



Introduction
Bats, a species-rich group with over 1454 species (Simmons, Reference Simmons2005; Miller-Butterworth et al., Reference Miller-Butterworth, Murphy, O'Brien, Jacobs, Springer and Teeling2007; Lack et al., Reference Lack, Roehrs, Stanley, Ruedi and Van Den Bussche2010) are mammalian hosts to a large diversity of eukaryotic protozoan blood parasites. These comprise different genera of haemosporidian parasites and diverse species of trypanosomes (e.g. Lima et al., Reference Lima, Espinosa-Alvarez, Hamilton, Neves, Takata, Campaner, Attias, de Souza, Camargo and Teixeira2013; Schaer et al., Reference Schaer, Perkins, Decher, Leendertz, Fahr, Weber and Matuschewski2013; Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020).
The apicomplexan parasites of the order Haemosporida infect diverse vertebrate hosts, comprising mammals, squamates and birds and use different dipteran hosts as vectors (Garnham, Reference Garnham1966; Levine, Reference Levine1988). The over 500 described haemosporidian parasites are classified in the genus Plasmodium that includes the human-infecting species that cause the malaria disease, plus 9 additional genera (Martinsen and Perkins, Reference Martinsen, Perkins, Carlton, Perkins and Deitsch2013). Among mammals, bats harbour the most diverse set of haemosporidian parasites and results of previous phylogenetic studies suggest that bats have played an important role in the evolutionary history of haemosporidian parasites (Duval et al., Reference Duval, Robert, Csorba, Hassanin, Randrianarivelojosia, Walston, Nhim, Goodman and Ariey2007; Schaer et al., Reference Schaer, Perkins, Decher, Leendertz, Fahr, Weber and Matuschewski2013; Perkins and Schaer, Reference Perkins and Schaer2016; Galen et al., Reference Galen, Borner, Martinsen, Schaer, Austin, West and Perkins2018).
The kinetoplastid flagellated Trypanosoma parasites feature complex lifecycles and are mainly transmitted to the vertebrate host by haematophagous arthropods or leeches (Simpson et al., Reference Simpson, Stevens and Lukeš2006). In Africa, the Trypanosoma species Trypanosoma brucei sensu lato, Trypanosoma vivax and Trypanosoma congolense present a threat to both humans and livestock (Morrison et al., Reference Morrison, Vezza, Rowan and Hope2016; Büscher et al., Reference Büscher, Cecchi, Jamonneau and Priotto2017). Previous studies have identified a high diversity of Trypanosoma species in bats and revealed that bats also played an important role in the evolutionary history of these parasites (e.g. Hamilton et al., Reference Hamilton, Cruickshank, Stevens, Teixeira and Mathews2012; Lima et al., Reference Lima, Silva, Neves, Attias, Takata, Campaner, de Souza, Hamilton and Teixeira2012, Reference Lima, Espinosa-Alvarez, Hamilton, Neves, Takata, Campaner, Attias, de Souza, Camargo and Teixeira2013; Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020; Austen and Barbosa, Reference Austen and Barbosa2021). The investigation of the diversity, distribution and phylogenetic relationships of trypanosome species of wildlife, including bats, is essential for a better understanding of the evolution of the whole parasite group.
With about 85 species of bats, Nigeria represents a diversity hotspot in Africa (Simmons and Cirranello, Reference Simmons and Cirranello2022). However, the knowledge about haemosporidian parasites and trypanosomes in Nigerian bats is scarce (Atama et al., Reference Atama, Manu, Ivande, Rosskopf, Matuschewski and Schaer2019). Here, we investigated the prevalence and phylogenetic relationships of haemosporidian and trypanosome parasites in different bat species in Northern Nigeria using molecular methods.
Materials and methods
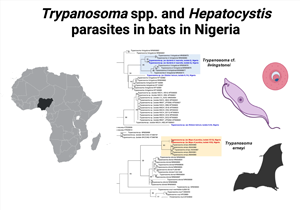
Bats were sampled in Northern Nigeria between December 2018 and March 2019 in the states of Bauchi, Benue, Katsina, Nasarawa and Plateau. The sampling sites comprised 10 peri-domestic roosting sites (trees, caves, ceiling/eaves of residential buildings) (Fig. 1).

Fig. 1. Map of Nigeria. Study areas are highlighted (in grey) and sampling sites are depicted.
