



Introduction
Leishmaniases are diseases caused by trypanosomatids of the Leishmania genus, with diverse clinical presentations ranging from self-healing cutaneous ulcers to a visceral form, potentially fatal if untreated (Reithinger et al., Reference Reithinger, Dujardin, Louzir, Pirmez, Alexander and Brooker2007). Approximately 12 million people are presently estimated to be suffering from this protozoal disease in Africa, Asia, America and Europe (Alvar et al., Reference Alvar, Croft, Kaye, Khamesipour, Sundar and Reed2013), and about 20 different species of Leishmania may cause leishmaniasis in humans (Akhoundi et al., Reference Akhoundi, Kuhls, Cannet, Votypka, Marty, Delaunay and Sereno2016). The clinical form of the disease depends on parasite species and on host immune response (McMahon-Pratt and Alexander, Reference McMahon-Pratt and Alexander2004).
The parasite's life cycle involves the extracellular promastigote stage in the intestine of the phlebotomine sand fly vector, and the intracellular amastigote in the vertebrate host (Bates, Reference Bates2007). Promastigotes transmitted by the vector are phagocytosed by different cells but reside and multiply mainly in parasitophorous vacuoles of macrophages (Naderer and McConville, Reference Naderer and McConville2011). Both stages of the parasite are phagocytosed after adhesion to different macrophage receptors, which are crucial in determining the parasite's intracellular fate (Ueno and Wilson, Reference Ueno and Wilson2012). The parasite intake process also involves components of the endoplasmic reticulum (ER) (Gagnon et al., Reference Gagnon, Duclos, Rondeau, Chevet, Cameron, Steele-Mortimer, Paiement, Bergeron and Desjardins2002). Indeed, Leishmania parasitophorous vacuoles interact continuously with ER compartments and can recruit components that are important for the intracellular survival of the parasite (Ndjamen et al., Reference Ndjamen, Kang, Hatsuzawa and Kima2010), and inhibition of this fusion leads to a reduction of the infection (Canton et al., Reference Canton, Ndjamen, Hatsuzawa and Kima2012; Dias-Teixeira et al., Reference Dias-Teixeira, Pereira, Silva, Fasel, Aktas and Lopes2016). In fact, a proteome analysis identified several ER-resident proteins in phagosomes such as calreticulin, calnexin, 78 kDa glucose-regulated protein (GRP78), endoplasmin and protein disulfide (PDI) (Garin et al., Reference Garin, Diez, Kieffer, Dermine, Duclos, Gagnon, Sadoul, Rondeau and Desjardins2001), suggesting that components of the ER may fuse with the macrophage plasma membrane in the process known as ER-mediated phagocytosis (Desjardins, Reference Desjardins2003; Canton et al., Reference Canton, Ndjamen, Hatsuzawa and Kima2012; Dias-Teixeira et al., Reference Dias-Teixeira, Pereira, Silva, Fasel, Aktas and Lopes2016).
The parasite uses multiple strategies to overcome the innate and acquired immune defence mechanisms and survive inside the macrophage (Lambertz et al., Reference Lambertz, Silverman, Nandan, McMaster, Clos, Foster and Reiner2012; Bifeld and Clos, Reference Bifeld and Clos2015). For example, the interaction of Leishmania surface molecules and macrophages leads to an inhibition of reactive oxygen species production and a decrease in the production of pro-inflammatory cytokines (Lambertz et al., Reference Lambertz, Silverman, Nandan, McMaster, Clos, Foster and Reiner2012; Bifeld and Clos, Reference Bifeld and Clos2015). The subversion of the host cell response is most likely dependent on parasite's products that find access to the host cell and interact with macrophage proteins or receptors, disrupting signalling pathways (Podinovskaia and Descoteaux, Reference Podinovskaia and Descoteaux2015). While it is known that Leishmania secretes a diverse repertoire of molecules, few of them have been functionally characterized. Lipophosphoglycan and GP63 are the most studied virulence factors in Leishmania (Isnard et al., Reference Isnard, Shio and Olivier2012; Podinovskaia and Descoteaux, Reference Podinovskaia and Descoteaux2015). Many virulence factors such as GP63, secreted acid phosphatase, heat shock proteins (HSPs 70, 90 and 100) and tryparedoxin peroxidase are secreted by the parasite (Silverman et al., Reference Silverman, Clos, de'Oliveira, Shirvani, Fang, Wang, Foster and Reiner2010; Lambertz et al., Reference Lambertz, Silverman, Nandan, McMaster, Clos, Foster and Reiner2012).
Part of Leishmania virulence factors is stage regulated. In a search for virulence factors, our group identified 2 genes in chromosome 17, named META1 and META2, which were more expressed in metacyclic than procyclic promastigotes (Uliana et al., Reference Uliana, Goyal, Freymuller and Smith1999; Ramos et al., Reference Ramos, Yokoyama-Yasunaka, Guerra-Giraldez, Price, Mortara, Smith and Uliana2011). While studying these genes, we identified an open reading frame (ORF) near the Leishmania (L.) amazonensis META1 gene that encodes a protein containing 6 leucine-rich repeat (LRR), which was named LaLRR17.
LRR is one of the most common protein motifs involved in protein–protein interactions (Kobe and Kajava, Reference Kobe and Kajava2001). The repeat is composed of 20–30 amino acids with an 11-residue core element; several copies in tandem lead to a horseshoe structure that is believed to be at the source of the interactive properties of these proteins (Kobe and Kajava, Reference Kobe and Kajava2001). The LRR family includes thousands of proteins, found from mammals to plants, taking part in interactions of various orders, including between parasites and host cells (Kobe and Kajava, Reference Kobe and Kajava2001; Kedzierski et al., Reference Kedzierski, Montgomery, Curtis and Handman2004b). In fact, LRRs are present in extracellular regions of Toll-like receptors and in intracellular nucleotide-binding and oligomerization domain (NOD) receptors (Girardin et al., Reference Girardin, Sansonetti and Philpott2002; McGuinness et al., Reference McGuinness, Dehal and Pleass2003), which are important for pathogen recognition. LRRs also play important roles in Listeria monocytogenes infection. Internalins A and B are surface proteins necessary for Listeria internalization by several cells, and internalin LRRs and inter repeat regions were shown to be necessary and sufficient for host cell invasion (Lecuit et al., Reference Lecuit, Ohayon, Braun, Mengaud and Cossart1997).
