reference Lundberg, Weitzberg and Gladwin74). Moreover, the remaining NO,
${\rm NO}_2^ - $ is protonated to form nitrous acid (HNO2)(Reference Bailey, Feelisch and Horowitz76). Nitrous acid can spontaneously give rise to the generation of NO through the following sequence of reactions: 2HNO2 → H2O + N2O3 and N2O3 ↔ NO + NO2(Reference Butler and Feelisch77). The liberated NO has been found to be protective for the gastric mucosa, i.e. enhances blood supply(Reference Lundberg, Weitzberg and Gladwin74). Moreover, the remaining NO, 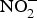 ${\rm NO}_2^ - $ and
${\rm NO}_2^ - $ and 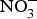 ${\rm NO}_3^ - $ diffuse to the general circulation and contribute to NO pool(Reference Lundberg, Feelisch and Bjorne78).
${\rm NO}_3^ - $ diffuse to the general circulation and contribute to NO pool(Reference Lundberg, Feelisch and Bjorne78).
In the circulation, 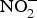 ${\rm NO}_2^ - $ may function as a source of NO that is activated in hypoxia and acidic conditions to increase blood flow and regulate BP(Reference Kevil, Kolluru and Pattillo79, Reference Zweier, Li and Samouilov80). There are many mechanisms involved in the bioconversion of
${\rm NO}_2^ - $ may function as a source of NO that is activated in hypoxia and acidic conditions to increase blood flow and regulate BP(Reference Kevil, Kolluru and Pattillo79, Reference Zweier, Li and Samouilov80). There are many mechanisms involved in the bioconversion of 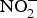 ${\rm NO}_2^ - $ to NO in the blood(Reference Kim-Shapiro and Gladwin81). The most common is the reaction of deoxyhaemoglobin (HbFe2+) with
${\rm NO}_2^ - $ to NO in the blood(Reference Kim-Shapiro and Gladwin81). The most common is the reaction of deoxyhaemoglobin (HbFe2+) with 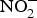 ${\rm NO}_2^ - $ in acidic environment, which will liberate NO (
${\rm NO}_2^ - $ in acidic environment, which will liberate NO (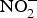 ${\rm NO}_2^ - $ + HbFe2+ + H+ → NO + HbFe3+ + OH−)(Reference Kim-Shapiro and Gladwin81). In addition to HbFe2+, there are many enzymes and proteins that enhance the conversion of
${\rm NO}_2^ - $ + HbFe2+ + H+ → NO + HbFe3+ + OH−)(Reference Kim-Shapiro and Gladwin81). In addition to HbFe2+, there are many enzymes and proteins that enhance the conversion of 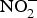 ${\rm NO}_2^ - $ to NO such as myoglobin, cytochrome C oxidase, eNOS and xanthine oxidoreductases(Reference Kim-Shapiro and Gladwin81).
${\rm NO}_2^ - $ to NO such as myoglobin, cytochrome C oxidase, eNOS and xanthine oxidoreductases(Reference Kim-Shapiro and Gladwin81).
Therapeutic effects of inorganic nitrate in patients with CVD
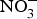 ${\rm NO}_3^ - $ has been used in the treatment of CVD including angina and digital ischaemia since medieval times(Reference Butler and Feelisch77, Reference Machha and Schechter82). In the past 30 years, since the discovery of the
${\rm NO}_3^ - $ has been used in the treatment of CVD including angina and digital ischaemia since medieval times(Reference Butler and Feelisch77, Reference Machha and Schechter82). In the past 30 years, since the discovery of the 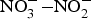 ${\rm NO}_3^ - {\rm -} {\rm NO}_2^ - $−NO pathway and its contribution to the overall NO pool(Reference Kapil, Weitzberg and Lundberg83), there has been a renewal in using
${\rm NO}_3^ - {\rm -} {\rm NO}_2^ - $−NO pathway and its contribution to the overall NO pool(Reference Kapil, Weitzberg and Lundberg83), there has been a renewal in using 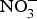 ${\rm NO}_3^ - $ and
