


INTRODUCTION
Apicomplexans are parasitic protozoa, which possess an apical complex, a unique set of organelles involved in invasion of host cells. The phylum includes the malaria parasite genus Plasmodium, Toxoplasma gondii – a parasite capable of infecting virtually all cell types in all warm-blooded animals – and a plethora of other parasites of humans and livestock: Cryptosporidium, Eimeria, Neospora, Theileria and Babesia. Thanks to improving technologies, large financial investment and much hard work over the past decade we have been furnished with annotated genome assemblies for the majority of important apicomplexans. Many of their genes are conserved across the phylum, representing the core genome of an apicomplexan parasite. However each genome has revealed unique genes and gene families, which should to tell us about the environmental niche of each parasite and its survival strategy. A gene family is a set of genes with similar sequences, domain structure and function that are thought to have a common ancestor.
By infecting the bodies and invading the cells of complex organisms, apicomplexan parasites endanger the life of their host and open themselves to attack from its immune system (Turner, 2002 #515). To be successful the parasite must optimize its chances of passing into a new host. This requires the parasite to minimize damage to its host until it finds a new host, while preventing the immune system from killing it. Good strategies for achieving these goals will differ in different tissues and in different hosts. Thus, in each parasite, specialized genes should have evolved to satisfy these needs. Evidence points to the large, rapidly mutating, clade-specific gene families having a large role to play in these processes.
These gene families are sometimes referred to as contingency gene families. Contingency genes have been defined as those which allow enhanced phenotypic variation (Bayliss et al. Reference Bayliss, Field and Moxon2001). These genes are found in large families and are thought to be subject to higher mutation rates than other genes, by processes such as gene conversion, recombination, duplication and deletion. They often reside in specialized regions of the genome, which may facilitate mutation and correct gene expression. Many examples encode surface antigens: membrane-bound proteins to which the host immune system develops antibodies. The classical example in Apicomplexa is the var family from Plasmodium falciparum, which is involved in antigenic variation (Guizetti and Scherf, Reference Guizetti and Scherf2013). However, other families encode products which are secreted into the host cell. These perform roles such as remodelling the host cell to favour the parasite (e.g. ROP kinases of the Coccidia, FIKK kinases in P. falciparum and SVSPs in Theileria spp.). Each new group of apicomplexan parasites investigated reveals several unique multi-gene families. In this review I will discuss these large gene families in apicomplexan parasites: how genome sequencing has been used to reveal their full extent in each genome, their genomic context, functions and how those functions relate to different parasitic niches.
IDENTIFYING GENE REPERTOIRES
Whole genome sequencing has dramatically improved our understanding of large variant gene families, their extent, diversity and genomic context. However, it has remained challenging to accurately determine their full repertoires. While contingency gene families make it difficult for the human immune system to battle parasites, they also make it difficult for us to produce complete genome assemblies, frustrating our attempts to better understand them. This is because these families are inherently repetitive. The whole or part of one gene may look much like that of another gene. This is likely key to their functional role, but also makes it difficult to piece together the genome.
The var gene family of P. falciparum, involved in antigenic variation and sequestration, is a prime example. Only using a variety of genome sequencing technologies, as well as manual finishing work, over several years have we become fairly sure that the P. falciparum 3D7 reference genome sequence is complete and describes the full complement of var genes (M. Berriman, personal communication). The problem is that each new parasite strain sequenced has a largely distinct set of var genes, variously recombined and mutated. This makes it difficult to determine the set of var genes in each new strain using current technologies without laboriously generating a completely new genome assembly. Furthermore, repetitive parts of these genes are sometimes longer than sequencing reads, meaning that de novo assembly approaches will fail to resolve them. Instead, researchers have been exploring targeted de novo assembly strategies to explore var gene repertoires in worldwide strains of P. falciparum (Assefa, Reference Assefa2013). Similarly, it was found that the T. gondii genome sequence contained collapsed repeats of the rop5 gene, a member of the ropk family (Reese et al. Reference Reese, Zeiner, Saeij, Boothroyd and Boyle2011). A genetic linkage analysis had identified the locus of this gene as important for parasite virulence. Detailed analysis of the locus identified the collapsed repeat, which was found to vary in copy number between parasites with differing virulence. Without continued improvement of genome sequences, using a variety of technologies, it is likely that key features of parasite biology will be missed.