In various Leishmania genomes already characterized there are 70–80 LRR-containing protein sequences. These include genes encoding surface molecules such as parasite surface antigen (PSA), gp46, various proteophosphoglycans (ppg3, ppg4, ppg5) and several putative proteins. Two proteins, proteophosphoglycan (PPG) and PSA-2, are important molecules involved in binding and phagocytosis of Leishmania by the macrophage (Kedzierski et al., Reference Kedzierski, Montgomery, Bullen, Curtis, Gardiner, Jimenez-Ruiz and Handman2004a). PSA-2 was detected in the extracellular region of the promastigote glycocalyx and contains 13 LRRs that interact with macrophage receptors (Kedzierski et al., Reference Kedzierski, Montgomery, Curtis and Handman2004b). A protein identified in Leishmania (L.) donovani similar to the Internalin-A-like (Inl-A) from L. monocytogenes contains an LRR region that interacts with E-cadherin present in the host cell membrane (Mukherjee et al., Reference Mukherjee, Chakraborty and Chakrabarti2016).
Here we report the characterization of the LRR17 gene and its expressed product. We also show that LaLRR17 increases adhesion of the parasite to the macrophage and in vitro infection by L. (L.) amazonensis. This effect is dependent on the binding to GRP78 in the macrophage surface. This is, to our knowledge, the first description of GRP78 involvement in Leishmania infection.
Materials and methods
Leishmania (L.) amazonensis promastigotes
Promastigotes of L. (L.) amazonensis from the MHOM/BR/73/M2269 strain were cultured at 24°C in M199 medium supplemented with 10% fetal calf serum (FCS). Transgenic L. (L.) amazonensis (pXG1 NEO LaLRR17::myc/His) and (pXG1 NEO) lines were cultured under the same conditions but in the presence of 150 μg mL−1 G418. Parasites were subcultured every 7 days to inoculums of 2 × 106 mL−1.
Leishmania transfection and selection of mutants
The LaLRR17 ORF was amplified and cloned in a modified phosphate-buffered saline (PBS) vector engineered to contain the sequence encoding the myc epitope and a tail of 6 histidines (a kind gift from Dr Carmen Fernandez-Becerra, Institute for Global Health, Barcelona, Spain). The LaLRR17 ORF in frame with the myc epitope and histidine tail was then excised and cloned into pXG1 (provided by Dr Steve Beverley, Washington University, St Louis, USA). DNA electroporation in L. (L.) amazonensis was performed as previously described (Kapler et al., Reference Kapler, Coburn and Beverley1990) using the construct pXG1 LaLRR17::myc or with the empty plasmid pXG1. Cells were then plated in medium 199–1% agar with G418 (20 mg mL−1). After 2 weeks, colonies were picked, expanded in liquid media and G418 concentrations were gradually increased to 160 mg mL−1.
Production of LaLRR17 and GRP78 by Escherichia coli
A fragment of 2016 pb containing the coding sequence of LaLRR17 was cloned in the pAE vector (Ramos et al., Reference Ramos, Abreu, Nascimento and Ho2004a) and used to transform E. coli BL21(DE3) pLysS. Bacteria were grown in 200 mL Luria-Bertani (LB) with 100 μg mL−1 ampicillin and expression was induced with 0.5 mm isopropyl-β-D-thiogalactopyranoside (IPTG) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 4 h at 37°C. Bacteria were sedimented by centrifugation and suspended in 20 mL buffer A [100 mm Tris-HCl, pH 8.0, 12 mm sodium chloride (NaCl), 1 mm phenylmethylsulfonyl fluoride (PMSF)]. Cells were disrupted by sonication (Unique Ultrasonic DES500) and lysates were centrifuged. The pellet was suspended in 20 mL buffer B (100 mm Tris-HCl, pH 8.0, 500 mm NaCl, 8 m urea) and incubated at 4°C under rotation for 24 h. The lysate was again centrifuged, the pellet was discarded and 2 mL of the supernatant were added drop by drop to 10 mL buffer A (250 μL min−1). β-Mercaptoethanol was added to 5 mm and the sample was transferred to Ni-NTA column (Thermo Fisher Scientific). After washing the column with 20 mL buffer A, proteins were eluted with 50, 100 and 500 mm imidazole in buffer A and dialysed against PBS. GRP78 was produced using pQE10-GRP78 plasmid gently provided by Linda Hendershot, St. Jude's Children Hospital, USA, following a published protocol (Gaut and Hendershot, Reference Gaut and Hendershot1993). Protein concentration was determined using Bradford (BioRad, Hercules, California, USA), and lipopolysaccharide (LPS) content was calculated using limulus amebocyte lysate method (Pharma & Biotech Lonza, Bella Vista, Sidney, Australia), following the manufacturer's instructions.
Isolation of bone marrow-derived macrophages (BMDMs)
BALB/c mice were euthanized and bone-marrow cells from femurs and tibias were collected in 10 mL R2030 medium (RPMI 1640 with 20% FCS and 30% supernatant from L929 cells). Cells were grown for 4 days, when 10 mL R2030 medium were added. After 3 days, cells were washed with PBS, detached with a cell scraper, counted and plated according to the experiment.
Macrophage infection with Leishmania
Peritoneal macrophages were isolated as previously described (Velasquez et al., Reference Velasquez, Galuppo, Rezende, Brandao, Peron, Uliana, Duarte and Stolf2016). Peritoneal or BMDMs (8 × 105 per well) in RPMI with (BMDM) or without (peritoneal macrophages) 10% FCS were transferred to 24-well plates covered with 13 mm circular coverslips. The choice of macrophage type depended on the number of cells required for each experiment, due to CEUA requests for reduction in the number of animals. After incubation at 37°C and 5% carbon dioxide (CO2) for 2 h (peritoneal cells) or overnight (BMDM), the medium was changed to RPMI with 10% FCS. When indicated, macrophages were pre-incubated for 1 h with anti-GRP78 1:150 (#3183, Cell Signaling). Infection was caused with L. (L.) amazonensis promastigotes at the beginning of the stationary phase (day 4) using a multiplicity of infection (MOI) of 5:1 in the presence or not of LaLRR17 (12.5, 25, 50 or 100 ng mL−1), LPS (2 ng mL−1, concentration found in 100 ng mL−1 LaLRR17) or GRP78 (4.6 μg mL−1). After removing non-internalized parasites by washing, cells were further incubated for 20 h. Cells were fixed with methanol, stained with InstantProv (NewProv, Pinhais, Paraná, Brazil) and mounted with Entellan (Merck, Darmstadt, Germany). One hundred macrophages were analysed per glass slide to determine the proportion of infected cells (IM) and amastigotes/infected macrophage, 3 coverslips were prepared for each condition, and 3 biological replicates (independent macrophages and Leishmania cultures) were performed for each experiment.