${\rm NO}_3^ - $ and 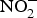 ${\rm NO}_2^ - $ in experiments and in clinical trials focused on the prevention of CVD. Larsen et al. (Reference Larsen, Ekblom and Sahlin84) in a pioneer study demonstrated the beneficial effect of inorganic
${\rm NO}_2^ - $ in experiments and in clinical trials focused on the prevention of CVD. Larsen et al. (Reference Larsen, Ekblom and Sahlin84) in a pioneer study demonstrated the beneficial effect of inorganic 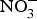 ${\rm NO}_3^ - $ in BP reduction; the investigators administered 0·1 mmol sodium
${\rm NO}_3^ - $ in BP reduction; the investigators administered 0·1 mmol sodium 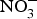 ${\rm NO}_3^ - $/kg body weight daily to healthy participants (which corresponds to an intake of 100–300 g of
${\rm NO}_3^ - $/kg body weight daily to healthy participants (which corresponds to an intake of 100–300 g of 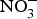 ${\rm NO}_3^ - $-rich vegetables daily) and found after 3 d of
${\rm NO}_3^ - $-rich vegetables daily) and found after 3 d of 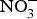 ${\rm NO}_3^ - $ supplementation, a 4 mmHg reduction in diastolic BP(Reference Larsen, Ekblom and Sahlin84). Administration of the same dose of
${\rm NO}_3^ - $ supplementation, a 4 mmHg reduction in diastolic BP(Reference Larsen, Ekblom and Sahlin84). Administration of the same dose of 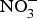 ${\rm NO}_3^ - $ to a larger group of individuals produced significant reductions in both systolic and diastolic BP(Reference Larsen, Weitzberg and Lundberg85). After the publication of these seminal studies, there has been a growing interest in the protective effects of dietary
${\rm NO}_3^ - $ to a larger group of individuals produced significant reductions in both systolic and diastolic BP(Reference Larsen, Weitzberg and Lundberg85). After the publication of these seminal studies, there has been a growing interest in the protective effects of dietary 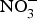 ${\rm NO}_3^ - $ on cardio-metabolic outcomes. However, the majority of the studies have been conducted in healthy populations and the evidence on the effects of dietary
${\rm NO}_3^ - $ on cardio-metabolic outcomes. However, the majority of the studies have been conducted in healthy populations and the evidence on the effects of dietary 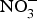 ${\rm NO}_3^ - $ supplementation in patients with CVD is still limited. A summary of the dietary
${\rm NO}_3^ - $ supplementation in patients with CVD is still limited. A summary of the dietary 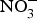 ${\rm NO}_3^ - $ and
${\rm NO}_3^ - $ and 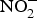 ${\rm NO}_2^ - $ interventions conducted in patients with CVD is provided in Table 1.
${\rm NO}_2^ - $ interventions conducted in patients with CVD is provided in Table 1.
Table 1. Summary of nutritional interventions testing the effects of dietary nitrate supplementations in patients with CVD

R, randomised; CO, cross-over; P, placebo; BJ, beetroot juice; BP, blood pressure; SBP, systolic blood pressure; ABPM, ambulatory blood pressure monitoring; PAR, parallel; DB, double blind; FMD, flow-mediated dilation; DBP, diastolic blood pressure; PWV, pulse wave velocity; CHF, chronic heart failure; PAD, peripheral arterial disease; CKD, chronic kidney disease.
Electronic search conducted on PubMed on 24 January 2018 using the following algorithm: ‘dietary nitrate’ OR beetroot OR beet root OR ‘inorganic nitrate’. Number of articles retrieved by primary search: 1649. One author screened all articles to include studies that investigated effects of dietary nitrate supplementation in patients with CVD.
* Placebo was not defined.