Although it can be difficult to define every gene in a repetitive gene family, straightforward analysis of even a draft assembly will highlight much of their repertoire. Each new genome assembly of sufficient phylogenetic distance from those we have will likely identify new such families. Furthermore, sequencing more members of well-studied genera will be useful in understanding the evolution of these families. Table 1 summarizes our knowledge of these large families (those with greater than 20 members) in apicomplexan species sequenced to date.
Table 1. Contingency gene family repertoires of the Apicomplexa
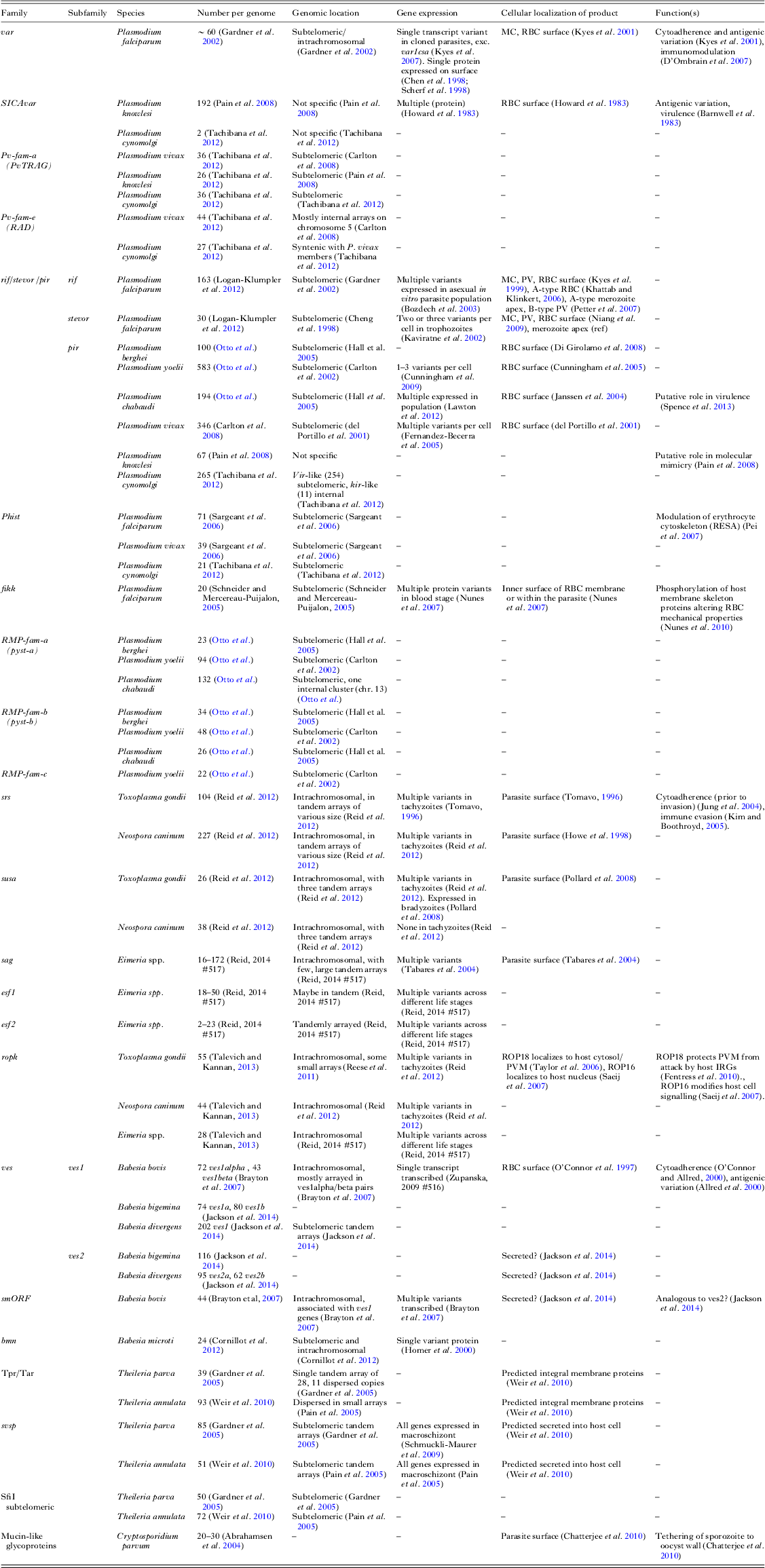
Here we show repertoires of large gene families (>20 members) for species with published genome sequences. Additional information on expression, localization and function is included where available. Some families may be missing in some species where they have not been described in the accompanying publication. Abbreviations: RBC, red blood cell; MC, Maurer's cleft; PV, parasitophorus vacuole.