Phagocytosis assay
For analysis of phagocytosis, 4 × 105 BMDMs were transferred to 24-well plates covered with 13 mm circular coverslips. On the following day, cells were incubated with wild-type L. (L.) amazonensis in the presence of LaLRR17 or LPS using an MOI of 10. The plate was maintained on ice for 2 h and then incubated for 5, 30 or 60 min at 33°C and 5% CO2. Cells were then fixed with 4% paraformaldehyde for 10 min, washed with PBS and incubated with 50 mm of NH4Cl for 10 min, washed and blocked with 5% bovine serum albumin (BSA) in PBS for 2 h. Glass slides were incubated overnight with anti-Leishmania serum diluted 1:500 in PBS, washed with PBS and incubated for 1 h with a mix containing anti-mouse immunoglobulin G (IgG) (H + L) 488 Alexa fluor 1:1000, Phalloidin 568 Alexa fluor 1:100 and diamidino-2-phenylindole (DAPI) 1:1000. After 5 washing cycles, cells were mounted in ProLong (Thermo Fisher Scientific). For calculation of the phagocytosis index, 500 macrophages were analysed, and promastigotes were classified and quantified as attached (labelled in green and blue) or internalized (labelled only with blue by DAPI).
Anti-Leishmania serum was obtained by infecting BALB/c mice with L. (L.) amazonensis. Blood was collected around 10 weeks after infection; serum was obtained after centrifugation, and was stored in frozen aliquots.
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot
To obtain total protein extracts, parasites were washed twice with PBS, suspended at 109 cells/300 μL in PBS + Proteoblock 1× (Fermentas, Waltham, Massachusetts, USA) and lysed by 8 freeze/thaw cycles (liquid nitrogen and 42°C). Soluble proteins were obtained after centrifugation at 12 000 g for 3 min and quantified by Bradford (BioRad).
SDS-PAGE (10% acrylamide:bisacrylamide separating gels) and western blots were performed as we previously described (Teixeira et al., Reference Teixeira, Velasquez, Lepique, de Rezende, Bonatto, Barcinski, Cunha-Neto and Stolf2015), using 10 μg of promastigote proteins and 1 μg of recombinant LaLRR17 or GRP78. Blocking was done overnight with PBS with 5% milk and 0.1% Tween 20, followed by incubation with primary antibody for 2 h and with secondary antibody for 1 h, both in PBS with 2.5% milk and 0.1% Tween 20. Three washing cycles (for 10 min) with 0.05% Tween 20 in PBS were performed after the incubations with primary and secondary antibodies. The following antibodies were used: anti-myc 1:3000 (Thermo Fisher Scientific) and anti-mouse horseradish peroxidase (HRP) 1:3000 (KPL Seracare, Milford, Massachusetts, USA) (for 1 h), anti-GRP78 ab32618 (Abcam, Cambridge, United Kingdom) 1:1000 and anti-rabbit HRP (Cell Signaling) 1:1000, anti-His 1:2500 (Thermo Fisher Scientific) and anti-mouse HRP 1:1000 (Sigma-Aldrich).
Affinity chromatography
Affinity chromatography was performed using CNBr-activated Sepharose™ 4B (GE Healthcare, Chicago, Illinois, EUA), following the manufacturer's protocols. Briefly, 5 μg of recombinant LaLRR17 or BSA (control) were immobilized by incubation with the resin in 0.1 m NaHCO3 (pH 8.3), 0.5 m NaCl for 18 h at 4°C, and in 0.1 m Tris-HCl, pH 8.0 for 2 h at room temperature (RT). After sequential washing cycles with 0.1 m acetic acid, pH 4.0, with 0.5 m NaCl and with 0.1 m Tris-HCl (pH 8)–0.5 m NaCl (5 washing cycles with each solution), resins were incubated for 18 h at 4° C with 0.5 mg macrophage extract. This extract was prepared by lysis of cells in PBS with 1% NP40, centrifugation and recovery of the supernatant. After washing resins with PBS, bound proteins were sequentially eluted with 10% SDS in PBS, 8 m urea in PBS and 1 m NaCl.
Mass spectrometry identification of LaLRR17-binding proteins
Eluted proteins from affinity chromatography were separated by SDS-PAGE and visualized by Coomassie blue G250 staining. Gel bands were extracted and transferred to microtubes. Trypsin digestion directly from the gel fragments (in gel digestion) and further processing for analysis by liquid chromatography coupled to sequential mass spectrometry (LC-MS/MS) in the nanoLC Easy-LTQ Orbitrap Velos-ETD equipment were performed in Centro de Facilidades para a Pesquisa, ICB-USP (CEFAP), according to CEFAP protocols. The proteins in the gel bands were identified by correlating the peptide masses in the MS/MS spectra with the protein sequences in the UniProt database.
Immunofluorescence for GRP78
BMDMs were plated at 3 × 105 cells per well over coverslips in 48-well plates for 24 h. Cells were then fixed by incubation with 4% paraformaldehyde (PFA) for 10 min, washed twice with PBS and incubated with 50 mm (NH4)Cl for 10 min. After 3 washing cycles with PBS and blocking with PBS containing 5% BSA for 2 h, coverslips were incubated for 2 h with anti-GRP78 ab32618 (Abcam) diluted 1:50 in PBS with 5% FCS.
Alternatively, BMDMs were washed twice with PBS and incubated for 30 min at RT with Fc Block (BD Biosciences, Franklin Lakes, New Jersey, USA) 1:100 in PBS with 5% FCS. Blocking solution was removed and cells were incubated with anti-GRP78 antibody for 30 min at RT. Wells were washed twice with PBS and cells were fixed by incubation with 4% PFA for 10 min. After 2 washing cycles with PBS, cells were incubated with 50 mm (NH4)Cl for 10 min.
For both protocols, coverslips were then washed with PBS and incubated for 30 min with anti-rabbit IgG (H + L) Alexa Fluor 488 (1:1000) and DAPI (1:1000) in PBS with 5% FCS. After washing cycles, coverslips were mounted in ProLong (Thermo Fisher Scientific). Images were acquired using a DMI6000B/AF6000 (Leica, Wetzlar, Germany) fluorescence microscope coupled to a digital camera system (DFC 365 FX) (Leica).