Four weeks of 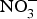 ${\rm NO}_3^ - $ supplementation (9 mg/kg) to older individuals at higher CVD risk significantly lowered systolic BP by 8 mmHg in comparison with placebo(Reference Rammos, Hendgen-Cotta and Sobierajski86). Supplementing beetroot juice (providing a
${\rm NO}_3^ - $ supplementation (9 mg/kg) to older individuals at higher CVD risk significantly lowered systolic BP by 8 mmHg in comparison with placebo(Reference Rammos, Hendgen-Cotta and Sobierajski86). Supplementing beetroot juice (providing a 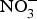 ${\rm NO}_3^ - $ dose of 300–400 mg) to older overweight, but otherwise healthy, participants for 3 weeks lowered daily home-measured systolic BP by 7 mmHg(Reference Jajja, Sutyarjoko and Lara87). However, BP values were found to have returned to pre-intervention values, 1 week after stopping the beetroot supplementation. Kapil et al. (Reference Kapil, Khambata and Robertson88) conducted the largest and longest trial in stage 1 hypertensive patients and found that dietary
${\rm NO}_3^ - $ dose of 300–400 mg) to older overweight, but otherwise healthy, participants for 3 weeks lowered daily home-measured systolic BP by 7 mmHg(Reference Jajja, Sutyarjoko and Lara87). However, BP values were found to have returned to pre-intervention values, 1 week after stopping the beetroot supplementation. Kapil et al. (Reference Kapil, Khambata and Robertson88) conducted the largest and longest trial in stage 1 hypertensive patients and found that dietary 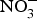 ${\rm NO}_3^ - $ improved both systolic and diastolic BP (measured by 24 h monitoring, home monitoring and clinic resting) and EF (measured by flow-mediated dilation and arterial stiffness). In contrast, studies in treated hypertensive patients did not show significant improvement of BP with beetroot administration(Reference Bondonno, Liu and Croft89). Moreover, an individual participant meta-analysis (eighty-five participants) showed that beetroot supplementation lowered 24 h ambulatory BP significantly in younger participants only (<65 years)(Reference Siervo, Lara and Jajja90). Two meta-analyses have demonstrated a significant reduction of systolic BP (−4·4 mm Hg)(Reference Siervo, Lara and Ogbonmwan91) and a significant improvement of EF(Reference Lara, Ashor and Oggioni92) with inorganic
${\rm NO}_3^ - $ improved both systolic and diastolic BP (measured by 24 h monitoring, home monitoring and clinic resting) and EF (measured by flow-mediated dilation and arterial stiffness). In contrast, studies in treated hypertensive patients did not show significant improvement of BP with beetroot administration(Reference Bondonno, Liu and Croft89). Moreover, an individual participant meta-analysis (eighty-five participants) showed that beetroot supplementation lowered 24 h ambulatory BP significantly in younger participants only (<65 years)(Reference Siervo, Lara and Jajja90). Two meta-analyses have demonstrated a significant reduction of systolic BP (−4·4 mm Hg)(Reference Siervo, Lara and Ogbonmwan91) and a significant improvement of EF(Reference Lara, Ashor and Oggioni92) with inorganic 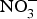 ${\rm NO}_3^ - $ or beetroot consumption.
${\rm NO}_3^ - $ or beetroot consumption.
The discovery of the contribution of dietary 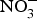 ${\rm NO}_3^ - $ to NO bioavailability has provided a rationale for the use of
${\rm NO}_3^ - $ to NO bioavailability has provided a rationale for the use of 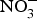 ${\rm NO}_3^ - $ to reverse ED secondary to NO insufficiency in cardiovascular and metabolic diseases(Reference Machha and Schechter82). Inorganic
${\rm NO}_3^ - $ to reverse ED secondary to NO insufficiency in cardiovascular and metabolic diseases(Reference Machha and Schechter82). Inorganic 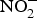 ${\rm NO}_2^ - $ supplementation reversed ED significantly in a murine model of hypercholesterolaemia(Reference Stokes, Dugas and Tang93). In human subjects, dietary
${\rm NO}_2^ - $ supplementation reversed ED significantly in a murine model of hypercholesterolaemia(Reference Stokes, Dugas and Tang93). In human subjects, dietary 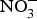 ${\rm NO}_3^ - $ supplementation has been found to improve flow-mediated dilation and arterial stiffness in hypercholesterolaemic patients(Reference Velmurugan, Gan and Rathod94) and reduce TAG concentrations in patients at higher CVD risk(Reference Zand, Lanza and Garg95).
${\rm NO}_3^ - $ supplementation has been found to improve flow-mediated dilation and arterial stiffness in hypercholesterolaemic patients(Reference Velmurugan, Gan and Rathod94) and reduce TAG concentrations in patients at higher CVD risk(Reference Zand, Lanza and Garg95).