Field sampling and microscopy
Bats were captured using improvised traps produced from fishing nets and wooden poles. The nets were set every capture night in some distance to their roosting sites. The bats were gently removed from the nets and transferred into individual bags and genus and/or species were identified following the keys available in Patterson and Webala (Patterson and Webala, Reference Patterson and Webala2012). Bats were captured and sacrificed within the scope of a larger project focusing on viral and bacterial infections in bats (e.g. Kamani et al., Reference Kamani, Harrus, Ocholi, Yague, Nyango, González-Miguel and Koizumi2021). The bats were anaesthetized using a combination of 5 mg kg−1 ketamine plus 2 mg kg−1 of xylazine injected intramuscularly. The voucher specimens were preserved in 70% (vol vol−1) ethanol and stored in the Parasitology Division NVRI Vom, in Nigeria. Blood samples were collected by venous puncture of the brachial vein and collected in capillary tubes. For the investigation of blood parasites, blood samples were used to spot blood dots onto DNA FTA cards (Whatman) and to prepare thin blood smears on slides. The blood smears were dried and fixed in 99–100% (vol vol−1) methanol solution for 3 s and later stained with 10% Giemsa solution for 50 min. Giemsa-stained blood smears were examined for the presence of haemosporidian and trypanosome parasites using light microscopy at a magnification of ×1000 with immersion oil.
Molecular methods
Whole genomic DNA was extracted from the dried blood dots on DNA FTA cards using the DNeasy extraction kit (Qiagen, Hilden, Germany) (e.g. Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017). The protocol for animal tissues was performed and samples were eluted in 60 μL AE buffer. Polymerase chain reactions (PCRs) were performed using the AllTaq Master Mix Kit (QIAGEN) with 4–5 μL of genomic DNA as the template, and 1 μL of each primer (10 mm).
To confirm morphological bat identifications, most of the samples were genotyped using 1 or more of the following genetic markers for the bat hosts: the mitochondrial cytochrome b (cytb) and the nuclear introns Acyl-CoA oxidase 2, intron 3 (acox2), and Beta-fibrinogen gene, intron 7 (fgb) (Table S1). A part of the cytb gene was sequenced for 47 of the 60 E. helvum individuals, which featured 100% nucleotide identity with E. helvum reference sequences in NCBI GenBank. For 11 E. helvum individuals, 650 bp of the fgb were additionally or instead sequenced, which again resulted in a nucleotide identity of 100% with published E. helvum reference sequences. To identify 3 individuals that were morphologically identified as Tadarida sp. to species level, cytb sequences were generated which resulted in highest nucleotide identities of 99.2% with reference sequences of Mops condylurus (e.g. GenBank accession number EF474030) and thus might represent M. condylurus or a close related species and are therefore termed M. cf. condylurus. Five out of 8 individuals that were morphologically identified as Chaerephon sp. were genotyped with cytb (featuring identical cytb sequences) and highest nucleotide identities of 98.4% were recovered with reference sequences for Mops pumilus (e.g. GQ489157). Again, these bats might represent the species M. pumilus or a close related species and are termed M. cf. pumilus. Twelve out of 16 Nycteris sp. individuals were genotyped with cytb and highest nucleotide identities of 94.7% (sample number KJ-M), 96.98% (sample numbers KJ-B, KJ-C, KJ-D, KJ-J) and 97.3% (remaining samples) were recovered with the reference sequence for Nycteris macrotis (accession number MT586790). The sequences of the nuclear acox2 for 2 of the Nycteris sp. samples (samples numbers KJ-E, KJ-F) featured a 98.7% nucleotide identity with the reference sequence for Nycteris macrotis complex sp.1 (MK837356). We, therefore, tentatively refer to the individuals of the study as Nycteris cf. macrotis.
The mitochondrial cytochrome b (cytb) gene was targeted for detection and subsequent phylogenetic analysis of haemosporidian parasites (e.g. Schaer et al., Reference Schaer, Perkins, Decher, Leendertz, Fahr, Weber and Matuschewski2013). Four genes were used for molecular detection of haemosporidian parasites: the mitochondrial genes cytochrome b (cytb) and cytochrome oxidase 1 (cox1); the apicoplast caseinolytic protease gene (clpC); and the nuclear gene elongation factor 2 (ef2) (Table S1) (Schaer et al., Reference Schaer, Perkins, Decher, Leendertz, Fahr, Weber and Matuschewski2013).