Binding assay with LaLRR17 and GRP78
For analysis of binding of LaLRR17 to GRP78, 1 μg LaLRR17 or 1 μg BSA in 100 μL PBS were incubated in 96-well plates (Costar EIA/RIA high binding) for 18 h at 4°C. Wells were blocked with 150 μL PBS with 5% BSA for 2 h, washed 6 times with PBS and incubated with 4.6 μg mL−1 GRP78 for 18 h at 4°C. Wells were incubated with anti-GRP78 1:500 (Abcam ab32618) for 2 h, washed 6 times with PBS and incubated with anti-rabbit antibody HRP (Cell Signaling) diluted 1:1000 for 1 h. After 6 washing cycles with PBS, wells were incubated with TMB microwell peroxidase substrate (KPL Inc.) following the manufacturer's guidelines and read at 450 nm.
Molecular docking
Molecular docking was used to test the binding affinity of LaLRR17 using a structure created by AlphaFold software, against the structures of GRP78 (AlphaFoldDB-P20029), GRP75 (AlphaFoldDB-P38647) and HSP-71 (AlphaFoldDB-AF-P63017-F1). The solvated docking software HADDOCK was used to model LaLRR1 regions against the solved structure of GRP78, GRP75 and HSP-71; the easy interface was used as there are no constraints set. PRODIGY software was used to predict the binding affinity for each region of the LaLRR17 to GRP78.
Statistical analysis
GraphPad software (San Diego, CA, USA) was used to perform all analyses. We employed one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test (for 3 or more samples), or t-test (for comparison of 2 conditions).
Results
Identification of L. (L.) amazonensis LaLRR17
The characterization and sequencing of a segment of chromosome 17 of L. (L.) amazonensis containing the META1 and META2 genes (Uliana et al., Reference Uliana, Goyal, Freymuller and Smith1999; Ramos et al., Reference Ramos, Yokoyama-Yasunaka, Guerra-Giraldez, Price, Mortara, Smith and Uliana2011) led to the identification of a neighbouring ORF encoding a protein containing 6 region LRRs that was named LaLRR17 (Fig. 1). The 5′ and 3′ untranslated region (UTR) regions were mapped through RACE, allowing determination of the complete ORF (original sequence deposited in GenBank under EU906911) (Ramos et al., Reference Ramos, Franco, Smith and Uliana2004b). The predicted polypeptide sequence contains no signal peptide or glycosylphosphatidylinositol (GPI)-anchoring sites. Analysis of the LaLRR17 peptide sequence (https://www.ebi.ac.uk/interpro/) (Mitchell et al., Reference Mitchell, Attwood, Babbitt, Blum, Bork, Bridge, Brown, Chang, El-Gebali, Fraser, Gough, Haft, Huang, Letunic, Lopez, Luciani, Madeira, Marchler-Bauer, Mi, Natale, Necci, Nuka, Orengo, Pandurangan, Paysan-Lafosse, Pesseat, Potter, Qureshi, Rawlings, Redaschi, Richardson, Rivoire, Salazar, Sangrador-Vegas, Sigrist, Sillitoe, Sutton, Thanki, Thomas, Tosatto, Yong and Finn2019) identified a region with homology with the superfamily of LRR domains (IPR032675). The structure predicted by the server AlphaFold indicates that the LRR motifs (with 28–30 amino acids in length) are mostly located in the central region of the protein (Fig. 1C).

Figure 1. LRR17 protein sequence in Leishmania. (A) Multiple alignment of the Leishmania (L.) amazonensis LRR17 translated sequence (GenBank EU906911.1) with sequences from several Leishmania species performed using MUSCLE. Identical amino acids in all sequences are shaded in black; amino acids conserved in 50% of the sequences are shaded in grey. Black lines indicate the LRR units. (B) Phylogenetic tree constructed with protein sequences using the ML method. Bar scale indicates 5% amino acid divergence. (C) Model of LaLRR17 structure predicted by the server AlphaFold. (D) Sequence coverage plot showing the number of homologues identified across the representative sequence and coloured by the sequence identity of the homologues. (E) A plot of the pLDDT score per position for each of the 5 AlphaFold models predicted.
Similar ORFs were identified in several other Leishmania species by searching published genome data (Fig. 1A). UniProt Knowledge base (UniProtKB) identified homologues in Leishmania major, Leishmania infantum and Leishmania braziliensis, identified as putative hypothetical proteins, sharing 92, 92 and 78% identity, respectively, with LaLRR17. Interestingly, in a phylogenetic analysis using the maximum-likelihood (ML) method, LRR17 sequences from Viannia subgenus cluster separately from Leishmania and Sauroleishmania subgenera branch (Fig. 1B).
Characterization of L. (L.) amazonensis line overexpressing LaLRR17
To gather functional information on LaLRR17 protein, we produced a transgenic strain overexpressing the protein, named (pXG1 NEO LaLRR17::myc/His). Western blot confirmed the expression of exogenous myc-tagged LaLRR17 in the transgenic overexpressor (pXG1 NEO LaLRR17::myc/His), but not in the transgenic control (pXG1 NEO) promastigotes (Fig. 2A). These transgenic parasites were then used for infection of peritoneal macrophages from BALB/c mice for 24 h, to analyse if LRR17 affects parasite entry and/or survival in the initial phase, before parasite multiplication. Significantly higher percentages of infected cells (P ⩽ 0.05) were observed for LaLRR17 overexpressing parasites, and a non-significant increase in the number of parasites per macrophage was also observed for this line (Fig. 2B). Independent clones selected after transfection exhibited the same phenotype (data not shown). These results suggest that LaLRR17 contributed to macrophage infection.

Figure 2. Infection of macrophages with transgenic L. (L.) amazonensis overexpressing LaLRR17-myc and control lines. (A) Immunoblotting of total protein extracts from L. (L.) amazonensis log phase promastigotes probed with a monoclonal anti-myc antibody. Equal numbers of parasites (2 × 107) were loaded per track: (1) wild-type line; (2) L. (L.) amazonensis (pXG1 NEO) and (3) L. (L.) amazonensis (pXG1 NEO LaLRR17). The bottom panels show the same blot incubated with an anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. (B) Peritoneal macrophages from BALB/c mice were infected with L. (L) amazonensis (pXG1 NEO LaLRR17::myc/His) and (pXG1 NEO) for 24 h at an MOI of 5. Percentage of infected cells (top graph) and numbers of amastigotes per infected macrophage (bottom graph); t-test, *P ⩽ 0.05. A representative experiment of 3 with similar profiles is shown.