GENOMIC CONTEXT
Complete genome sequences have allowed us to analyse the genomic context of gene families in unprecedented detail. Genomic context appears to be important for regulation of gene expression and generation of diversity in large apicomplexan gene families. In P. falciparum the var, rif and stevor families cluster together, proximal to each telomere (Fig. 1). The phist and fikk families are also found in these subtelomeric regions but further towards the centromeres.

Fig. 1. Genomic context of large gene families in Apicomplexa. Gene families from several species are shown in their genomic context using example chromosomes. Only those gene families described in Table 1 are shown, with dashed lines representing gaps in between. The figure is not to scale. Genes are shown on their coding strand. Colours are specific to each species and are not meant to imply any homology between families in different species, even where this exists. Telomeric sequences are highlighted where they are present in the genomic assemblies. Subtelomeres are highlighted where genes families proximal to them are those described in Table 1. A cladogram shows the known relationships between species and highlights those, which specifically organize their gene families at telomeres and those, which do not. Genome sequences were downloaded from either GeneDB (Logan-Klumpler et al. Reference Logan-Klumpler, De Silva, Boehme, Rogers, Velarde, McQuillan, Carver, Aslett, Olsen, Subramanian, Phan, Farris, Mitra, Ramasamy, Wang, Tivey, Jackson, Houston, Parkhill, Holden, Harb, Brunk, Myler, Roos, Carrington, Smith, Hertz-Fowler and Berriman2012) or EuPathDB (Aurrecoechea et al. Reference Aurrecoechea, Brestelli, Brunk, Dommer, Fischer, Gajria, Gao, Gingle, Grant, Harb, Heiges, Innamorato, Iodice, Kissinger, Kraemer, Li, Miller, Nayak, Pennington, Pinney, Roos, Ross, Stoeckert, Treatman and Wang2009): P. falciparum (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya and Martin2002), Plasmodium chabaudi (Reference Otto, Boehme, Jackson, Hunt, Franke-Fayard, Hoeijmakers, Religa, Robertson, Sanders, Ogun, Cunningham, Erhart, Billker, Khan, Stunnenberg, Langhorne, Holder, Waters, Newbold, Pain, Berriman and JanseOtto et al. ), P. knowlesi (Pain et al. Reference Pain, Bohme, Berry, Mungall, Finn, Jackson, Mourier, Mistry, Pasini, Aslett, Balasubrammaniam, Borgwardt, Brooks, Carret, Carver, Cherevach, Chillingworth, Clark, Galinski, Hall, Harper, Harris, Hauser, Ivens, Janssen, Keane, Larke, Lapp, Marti and Moule2008), B. bovis (Brayton et al. Reference Brayton, Lau, Herndon, Hannick, Kappmeyer, Berens, Bidwell, Brown, Crabtree, Fadrosh, Feldblum, Forberger, Haas, Howell, Khouri, Koo, Mann, Norimine, Paulsen, Radune, Ren, Smith, Suarez, White, Wortman, Knowles, McElwain and Nene2007), T. parva (Gardner et al. Reference Gardner, Bishop, Shah, de Villiers, Carlton, Hall, Ren, Paulsen, Pain, Berriman, Wilson, Sato, Ralph, Mann, Xiong, Shallom, Weidman, Jiang, Lynn, Weaver, Shoaibi, Domingo, Wasawo, Crabtree, Wortman, Haas, Angiuoli, Creasy, Lu and Suh2005), T. gondii (Gajria et al. Reference Gajria, Bahl, Brestelli, Dommer, Fischer, Gao, Heiges, Iodice, Kissinger, Mackey, Pinney, Roos, Stoeckert, Wang and Brunk2008), Eimeria tenella (Reid, Reference Reid, Blake, Ansari, Billington, Browne, Bryant, Dunn, Hung, Kawahara, Miranda-Saavedra, Malas, Mourier, Naghra, Nair, Otto, Rawlings, Rivailler, Sanchez-Flores, Sanders, Subramanian, Tay, Woo, Wu, Barrell, Dear, Doerig, Gruber, Ivens, Parkinson, Rajandream, Shirley, Wan, Berriman, Tomley and Pain2014 #517).