For the detection of trypanosomes, a nested-PCR approach was carried out for the amplification of the partial sequence of about 640 bp of the small subunit 18S ribosomal RNA gene (18S rRNA) following Noyes et al. (Reference Noyes, Stevens, Teixeira, Phelan and Holz1999). A second gene, the nuclear glycosomal glyceraldehyde phosphate dehydrogenase (gGAPDH), was subsequently targeted for all positive samples in a nested-PCR approach following Clement et al. (Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020). All primers are listed in Table S1. All positive PCR products were sequenced with the amplification primers and run on an ABI-373 sequencer. The software Geneious Prime 2022.1.1 (https://www.geneious.com) was used to quality-check and manually edit all nucleotide sequences. Ambiguous base calls or missing data were coded with N's or the corresponding ambiguity code. Sequences were aligned using the MAFFT algorithm (Katoh et al., Reference Katoh, Misawa, Kuma and Miyata2002; Katoh and Standley, Reference Katoh and Standley2013). Sequences for the analysis of the phylogenetic relationships of Hepatocystis comprised 531 nucleotides (nt) of the partial cytb gene. Sequence alignments for the analysis of Trypanosoma spp. included 1022 nt for 18S rRNA and 894 nt for gGAPDH, respectively. Reference sequences were retrieved from GenBank and added to the alignments (all accession numbers for analysis of Hepatocystis parasites are listed in Table S2, all accession numbers for 18S rRNA and gGAPDH analyses are listed in the corresponding phylogenetic tree figures and Table S3). The software ModelTest-NG was used to test different DNA substitution models (Darriba et al., Reference Darriba, Posada, Kozlov, Stamatakis, Morel and Flouri2020). Phylogenetic relationships were evaluated with maximum likelihood (ML) analysis and carried out in the software raxmlGUI version 2.0.6 (Edler et al., Reference Edler, Klein, Antonelli and Silvestro2021). The Hepatocystis analysis of the partial cytb was carried out using the substitution model GTR + I (proportion of invariant sites) + Gamma (rate heterogeneity) and the taxon Leucocytozoon as outgroup. Nodal support was evaluated using 1000 replicates (thorough bootstrap). The trypanosome ML analysis of the 18S rRNA and gGAPDH gene were carried out using the models TIM3 + I + G and GTR + I + G, respectively, and 10 000 replicates and Trypanosoma lewisi as outgroup taxon. Phylogenetic trees were displayed in FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Results
A total of 95 bats belonging to 3 bat families were investigated. The greater part of the captured bats (n = 68) belonged to the African fruit bat species Eidolon helvum. Further, 16 Nycteris cf. macrotis of the family Nycteridae, 8 Mops cf. pumilus and 3 Mops cf. condylurus (Molossidae) were screened for haemosporidian parasite and trypanosome parasite infections (Table 1).
Table 1. Investigated bat species and prevalences of haemosporidian and trypanosome parasite infections

Prevalence and phylogenetic characterization of haemosporidian parasites
Haemosporidian parasite infections were limited to only 4 individuals of E. helvum, which correspond to a prevalence of 5.9% (4/68) in E. helvum and an overall prevalence of 4.2% (4/95). No infections were detected in the species Nycteris cf. macrotis, Mops cf. condylurus and Mops cf. pumilus (Tables 1 and S4). The molecular analysis identified the parasite infections in E. helvum as Hepatocystis parasites. Microscopic screening of the corresponding Giemsa-stained thin blood smears of the 4 PCR-positive samples did not result in detection of parasite stages which points to subpatent infections with a very low parasitaemia. The partial cytb sequences of Hepatocystis sp. of 2 samples (sample numbers KJ72, KJ82) were identical, the third sample (sample number J53) featured a few ambiguous base calls which point to a mixed haplotype infection but was otherwise identical to the other 2 samples. The Hepatocystis sequences are identical to published parasite sequences from fruit bat hosts in Nigeria and South Sudan (Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017; Atama et al., Reference Atama, Manu, Ivande, Rosskopf, Matuschewski and Schaer2019). The 4th sequence (sample number KJ63) featured about 96% identity with the other 3 sequences that were recovered from Hepatocystis parasite infections in E. helvum in this study. Interestingly, all 4 sequences showed higher similarities to parasite sequences from other bat host species than to the sole published sequence from Hepatocystis sp. from E. helvum in Gabon. The phylogenetic analysis that was performed to assess the phylogenetic relationships and possible geographic and/or host specific patterns of Hepatocystis parasites from Nigeria confirmed Hepatocystis as a monophyletic group and as sister clade to the rodent- and bat-infecting Plasmodium (Vinckeia) parasite clade as shown before (Fig. 2) (e.g. Galen et al., Reference Galen, Borner, Martinsen, Schaer, Austin, West and Perkins2018). Within the African bat Hepatocystis clade, the Hepatocystis sequence from sample KJ63 groups basal, whereas the other 3 sequences fall within the main clade (Fig. 2). Overall, the Hepatocystis sp. sequences of different African bat species from several distant related African countries neither show geographic nor host specificity patterns confirming previous analysis (e.g. Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017).