Soluble LaLRR17 increases phagocytosis of L. (L.) amazonensis
As previously mentioned, LaLRR17 has no domains for membrane anchoring or secretion. However, many virulence factors of Leishmania are present in extracellular space, after secretion by non-classical pathways. To test whether the soluble protein would affect infection, we produced the recombinant His-tagged protein in the BL21(DE3) pLysS system using the pAE plasmid LaLRR17 (Ramos et al., Reference Ramos, Abreu, Nascimento and Ho2004a). The recombinant LaLRR17 protein, which was not soluble after induction, was obtained from 8 m urea pellet and refolded by drop dilution (Fig. 3A, SDS-PAGE and western blot). Soluble LaLRR17 was then used in macrophage infections for 24 h with wild-type L. (L.) amazonensis.
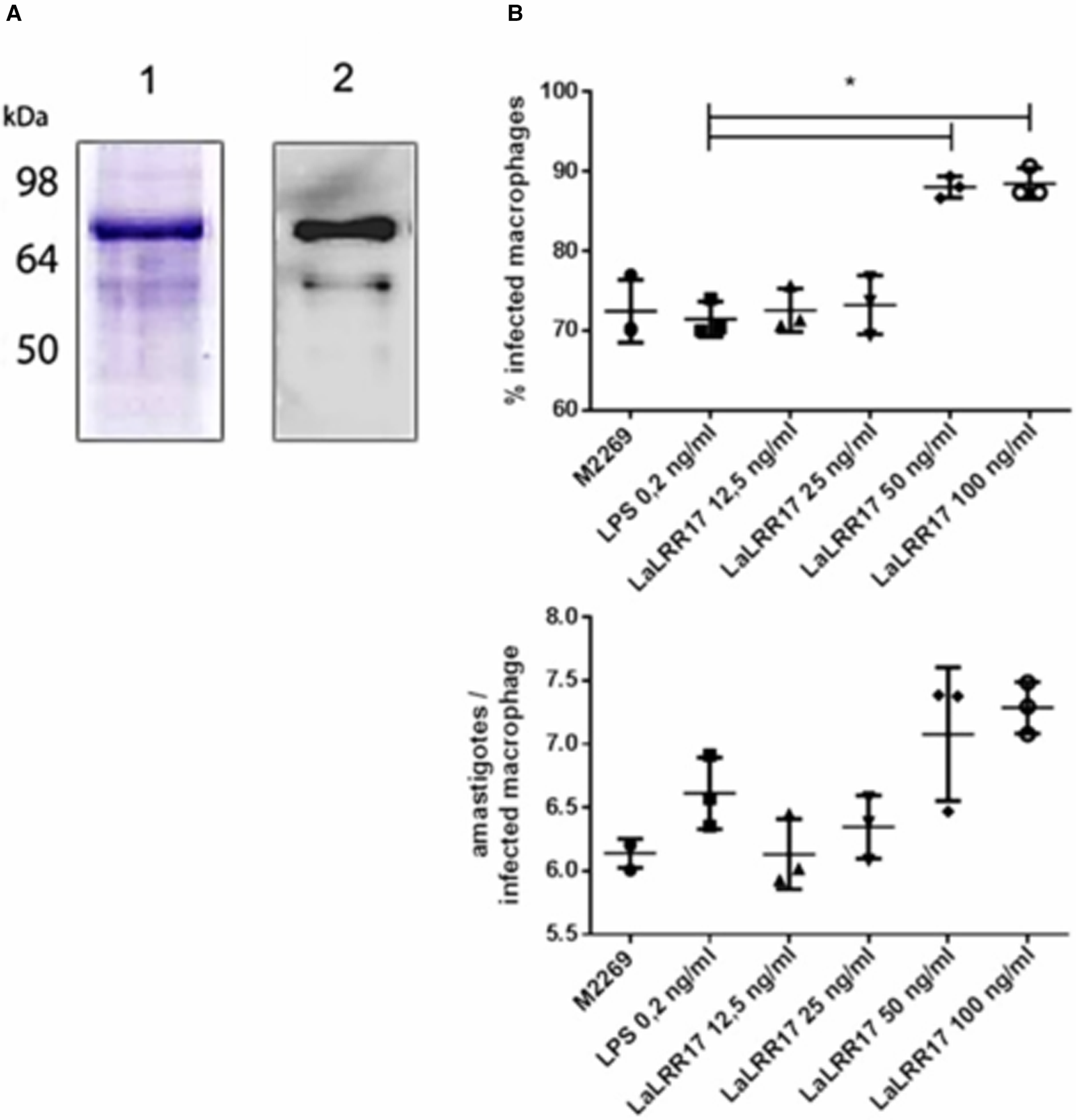
Figure 3. Production of recombinant LaLRR17 and its effect on macrophage infection by L. (L.) amazonensis. (A) SDS-PAGE (1) and western blot with anti-His 1:2500 (2) showing recombinant LaLRR17 purified from bacteria. (B) Peritoneal macrophages from BALB/c mice were infected with wild-type L. (L) amazonensis for 24 h at an MOI of 5 in the presence of LPS or 12.5, 25, 50 or 100 ng mL−1 of LaLRR17. Percentage of infected cells (top graph) and numbers of amastigotes per infected macrophage (bottom graph). ANOVA followed by post-test of Tukey's, *P ⩽ 0.05. Means and standard deviations of 3 independent experiments.
Since LaLRR17 produced in bacterial systems is expected to contain small traces of LPS, which may activate macrophages and reduce infection (Meng and Lowell, Reference Meng and Lowell1997), we added LPS at the highest concentration found in our LaLRR17 preparations (2 EU mL−1, corresponding to 0.2–0.4 ng endotoxin mL−1) as the control condition. Different concentrations of LaLRR17 were assayed in macrophage infections for 24 h (4 h of contact with the parasite and 20 h of incubation at 34°C), and a significant difference (P ⩽ 0.05) in the proportion of infected cells was observed in the presence of 50 and 100 ng mL−1 of LaLRR17, but not in lower concentrations (Fig. 3B). A non-significant increase was noted in the number of parasites per infected macrophage in the presence of LaLRR17 (Fig. 3B). LPS did not affect any of the parameters analysed.
The results obtained with the recombinant protein were similar to those observed with the parasites overexpressing LaLRR17, indicating that the recombinant protein is active. Besides, these results suggest that if LaLRR17 is present in extracellular space, shed by parasites or made available after neighbouring parasites death, it probably exerts an effect on promastigote infection.