Subtelomeric location is common to contingency families in almost all species of Plasmodium examined. It has been suggested to play a role in regulating expression and generating diversity by promoting recombination (Scherf et al. Reference Scherf, Lopez-Rubio and Riviere2008). The occasionally zoonotic monkey malaria parasite Plasmodium knowlesi is an exception. In P. knowlesi, both SICAvars and pirs are spread throughout the chromosomes (Fig. 1). However, despite their internal location, they are associated with telomere-like repeats, which might play a role in promoting recombination (Pain et al. Reference Pain, Bohme, Berry, Mungall, Finn, Jackson, Mourier, Mistry, Pasini, Aslett, Balasubrammaniam, Borgwardt, Brooks, Carret, Carver, Cherevach, Chillingworth, Clark, Galinski, Hall, Harper, Harris, Hauser, Ivens, Janssen, Keane, Larke, Lapp, Marti and Moule2008). Var genes (excluding the highly conserved var1csa and var2csa) can be classified into three types based on their promoter sequences: upsA, upsB and upsC (Lavstsen et al. Reference Lavstsen, Salanti, Jensen, Arnot and Theander2003). These distinct promoters are associated with distinct contexts. UpsB var genes are subtelomeric and transcribed away from the telomere, upsA are subtelomeric, but transcribed towards the telomere. UpsC var genes are found in core chromosomal regions. The telomeres of P. falciparum chromosomes (Freitas-Junior et al. Reference Freitas-Junior, Bottius, Pirrit, Deitsch, Scheidig, Guinet, Nehrbass, Wellems and Scherf2000) and also internal var genes (Ralph et al. Reference Ralph, Bischoff, Mattei, Sismeiro, Dillies, Guigon, Coppee, David and Scherf2005) have been shown to localize in a small number of foci at the periphery of the nucleus. This also occurs in rodent malaria parasites and is thought to foster recombination, generating new variants (Figueiredo et al. Reference Figueiredo, Pirrit and Scherf2000).
Subtelomeric gene families are also a feature of Theileria spp. but not of Babesia or any Coccidia so far examined (Fig. 1). A much greater sampling of the Apicomplexa will be required to understand just how common this arrangement is and whether it has evolved multiple times in Apicomplexa.
Relative orientation of genes is important for expression of the ves1 gene family in Babesia bovis. Ves1/smORF units comprise a handful of genes, generally with at least one ves1a, one ves1b and a smORF (Brayton et al. Reference Brayton, Lau, Herndon, Hannick, Kappmeyer, Berens, Bidwell, Brown, Crabtree, Fadrosh, Feldblum, Forberger, Haas, Howell, Khouri, Koo, Mann, Norimine, Paulsen, Radune, Ren, Smith, Suarez, White, Wortman, Knowles, McElwain and Nene2007). Ves1α and ves1β together encode VESA proteins, involved in antigenic variation and cytoadherence (Allred et al. Reference Allred, Carlton, Satcher, Long, Brown, Patterson, O'Connor and Stroup2000; O'Connor and Allred, Reference O'Connor and Allred2000). The function of smORF is not known. The ves1 family is one of two known amongst apicomplexans, which shows true properties of antigenic variation, with only one variant expressed at a time. Transcription from the functional locus of active transcription (LAT) is thought to require a ves1α and a ves1β in divergent orientation, separated by a bidirectional promoter (Al-Khedery and Allred, Reference Al-Khedery and Allred2006). Ves1/smORF units are found scattered throughout Babesia chromosomes, not in subtelomeres or in tandem arrays (Fig. 1).
The srs and sag genes families, which encode unrelated, but structurally similar, surface antigens in Coccidia are found spread throughout chromosomes, mostly in tandem arrays (Fig. 1b). These arrays tend to contain genes, which are more similar to each other than other clusters, and tend to share domain subfamily architectures (Reid et al. Reference Reid, Vermont, Cotton, Harris, Hill-Cawthorne, Konen-Waisman, Latham, Mourier, Norton, Quail, Sanders, Shanmugam, Sohal, Wasmuth, Brunk, Grigg, Howard, Parkinson, Roos, Trees, Berriman, Pain and Wastling2012; Wasmuth et al. Reference Wasmuth, Pszenny, Haile, Jansen, Gast, Sher, Boyle, Boulanger, Parkinson and Grigg2012). This is probably maintained through local gene duplication as well as gene conversion (Reid et al. Reference Reid, Vermont, Cotton, Harris, Hill-Cawthorne, Konen-Waisman, Latham, Mourier, Norton, Quail, Sanders, Shanmugam, Sohal, Wasmuth, Brunk, Grigg, Howard, Parkinson, Roos, Trees, Berriman, Pain and Wastling2012).