Data from animal studies have also shown promising results regarding the effect of dietary 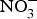 ${\rm NO}_3^ - $ on biomarkers of metabolic diseases. Supplementation of eNOS-deficient mice suffering from metabolic syndrome with inorganic
${\rm NO}_3^ - $ on biomarkers of metabolic diseases. Supplementation of eNOS-deficient mice suffering from metabolic syndrome with inorganic 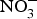 ${\rm NO}_3^ - $ for 10 weeks reduced visceral fat and circulating TAG concentration and reversed the pre-diabetic phenotype(Reference Carlstrom, Larsen and Nystrom96). Further, supplementing diabetic rats with sodium
${\rm NO}_3^ - $ for 10 weeks reduced visceral fat and circulating TAG concentration and reversed the pre-diabetic phenotype(Reference Carlstrom, Larsen and Nystrom96). Further, supplementing diabetic rats with sodium 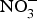 ${\rm NO}_3^ - $ for 2 months produced significant improvements in glucose homeostasis, lipid profile and oxidative stress markers(Reference Khalifi, Rahimipour and Jeddi97). However,
${\rm NO}_3^ - $ for 2 months produced significant improvements in glucose homeostasis, lipid profile and oxidative stress markers(Reference Khalifi, Rahimipour and Jeddi97). However, 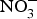 ${\rm NO}_3^ - $ supplementation in human subjects showed no evidence of improvement in glucose and insulin homeostasis in diabetics(Reference Cermak, Hansen and Kouw98–Reference Shepherd, Gilchrist and Winyard100) or non-diabetic participants(Reference Larsen, Schiffer and Ekblom101). Potassium
${\rm NO}_3^ - $ supplementation in human subjects showed no evidence of improvement in glucose and insulin homeostasis in diabetics(Reference Cermak, Hansen and Kouw98–Reference Shepherd, Gilchrist and Winyard100) or non-diabetic participants(Reference Larsen, Schiffer and Ekblom101). Potassium 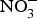 ${\rm NO}_3^ - $ supplementation did not improve glucose tolerance in young and older obese individuals but reduced oxidative stress during hyperglycaemia in older individuals(Reference Ashor, Chowdhury and Oggioni102).
${\rm NO}_3^ - $ supplementation did not improve glucose tolerance in young and older obese individuals but reduced oxidative stress during hyperglycaemia in older individuals(Reference Ashor, Chowdhury and Oggioni102).
Inorganic 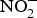 ${\rm NO}_2^ - $ reversed ageing-related arterial stiffness in older mice. In one study, the plasma
${\rm NO}_2^ - $ reversed ageing-related arterial stiffness in older mice. In one study, the plasma 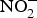 ${\rm NO}_2^ - $ concentration in older mice was found to be restored to youthful concentrations with inorganic
${\rm NO}_2^ - $ concentration in older mice was found to be restored to youthful concentrations with inorganic 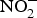 ${\rm NO}_2^ - $ supplementation(Reference Sindler, Fleenor and Calvert103). In healthy human subjects, Bahra et al. (Reference Bahra, Kapil and Pearl104) observed a significant reduction in arterial stiffness 3 h after
${\rm NO}_2^ - $ supplementation(Reference Sindler, Fleenor and Calvert103). In healthy human subjects, Bahra et al. (Reference Bahra, Kapil and Pearl104) observed a significant reduction in arterial stiffness 3 h after 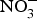 ${\rm NO}_3^ - $ ingestion. Further, one study found that daily consumption of
${\rm NO}_3^ - $ ingestion. Further, one study found that daily consumption of 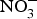 ${\rm NO}_3^ - $ (900 mg) for 4 weeks reduced pulse wave velocity in older people at increased CVD risk(Reference Rammos, Hendgen-Cotta and Sobierajski86). However, in another study, arterial compliance increased with no change in pulse wave velocity after 220 mg
${\rm NO}_3^ - $ (900 mg) for 4 weeks reduced pulse wave velocity in older people at increased CVD risk(Reference Rammos, Hendgen-Cotta and Sobierajski86). However, in another study, arterial compliance increased with no change in pulse wave velocity after 220 mg 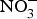 ${\rm NO}_3^ - $ supplementation in twenty-eight healthy participants(Reference Liu, Bondonno and Croft105).
${\rm NO}_3^ - $ supplementation in twenty-eight healthy participants(Reference Liu, Bondonno and Croft105).