We then analysed whether the increase observed in infection experiments (24 h) was due to an action of LRR17 in the initial stages of phagocytosis. To test this hypothesis, we performed a phagocytosis assay, in which parasites contact macrophages for very short periods. Promastigotes were incubated with macrophages (BMDM) in the presence or absence of LaLRR17 for 5, 30 or 60 min. Promastigotes adhered to the host cell, but not internalized, were visualized by fluorescence microscopy on non-permeabilized cells stained in green and blue (anti-Leishmania + secondary antibody Alexa 488 and DAPI, respectively), while internalized parasites were labelled only in blue (DAPI) (Fig. 4D). Phagocytosis of Leishmania was assessed quantitatively in terms of the number of promastigotes internalized per 100 macrophages, and we determined the number of parasites bound to these cells. Figure 4A–C shows 1 graph for each time point, and each graph shows bound and internalized parasites. Interestingly, LaLRR17 led to an increase in bound promastigotes after 5 min of macrophage–parasite contact, and in internalized promastigotes after 30 and 60 min contact, indicating that the protein affects the binding and further phagocytosis of Leishmania, probably by interaction with a ligand on the macrophage surface.

Figure 4. Effect of LaLRR17 on binding and on phagocytosis of L. (L.) amazonensis by BMDM. Each graph shows promastigotes bound (circles in red, left) and phagocytosed (triangles, right) by 100 BMDM (MOI of 10) in the continuous presence of 100 ng mL−1 LaLRR17 after (A) 5 min, (B) 30 min and (C) 60 min of contact between parasite and macrophage at 37°C. Experiment representative of 3 (A) or 2 (B, C) independent experiments; t-test, P ⩽ 0.05.
Identification of GRP78 as a ligand for LaLRR17 by affinity chromatography followed by mass spectrometry
In order to identify macrophage proteins that bind to LaLRR17 and can mediate Leishmania phagocytosis, we employed affinity chromatography with immobilized LaLRR17. The recombinant protein was coupled to CNBr-activated Sepharose™ 4B beads and incubated with soluble lysates of BMDM from BALB/c mice. BSA was coupled to the resin as a control and submitted to the same protocol. The eluates of the 2 columns were analysed on SDS-PAGE, and the differential band between 64 and 98 kDa retained in LaLRR17 but not on BSA column (Fig. 5A) was collected and submitted to mass spectrometry analysis (Supplementary Table 1). A region of the same size on the BSA lane was also collected and analysed as negative control. Peptide–spectrum matches (PSMs) were used to derive the relative abundance of each protein in LaLRR17 and BSA eluates, as previously described (Shteynberg et al., Reference Shteynberg, Nesvizhskii, Moritz and Deutsch2013). High PSM ratios for LaLRR17/BSA indicate potential affinity for LaLRR17. Among the identified proteins, 11 had ratios above 1, 5 of which were equal or above 2 and 4 of which were above 3 (Fig. 5B). Supplementary Table 1 lists the PSMs of all proteins identified in the affinity chromatography.

Figure 5. Identification of macrophage proteins that bind to LaLRR17. (A) SDS-PAGE 10% showing proteins eluted (with SDS or urea) from BSA and LaLRR17 columns. The arrow points to the differential band recovered from the gel and analysed in (B). (B) Proteins with PSM ratios above 1 for LaLRR17/BSA.
The GRP78, also known as binding immunoglobulin protein or heat shock 70 kDa protein 5, was the protein with the highest PSMs for LaLRR17 relative to BSA (Fig. 5B), suggesting its ability to bind to LaLRR17. However, other proteins similar to GRP78 also bound to LaLRR17, such as GRP75 and HSP cognate 71 kDa, which have 46.8 and 60.8% amino acid identity with GRP78, respectively (Supplementary Fig. 1).
Prediction of binding between LaLRR17 and GRP78, GRP75 and HSP-71
The docking of LaLRR17 and GRP78 proteins, whose structure was determined using AlphaFold, was performed using HADDOCK software (Ibrahim et al., Reference Ibrahim, Abdelmalek, Elshahat and Elfiky2020) setting no restrictions and therefore using the easy interface (Elfiky and Ibrahim, Reference Elfiky and Ibrahim2021). To define the surface residues of the LaLRR17 protein as active and passive, we employed the automatic definition mode. HADDOCK software is widely employed for protein docking, to investigate the molecular interactions between a target protein and its partner molecules such as ligands, substrates or other proteins. It is based on constraints and experimental information. It employs an integrative approach that combines structural and experimental data, such as nuclear magnetic resonance and mass spectrometry, to enhance the accuracy of the results.
For GRP78, we used the active residues characterized in previous studies (Almagro Armenteros et al., Reference Almagro Armenteros, Sonderby, Sonderby, Nielsen and Winther2017) and adapted to the sequence of mouse GRP78 as follows: I427, T429, V430, V433, T435, F452, S453, V462 and I464 (Fig. 6A). The best complex created by HADDOCK software achieved the score of −76.7 ± 9.1, with the predicted binding site of the LaLRR17 protein to GRP78 in good agreement with studies that have identified the binding domain of pathogens to membrane GRP78, such as proteins from Zika virus and severe acute respiratory syndrome coronavirus 2 (Ibrahim et al., Reference Ibrahim, Abdelmalek, Elshahat and Elfiky2020; Elfiky and Ibrahim, Reference Elfiky and Ibrahim2021).

Figure 6. Prediction of binding between LaLRR17 and GRP78. Images show the binding for each of the docking assays (the best-formed complexes from each docking experiment). Orange drawing represents GRP78, indicating SBD and nucleotide-binding domain (NBD) regions, and the green drawing represents LaLRR17, indicating the LRR region.
We have also analysed the docking of LaLRR17 and GRP75 or HSP-71, the 2 other chaperones similar to GRP78 already mentioned. The results of the predictions are shown in Supplementary Fig. 2. All chaperones bind, on average, to the first 25 amino acids of the N-terminal region of LaLRR17. The interaction with GRP78 occurs in the substrate binding domain (SBD) region, which usually binds to peptides/proteins.
To confirm that GRP78 interacts with LaLRR17, we produced a recombinant GRP78 and performed a binding assay using both proteins. The results shown in Fig. 7 indicate that GRP78 exhibited significantly higher binding to LaLRR17 than to BSA, indicating specific binding between the 2 proteins. Unfortunately, biophysical approaches could not be performed to validate these observations due to the low concentrations of LaLRR17 always obtained after induction and refolding.