Fig. 2. Maximum likelihood analysis of Hepatocystis parasites in African bats. The analysis is based on the mitochondrial gene cytb (1119 bp) and was run in the context of the major haemosporidian parasite clades comprising Leucocytozoon (used as outgroup taxon), Haemoproteus, Polychromophilus and the mammalian-infecting Plasmodium clades Plasmodium (Laverania) and Plasmodium (Vinckeia), with the latter representing the closest relative to Hepatocystis parasites. The phylogenetic analysis recovered the Hepatocystis sequences from E. helvum within the African bat Hepatocystis clade without any apparent geographic or host species specific pattern following previous findings [e.g. (Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017)]. Sequences of the study are highlighted in bold blue. Numbers at nodes are ML bootstrap value (> 70) using 1000 replicates.
Prevalence and phylogenetic characterization of Trypanosoma parasites
Trypanosome infections were detected in 16 individuals from 3 out of the 4 bat species of the study (16/95, overall prevalence of 16.8%) (Tables 1 and S4). Two infections were confirmed in E. helvum (2/68, prevalence of 2.9%), 3 infections in Mops cf. pumilus (3/8, 37.5%) and a high prevalence of 68.8% was found in Nycteris cf. macrotis (11/16). No infections were recorded for 3 individuals of Mops cf. condylurus (Table 1). The trypanosomes of Mops cf. pumilus (Molossidae) were identified as species Trypanosoma cf. erneyi based on the blastn (https://blast.ncbi.nlm.nih.gov/) search of the 18S and gGAPDH sequences and the phylogenetic analysis. All 3 infected individuals (sample numbers VC6, VC9, VC13) featured identical 18S rRNA sequences which shared highest sequence identities (96.5%) with a reference sequence of a trypanosome from Mops condylurus in Mozambique (JN040989). One gGAPDH sequence was successfully generated (for sample number VC13), which showed highest identity (95.2%) with a trypanosome reference sequence from Tadarida sp. (Molossidae) again from Mozambique (JN04095). The species Trypanosoma erneyi has been described from bat species of the family Molossidae in Africa and the sequences of this study group closely to the reference sequences of T. erneyi, but in their own separate clade, in the 18S rRNA phylogenetic analysis as well as in the gGAPDH analysis (Figs 3 and 4). This finding indicates that the sequences of this study might represent a distinct species or subspecies, however with the lack of morphological information of these parasites (no parasite stages were detected in the blood smears), they are tentatively referred to as T. cf. erneyi.

Fig. 3. Phylogeny of the Trypanosoma parasites inferred by maximum likelihood analysis from the 18S rRNA gene (1022 bp) using TIM3 + I + G and Trypanosoma lewisi as outgroup. Numbers at nodes are ML bootstrap values using 10 000 replicates. The parasites from Nycteris cf. macrotis hosts of the study group closely with T. cf. livingstonei parasites. One sequence from the parasites from E. helvum hosts groups basal to T. livingstonei parasites, whereas the other one was recovered as separate lineage to Trypanosoma sp. parasites. The parasite sequences of N. cf. macrotis and E. helvum of the study are highlighted in bold blue, the parasite sequences of Mops cf. pumilus in red. The trypanosome sequences from Mops cf. pumilus hosts of the study group as distinct lineage to T. erneyi parasites with high support (bootstrap value 93).

Fig. 4. Phylogeny of the Trypanosoma parasites inferred by maximum likelihood analysis from the gGAPDH gene (894 bp) using GTR + I + G and Trypanosoma lewisi as outgroup. Numbers at nodes are ML bootstrap values using 10 000 replicates. The parasites from Nycteris cf. macrotis hosts of the study group closely with T. cf. livingstonei parasites supporting the results of the 18S rRNA analysis (the sequences of the study are highlighted in bold blue). The trypanosome sequence from Mops cf. pumilus of the study (highlighted in bold red) groups as distinct lineage to T. erneyi parasites with high support (bootstrap value 92).