Inorganic 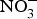 ${\rm NO}_3^ - $ administration inhibits platelet aggregation, and therefore, may reduce thrombotic events in both human subjects and experimental animals(Reference Park, Piknova and Huang106, Reference Richardson, Hicks and O'Byrne107). Two studies have found a positive effect of dietary
${\rm NO}_3^ - $ administration inhibits platelet aggregation, and therefore, may reduce thrombotic events in both human subjects and experimental animals(Reference Park, Piknova and Huang106, Reference Richardson, Hicks and O'Byrne107). Two studies have found a positive effect of dietary 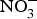 ${\rm NO}_3^ - $ supplementation on platelet aggregation in hypercholesterolaemic patients(Reference Velmurugan, Gan and Rathod94) and platelet-derived extracellular vesicles in coronary artery disease patients on clopidogrel therapy(Reference Burnley-Hall, Abdul and Androshchuk108). The restoration of blood to a tissue after a period of ischaemia is sometimes associated with severe tissue injury due to a high release of free radicals. Animal studies have demonstrated that the prior administration of inorganic
${\rm NO}_3^ - $ supplementation on platelet aggregation in hypercholesterolaemic patients(Reference Velmurugan, Gan and Rathod94) and platelet-derived extracellular vesicles in coronary artery disease patients on clopidogrel therapy(Reference Burnley-Hall, Abdul and Androshchuk108). The restoration of blood to a tissue after a period of ischaemia is sometimes associated with severe tissue injury due to a high release of free radicals. Animal studies have demonstrated that the prior administration of inorganic 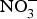 ${\rm NO}_3^ - $ reduces the infarct size in a model of ischaemic-reperfusion injury(Reference Lundberg, Carlstrom and Larsen109). Moreover, low-dose sodium
${\rm NO}_3^ - $ reduces the infarct size in a model of ischaemic-reperfusion injury(Reference Lundberg, Carlstrom and Larsen109). Moreover, low-dose sodium 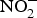 ${\rm NO}_2^ - $ attenuated myocardial ischaemia and vascular reperfusion injury in a human experimental study(Reference Ingram, Fraser and Bleasdale110). However, Schwarz et al. (Reference Schwarz, Singh and Parasuraman111) showed that supplementation with sodium
${\rm NO}_2^ - $ attenuated myocardial ischaemia and vascular reperfusion injury in a human experimental study(Reference Ingram, Fraser and Bleasdale110). However, Schwarz et al. (Reference Schwarz, Singh and Parasuraman111) showed that supplementation with sodium 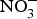 ${\rm NO}_3^ - $ marginally improved exercise performance in patients with chronic angina on prescribed medications. Conversely, positive effects of dietary
${\rm NO}_3^ - $ marginally improved exercise performance in patients with chronic angina on prescribed medications. Conversely, positive effects of dietary 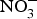 ${\rm NO}_3^ - $ supplementation were found on exercise tolerance and onset of claudication intermittens in eight patients with peripheral arterial disease(Reference Kenjale, Ham and Stabler112). Dietary
${\rm NO}_3^ - $ supplementation were found on exercise tolerance and onset of claudication intermittens in eight patients with peripheral arterial disease(Reference Kenjale, Ham and Stabler112). Dietary 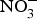 ${\rm NO}_3^ - $ supplementation appears to have positive effects on exercise performance and oxygen consumption in patients with chronic heart failure(Reference Coggan, Broadstreet and Mahmood113, Reference Zamani, Tan and Soto-Calderon114), whereas the effects on BP, at rest and during exercise, and cardiac haemodynamics are less replicable(Reference Kapil, Khambata and Robertson88–Reference Siervo, Lara and Ogbonmwan91, Reference Gilchrist, Winyard and Aizawa99, Reference Oggioni, Jakovljevic and Klonizakis115–Reference Ashor, Lara and Siervo117).
${\rm NO}_3^ - $ supplementation appears to have positive effects on exercise performance and oxygen consumption in patients with chronic heart failure(Reference Coggan, Broadstreet and Mahmood113, Reference Zamani, Tan and Soto-Calderon114), whereas the effects on BP, at rest and during exercise, and cardiac haemodynamics are less replicable(Reference Kapil, Khambata and Robertson88–Reference Siervo, Lara and Ogbonmwan91, Reference Gilchrist, Winyard and Aizawa99, Reference Oggioni, Jakovljevic and Klonizakis115–Reference Ashor, Lara and Siervo117).