Figure 7. Binding of recombinant GRP78 to LaLRR17. Binding of GRP78 to 1 μg LaLRR17 or BSA estimated by enzyme-linked immunoassay using anti-GRP78 (ab32618, Abcam) diluted 1:500. Results representative of 3 independent experiments, statistical analysis used: ANOVA and post-test of Tukey's, P ⩽ 0.05.
Since the literature is scarce on data about the expression of GRP78 by macrophages, we examined whether it was present on the macrophage surface. Positive staining with anti-GRP78 of non-permeabilized cells suggested the presence of GRP78 on the cell surface (Fig. 8A).

Figure 8. Analysis of GRP78 expression and effects on macrophage infection. (A) Immunofluorescence showing GRP78 labelling in live BMD macrophages (top image, anti-GRP78 and anti-rabbit 488 Alexa fluor) and no labelling with secondary antibody only (bottom image, anti-rabbit 488 Alexa fluor). (B) Macrophage infection in the presence or absence of LaLRR17 and anti-GRP78. Peritoneal BALB/c macrophages pre-incubated or not with anti-GRP78 were infected at an MOI of 5 with L. (L.) amazonensis in the presence or absence of 100 ng mL−1 of LaLRR17, for 24 h. Percentage of infected cells (top graph) and numbers of amastigotes per infected macrophage (bottom graph). ANOVA followed by post-test of Tukey's, P ⩽ 0.05. Results of 1 experiment representative of 3.
We then evaluated its participation in the infection enhancement promoted by LaLRR17. Blocking GRP78 with a specific antibody did not affect infection percentage in macrophages in the absence of soluble LRR17 (Fig. 8B). However, the increased infection observed in the presence of soluble LRR17 was reversed by the incubation with anti-GRP (Fig. 8B), reinforcing that the GRP78 protein on the macrophage surface mediated the effect of LaLRR17.
Discussion
LRR motifs present in several proteins of mammals and plants are involved in protein–protein interactions (Kobe and Kajava, Reference Kobe and Kajava2001). In this paper, we described the characterization of a Leishmania-conserved gene encoding LaLRR17, a leucine-rich protein, found in the vicinity of the META genes in chromosome 17. The study of protein abundance and cellular localization proved challenging since, despite several attempts to immunize mice or rabbits with recombinant proteins or with synthetic peptides, we could not generate antibodies that specifically detect LaLRR17 (data not shown).
Similar to what has been suggested for PPG, PSA-2 and Inl-A-like protein, we hypothesized that LaLRR17 may interact with macrophages. The LaLRR17 protein does not have any membrane-anchoring domain and is predicted to be soluble and localized in the cytosol (DeepLoc-1.0 value of 0.57, corresponding to soluble cytoplasm) (Almagro Armenteros et al., Reference Almagro Armenteros, Sonderby, Sonderby, Nielsen and Winther2017). It does not exhibit an N-terminal secretion signal peptide and therefore would not be secreted by the classical eukaryotic secretion pathway. Analyses of Leishmania secretomes show that only a few proteins in the extracellular environment do possess classical secretion signals (Silverman et al., Reference Silverman, Chan, Robinson, Dwyer, Nandan, Foster and Reiner2008; Pissarra et al., Reference Pissarra, Pagniez, Petitdidier, Seveno, Vigy, Bras-Goncalves, Lemesre and Holzmuller2022), with most proteins being secreted through extracellular vesicles (EVs). EVs released by Leishmania contribute to the immunomodulation of the host and consequently to the establishment of infection (Corrales et al., Reference Corrales, Sereno and Mathieu-Daude2010; Barbosa et al., Reference Barbosa, Dupin, Toledo, Reis, Ribeiro, Cronemberger-Andrade, Rugani, De Lorenzo, Novaes, Soares, Torrecilhas and Xander2018; Garg et al., Reference Garg, Ali, Singh, Gupta, Ganguly, Sahasrabuddhe and Das2019; Rodriguez-Vega et al., Reference Rodriguez-Vega, Losada-Barragan, Berbert, Mesquita-Rodrigues, Bombaca, Menna-Barreto, Aquino, Carvalho, Padron and de Jesus2021). In addition to secretion by EVs, LaLRR17 might be shed by a portion of promastigotes present in the initial inoculum that initiate programmed cell death in the mammalian host (El-Hani et al., Reference El-Hani, Borges, Wanderley and Barcinski2012), releasing cytosolic proteins to the extracellular environment (Santarem et al., Reference Santarem, Silvestre, Tavares, Silva, Cabral, Maciel and Cordeiro-da-Silva2007).
We have shown that parasites overexpressing LaLRR17 infected a higher percentage of macrophages than the wild-type strain. Similarly, the presence of the soluble recombinant LaLRR17 protein increased the proportion of infected cells. Moreover, soluble LaLRR17 led to an increase in the number of promastigotes bound to macrophages after incubations for only 5 min, and in the number of internalized parasites after 30 min or longer. These data indicated that LaLRR17 and its ligand in the macrophage enhance parasite binding and phagocytosis. The best-studied receptors related to Leishmania phagocytosis are complement receptors 3 (CR3) and 1 (CR1), mannose receptors, Fc gamma receptor (mainly FcyRII-B2) and fibronectin receptors (Guy and Belosevic, Reference Guy and Belosevic1993; Podinovskaia and Descoteaux, Reference Podinovskaia and Descoteaux2015; Podinovskaia and Russell, Reference Podinovskaia and Russell2015). As previously mentioned, the ER also contributes to phagocytosis, as attested by the presence of proteins such as calnexin and calreticulin on the cell surface (Muller-Taubenberger et al., Reference Muller-Taubenberger, Lupas, Li, Ecke, Simmeth and Gerisch2001). These 2 proteins appear to participate in Leishmania phagocytosis, as well as parasite GP63, which contains a Glc1Man6 GlcNac2 structure recognized by calnexin and calreticulin (Olafson et al., Reference Olafson, Thomas, Ferguson, Dwek, Chaudhuri, Chang and Rademacher1990; Garin et al., Reference Garin, Diez, Kieffer, Dermine, Duclos, Gagnon, Sadoul, Rondeau and Desjardins2001). None of these macrophage proteins, however, has been shown to interact with Leishmania proteins containing LRRs.