The trypanosome infections of the host species Nycteris cf. macrotis belong to 1 parasite haplotype of Trypanosoma cf. livingstonei, all 18S rRNA as well as the gGAPDH sequences were identical. The 18S rRNA parasite haplotype showed highest identity (98.3%) with a trypanosome reference sequence from Rhinolophus landeri from Mozambique (KF192981), the gGAPDH parasite haplotype of N. cf. macrotis featured highest identity (94.1%) with a reference sequence from a trypanosome isolated from Hipposideros caffer in Mozambique (KF192969). The phylogenetic analysis of the 18S rRNA as well as the nuclear gGAPDH sequences grouped the representative parasite sequences of N. cf. macrotis within the T. cf. livingstonei clades that comprise sequences from Nycteris hosts among other bat species from Africa (Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020) (Figs 3 and 4). Unfortunately, the quality of the blood smears did not allow a further analysis of the detected parasite stages in 2 samples of N. cf. macrotis (Fig. S1).
Trypanosome infections were further detected in 2 individuals of E. helvum. The first sample (number KJ14) featured 97.5% nucleotide identity (18S rRNA) with a reference sequence of T. livingstonei (KF19298) from Rhinolophus landeri in Mozambique and in the ML analysis, the sequence groups basal to the T. livingstonei clade, though with low support (Fig. 3). The phylogenetic analysis of the second infected E. helvum (sample number KJ2) recovered the trypanosome sequence as basal to a clade that comprises Trypanosoma sp. sequences from African bats (bootstrap value of 96) that together group as sister clade to the clade that comprises T. cf. livingstonei and T. livingstonei (bootstrap value of 96) and might therefore represent a different species (Fig. 3). Unfortunately, no gGAPDH sequences for further characterization could be amplified for the 2 Trypanosoma infections of E. helvum and no parasite blood stages were detected in the blood smears.
Discussion
This study adds information to the scarce knowledge about haemosporidian parasite infections in Nigerian bats. Hepatocystis parasites were detected in the straw-coloured fruit bat species E. helvum, which is only the second time that this bat species was recovered as host to Hepatocystis, the only other single evidence has been reported from Gabon (Boundenga et al., Reference Boundenga, Ngoubangoye, Mombo, Tsoubmou, Renaud, Rougeron and Prugnolle2018). The Hepatocystis prevalence in E. helvum in this study (4 infected individuals, prevalence of 6%, 4/68) was quite low, compared to reports of Hepatocystis infections in African epauletted fruit bat species where prevalences can range from 25% up to about 90% (e.g. Schaer et al., Reference Schaer, Perkins, Decher, Leendertz, Fahr, Weber and Matuschewski2013; Lutz et al., Reference Lutz, Patterson, Kerbis Peterhans, Stanley, Webala, Gnoske, Hackett and Stanhope2016; Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017; Boundenga et al., Reference Boundenga, Ngoubangoye, Mombo, Tsoubmou, Renaud, Rougeron and Prugnolle2018; Atama et al., Reference Atama, Manu, Ivande, Rosskopf, Matuschewski and Schaer2019). The phylogenetic analysis recovered the Hepatocystis sequences from E. helvum within the African bat Hepatocystis clade without any apparent geographic or host species specific pattern following previous findings (e.g. Schaer et al., Reference Schaer, Perkins, Ejotre, Vodzak, Matuschewski and Reeder2017). Yet, as the morphological verification of parasite stages in the blood of the vertebrate host (proof that a parasite has undergone asexual reproduction) is lacking for Hepatocystis parasites in E. helvum hosts, the findings must be considered with caution. In some cases, the sensitivity of PCR methods might verify haemosporidian parasite DNA (transmittable sporozoite stages) in wrong hosts, where the parasite life cycle has been aborted (Valkiunas et al., Reference Valkiunas, Iezhova, Loiseau and Sehgal2009). Interestingly, another African fruit bat species, Rousettus aegyptiacus that represents a true but perhaps rare host species of Hepatocystis (1 published record of a single infected individual) has been also documented from Nigeria (Atama et al., Reference Atama, Manu, Ivande, Rosskopf, Matuschewski and Schaer2019). Systematic serial surveys of the 2 specific African fruit bat species R. aegyptiacus and E. helvum (Pteropodidae) along with the commonly infected epauletted bat host species are needed to understand the different prevalences and transmission dynamics of Hepatocystis infections in African bats. Both R. aegyptiacus and E. helvum are common fruit bat species that form large colonies in caves and trees, respectively, and feature an extensive distribution across sub-Saharan Africa (e.g. Cooper-Bohannon, Reference Cooper-Bohannon2016; Korine, Reference Korine2016). With the reported bat host records, Nigeria might be a particular suitable country/area to investigate Hepatocystis infections in African fruit bats.