Directions for future research
There is currently limited evidence to support the protective effects of inorganic 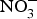 ${\rm NO}_3^ - $ supplementation on cardiovascular and metabolic outcomes in patients at higher CVD risk. Several studies have been conducted in patients with hypertension and chronic heart failure, but the results have been contrasting, whereas for other cardiovascular disorders such as diabetes, CHD or chronic kidney failure, there is simply a paucity of studies. In addition, the evidence is further weakened by the pilot nature of these studies both in terms of short duration (longest trial is 8 weeks)(Reference Kapil, Weitzberg and Lundberg83) of the interventions and small sample size (largest population is seventy patients)(Reference Schwarz, Singh and Parasuraman111). Future research efforts should be therefore directed at the conduction of more robust, confirmatory trials to provide strong and unbiased evidence on the effects of dietary
${\rm NO}_3^ - $ supplementation on cardiovascular and metabolic outcomes in patients at higher CVD risk. Several studies have been conducted in patients with hypertension and chronic heart failure, but the results have been contrasting, whereas for other cardiovascular disorders such as diabetes, CHD or chronic kidney failure, there is simply a paucity of studies. In addition, the evidence is further weakened by the pilot nature of these studies both in terms of short duration (longest trial is 8 weeks)(Reference Kapil, Weitzberg and Lundberg83) of the interventions and small sample size (largest population is seventy patients)(Reference Schwarz, Singh and Parasuraman111). Future research efforts should be therefore directed at the conduction of more robust, confirmatory trials to provide strong and unbiased evidence on the effects of dietary 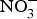 ${\rm NO}_3^ - $ on cardiovascular outcomes. In consideration of the larger number of studies and overall supportive effects of dietary
${\rm NO}_3^ - $ on cardiovascular outcomes. In consideration of the larger number of studies and overall supportive effects of dietary 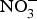 ${\rm NO}_3^ - $ on BP, priority might be given to the design of trials testing the effects of dietary
${\rm NO}_3^ - $ on BP, priority might be given to the design of trials testing the effects of dietary 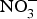 ${\rm NO}_3^ - $ in larger populations of hypertensive patients with and without anti-hypertensive medications to evaluate whether dietary
${\rm NO}_3^ - $ in larger populations of hypertensive patients with and without anti-hypertensive medications to evaluate whether dietary 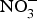 ${\rm NO}_3^ - $ provides additive effects to background pharmacological treatments of BP. These studies may also take into consideration the recruitment of patients with more severe hypertension (stage 2 or 3) and evaluate whether ethnicity could be a modifying factor of the BP response to dietary
${\rm NO}_3^ - $ provides additive effects to background pharmacological treatments of BP. These studies may also take into consideration the recruitment of patients with more severe hypertension (stage 2 or 3) and evaluate whether ethnicity could be a modifying factor of the BP response to dietary 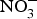 ${\rm NO}_3^ - $ supplementation.
${\rm NO}_3^ - $ supplementation.
Conclusions
NO influences several physiological functions involved in the pathogenesis of CVD such as ROS generation, inflammation and platelet aggregation. Increasing inorganic 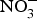 ${\rm NO}_3^ - $ intake, via supplementation of
${\rm NO}_3^ - $ intake, via supplementation of 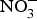 ${\rm NO}_3^ - $ salts or increased high-
${\rm NO}_3^ - $ salts or increased high-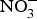 ${\rm NO}_3^ - $ food consumption (i.e. beetroot, green leafy vegetables) could represent a viable and effective strategy for the prevention of age-related chronic cardiovascular and metabolic diseases. The evidence from randomised clinical trials has so far suggested positive effects of systolic BP and EF but the size of the effect appears to be declining in older patients at higher cardiovascular risk. Therefore, until larger and more robust trials are conducted in patients at higher CVD risk, dietary
${\rm NO}_3^ - $ food consumption (i.e. beetroot, green leafy vegetables) could represent a viable and effective strategy for the prevention of age-related chronic cardiovascular and metabolic diseases. The evidence from randomised clinical trials has so far suggested positive effects of systolic BP and EF but the size of the effect appears to be declining in older patients at higher cardiovascular risk. Therefore, until larger and more robust trials are conducted in patients at higher CVD risk, dietary 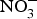 ${\rm NO}_3^ - $ supplementation cannot be recommended as a nutritional or clinical strategy for the primary and secondary prevention of CVD.
${\rm NO}_3^ - $ supplementation cannot be recommended as a nutritional or clinical strategy for the primary and secondary prevention of CVD.
Acknowledgements
The authors are very grateful to the reviewer of this manuscript for their helpful comments and swift feedback.
Financial Support
None.
Conflicts of Interest
None.
Authorship
M. S. is the guarantor of this work and takes responsibility for the integrity of the data. M. S., F. S. and A. W. A. wrote the manuscript; M. S. conducted the systematic search and completed the data extraction; O. M. S. and B. C. M contributed to the discussion and reviewed/edited the manuscript.