Affinity chromatography followed by mass spectrometry identified chaperones from the HSP-70 family including GRP78, GRP75 and HSP cognate 71 kDa as ligands for LaLRR17, with GRP78 being the most abundant. GRP78 is the most well-studied protein in the GRP family (Wang et al., Reference Wang, Wey, Zhang, Ye and Lee2009; Behnke et al., Reference Behnke, Feige and Hendershot2015; Pobre et al., Reference Pobre, Poet and Hendershot2019). Due to the presence of the KDEL (Lys–Asp–Glu–Leu) peptide for retention in the ER, most GRP78 is localized within this organelle, but in some circumstances, it can be redistributed to the cytosol, nucleus, mitochondria and plasma membrane, and can even be secreted (Suzuki et al., Reference Suzuki, Bonifacino, Lin, Davis and Klausner1991). A determinant factor for change in GRP78 cellular localization may be ER stress, when proteins that reside inside the ER can translocate to the cytosol, cell membrane or extracellular space (Pelham, Reference Pelham1990), contributing to various cellular pathologies (Arap et al., Reference Arap, Lahdenranta, Mintz, Hajitou, Sarkis, Arap and Pasqualini2004; Tsai et al., Reference Tsai, Zhang, Tseng, Stanciauskas, Pinaud and Lee2015; Wiersma et al., Reference Wiersma, Michalak, Abdullah, Bremer and Eggleton2015; Ha et al., Reference Ha, Van Krieken, Carlos and Lee2020). On the cell surface GRP78 functions as a signalling molecule and may play an important role in regulating pro-proliferative/anti-apoptotic, pro-migratory, signalling pathways and pathogen entry (Gonzalez-Gronow et al., Reference Gonzalez-Gronow, Selim, Papalas and Pizzo2009; Ibrahim et al., Reference Ibrahim, Abdelmalek and Elfiky2019; Lenin et al., Reference Lenin, Nagy, Jha and Gangaraju2019).
The mechanism by which L. (L.) amazonensis may induce ER stress is not well understood, but mild ER stress response induced by Leishmania infection may represent a common pathogenic mechanism among different Leishmania species and may be part of the strategies to survive in the host cell (Galluzzi et al., Reference Galluzzi, Diotallevi, De Santi, Ceccarelli, Vitale, Brandi and Magnani2016).
Some reports have described GRP78 on the surface of mouse macrophages (Misra et al., Reference Misra, Deedwania and Pizzo2005; Lu et al., Reference Lu, Lai, Yu, Huang, Hsieh and Yu2010) and its participation in signalling pathways triggered by α2-macroglobulin binding, leading to macrophage activation and chemotaxis (Misra and Pizzo, Reference Misra and Pizzo2008). We confirmed here that murine-resident peritoneal macrophages express GRP78 on their surface and that blocking this protein using a specific antibody prevented the increase in infection induced by LaLRR17. Interestingly, blocking GRP78 did not reduce infection in the absence of exogenous LaLRR17. We believe LRR17 expression is low in wild-type parasites and its presence is even lower under the conditions used in infection experiments: day 4 culture promastigotes, centrifuged and resuspended in medium, added to macrophages. Under such conditions, we do not expect to have considerable amounts of soluble endogenous LaLRR17. On the contrary, during the transmission of promastigotes to mammals, a part of the parasites in the inoculum die and release LRR17 in the lesion environment. Such a condition may be mimicked by in vitro infections in the presence of exogenous recombinant LRR17, in which LRR17 augments infection by binding to GRP78, and thus the blocking of GRP78 precludes this increase.
These findings suggest that LaLRR17 interacts with macrophage GRP78 and increases parasite binding and phagocytosis, enhancing infection. Interestingly, GRP78 is used as a receptor that mediates the binding of the fungus Rhizopus oryzae to its host cell (Liu et al., Reference Liu, Spellberg, Phan, Fu, Fu, Lee, Edwards, Filler and Ibrahim2010) and its endocytosis (Gebremariam et al., Reference Gebremariam, Liu, Luo, Bruno, Phan, Waring, Edwards, Filler, Yeaman and Ibrahim2014). In infections with Zika virus, the envelope protein is responsible for binding, entry and cell fusion, and one of the receptors responsible for virus endocytosis is GRP78 (Royle et al., Reference Royle, Ramirez-Santana, Akpunarlieva, Donald, Gestuveo, Anaya, Merits, Burchmore, Kohl and Varjak2020; Khongwichit et al., Reference Khongwichit, Sornjai, Jitobaom, Greenwood, Greenwood, Hitakarun, Wikan, Murphy and Smith2021). In Coxsackievirus A9, the virus invades the cell by binding to GRP78 and integrin αvβ3 (Triantafilou et al., Reference Triantafilou, Fradelizi, Wilson and Triantafilou2002), as well as to the ACE2 protein (Ibrahim et al., Reference Ibrahim, Abdelmalek, Elshahat and Elfiky2020; Carlos et al., Reference Carlos, Ha, Yeh, Van Krieken, Tseng, Zhang, Gill, Machida and Lee2021).
This study described for the first time the LaLRR17 protein as a possible virulence factor of Leishmania and its interaction with the principal ER chaperone and cell surface co-receptor, GRP78. Soluble LaLRR17 may be available after secretion by the parasite or parasite death and will increase phagocytosis of promastigotes. Although the lack of an anti-LRR17 antibody precluded precise analysis of LRR17 location and secretion, our findings suggest functions for this protein in parasite–host interaction. This is the first report of the involvement of GRP78 in a parasitic infection.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182023000720.
Author's contribution
M. S. P., S. R. B. U. and B. S. S. conceived and designed the study. M. S. P. performed most experiments mentioned in the paper. F. A. L. F. and A. T. R. performed initial experiments for LRR characterization. F. H. F. T. and R. J. G. were responsible for affinity chromatography assays. G. M. P. C. and T. L. S. A. performed GRP78 labelling in macrophages. M. M. C. involved in design of GRP78 inhibition assays. G. P. involved in designing and analysis mass spectrometry data. M. S. P. and B. S. S. wrote the article. S. R. B. U., R. J. G. and M. M. C. revised the paper.
Financial support
This study was supported by FAPESP (grants 2014/26777-4 and 2018/14972-8 for BSS and scholarship 2012/19135-0 for M. S. P.) and by CAPES.
Competing interests
None.
Ethical standards
All animals were used according to the Brazilian College of Animal Experimentation (CONEP) guidelines and the protocols were approved by the Institutional Animal Care and Use Committee (CEUA) of the University of São Paulo (protocol numbers 9829290419/2019 and 001/2009). Euthanasia was performed in CO2 camera.












 Open access
Open access