The trypanosome infections that were detected in the 3 bat species E. helvum, Mops cf. pumilus and Nycteris cf. macrotis belong to 2 or 3 different Trypanosoma species. The trypanosomes of Mops cf. pumilus (Molossidae) are related to T. erneyi, a species which is closely related to T. dionisii and T. cruzi and that has been described from different bat species exclusively of the family Molossidae in Africa (Lima et al., Reference Lima, Silva, Neves, Attias, Takata, Campaner, de Souza, Hamilton and Teixeira2012). The parasites of the study might represent a subspecies of T. erneyi as the phylogenetic analysis recovered their sequences in their own separate clade (Figs 3 and 4). The trypanosome parasites of the host species Nycteris cf. macrotis are closely related to the lineage of T. cf. livingstonei parasites that has been reported from e.g. Nycteris thebaica bats from South Africa (Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020). T. cf. livingstonei infections appear to be common in different African insectivorous bat species and future investigations will determine whether the divergent lineage T. cf. livingstonei might represent a distinct species or a subspecies of T. livingstonei (Lima et al., Reference Lima, Espinosa-Alvarez, Hamilton, Neves, Takata, Campaner, Attias, de Souza, Camargo and Teixeira2013; Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020). Lacking the gGAPDH sequence for the trypanosome infections in the 2 individuals of E. helvum in this study did not allow an unambiguous assignment to 1 trypanosome species. One sample showed highest 18S rRNA nucleotide identity with T. livingstonei, whereas the other sample was recovered as basal to a clade that comprises Trypanosoma sp. sequences from different African bats, that are closely related to T. cf. livingstonei and T. livingstonei parasites (Fig. 3). The findings of this study confirm the notion that trypanosomes of African bats are phylogenetically diverse and might harbour a variety of yet undescribed species (Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020). Understanding the diversity and the phylogenetic relationships of bat trypanosomes are necessary to improve our understanding of the whole group of parasites that comprise species that are a threat to humans (Hamilton et al., Reference Hamilton, Adams, Njiokou, Gibson, Cuny and Herder2009; Lima et al., Reference Lima, Silva, Neves, Attias, Takata, Campaner, de Souza, Hamilton and Teixeira2012). Many trypanosome lineages of the T. cruzi clade might have evolved in African bat species (e.g. Lima et al., Reference Lima, Espinosa-Alvarez, Hamilton, Neves, Takata, Campaner, Attias, de Souza, Camargo and Teixeira2013; Clement et al., Reference Clement, Dietrich, Markotter, Fasel, Monadjem, Lopez-Baucells, Scaravelli, Theou, Pigeault, Ruedi and Christe2020) and therefore, a more targeted systematic sampling and a subsequent molecular characterization of trypanosome species from African bats is of particular importance.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182022000890.
Data availability
All Hepatocystis and trypanosome sequences of the study are available at GenBank (NCBI) with the accession numbers ON326584 – ON326587, ON332819 – ON332820, ON494560 – ON494563, ON571545 – ON571548.
Acknowledgements
We thank Kamani EV, Bishir Jibrin, Nyor Delahan and Adanu O. Ngbede for assistance during field sampling. We thank Lara Palm for helping with the molecular work in the laboratory.
Author contributions
J. K. conceived and designed the study. J. K., A. Y. J., S. A. and O. D. A. carried out field work and bat sampling. J. S., J. K., O. W., I. E. performed molecular work and phylogenetic analysis. All authors conducted data gathering and wrote the article.
Financial support
J. S. is funded by an individual research grant from the German Research Foundation (DFG; project number 437846632). I. E. is supported by a PhD scholarship of the German Academic Exchange Service (DAAD).
Conflict of interest
The authors declare there are no conflicts of interest.
Ethical standards
This study was approved by the Institutional Animal Use and Care Committee (IAUCC), National Veterinary Research Institute (NVRI), Vom, permission numbers AEC/02/59/18 and AEC/03/65/19. All work was performed in accordance with the relevant guidelines and regulations regarding care and use of animals.








 Open access
Open access