Tandem arrays of genes can easily arise through gene duplication but the maintenance of this pattern may have advantages for promoting gene conversion. In some cases genes within a tandem array may be more similar to each other than those in other arrays (e.g. Coccidia srs/sag). In other cases they are not e.g. Plasmodium pirs (Reference Otto, Boehme, Jackson, Hunt, Franke-Fayard, Hoeijmakers, Religa, Robertson, Sanders, Ogun, Cunningham, Erhart, Billker, Khan, Stunnenberg, Langhorne, Holder, Waters, Newbold, Pain, Berriman and JanseOtto et al. ) and Theileria SfiI sub-telomeric genes (Weir et al. Reference Weir, Karagenc, Baird, Tait and Shiels2010 ), suggesting recombination between arrays. Similarity within loci might be due to either recent divergence, through copying or a continued process of local gene conversion. Some genes families, present in tandem arrays, may preserve their orientation, all being transcribed towards centromere or telomere, in order that they can line up with homologous arrays from other chromosomes or other parts of the same chromosome for recombination.
Currently it is clear that subtelomere context is not a common feature of large apicomplexan gene families. Indeed there is no evidence for it in Coccidia. In the haemosporidia (Plasmodium) and piroplasms (Babesia, Theileria) it may have been lost or gained multiple times.
PROTEIN DOMAIN STRUCTURE
The most common feature of large apicomplexan gene family sequences is a signal peptide: a hydrophobic N-terminal sequence of 10–20 amino acids. This is an indication that such genes may be directly involved in host–parasite interactions as they can be exported from the parasite cell via the secretory pathway. Many of these gene families appear to be ultimately bound to membrane, using either transmembrane domains or glycophosphatidylinositol (GPI) anchors. In general, extracellular proteins tend to be rich in disulphide bonds to improve stability, and indeed apicomplexans are no exception (Fass, Reference Fass2012). Despite these similarities, gene families in different groups of parasites share no detectable homology and in some cases can be shown to derive from different ancestral families.
The var gene family exhibits a complex protein domain architecture. Plasmodium falciparum var genes are composed of Duffy binding-like (DBL), CIDR and C2 domains (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya and Martin2002). DBL domains have six subfamilies and CIDR two. There is a great diversity of domain architectures in the var family, although the N-termini are usually composed of a DBLα domain followed by a CIDR domain, suggesting this structure is key to their function (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya and Martin2002). DBL domains are also found in several other genes of P. falciparum, which are involved in binding host cells and are required for invasion by the merozoite, implying an ancestral role for this domain in host cell binding (Tolia et al. Reference Tolia, Enemark, Sim and Joshua-Tor2005). The C-terminus includes a transmembrane domain, which tethers the proteins to the erythrocyte surface.
Other protein families have much simpler architectures. Plasmodium falciparum rifs have an N-terminal signal peptide, a PEXEL motif and either one or two transmembrane domains depending on the subfamily (Bultrini et al. Reference Bultrini, Brick, Mukherjee, Zhang, Silvestrini, Alano and Pizzi2009). PEXEL motifs are required for export of proteins to the red blood cell surface (Hiller et al. Reference Hiller, Bhattacharjee, van Ooij, Liolios, Harrison, Lopez-Estrano and Haldar2004; Marti et al. Reference Marti, Good, Rug, Knuepfer and Cowman2004). Based on remote sequence similarity, conservation of intronic sequence motifs and predicted protein secondary structure, it has been proposed that rif genes are related to the pirs, a family present in all other species so far sequenced (Janssen et al. Reference Janssen, Phillips, Turner and Barrett2004). These families are an example of those which have diverged almost beyond recognition, and which may help to provide insights into evolution of gene families in malaria.
Coccidian surface antigen gene families also use relatively simple domain structures. Toxoplasma srs genes usually contain two SAG domains (Jung et al. Reference Jung, Lee and Grigg2004), while the short, consistent lengths of Eimeria sag genes suggest a single domain structure (Reid, Reference Reid, Blake, Ansari, Billington, Browne, Bryant, Dunn, Hung, Kawahara, Miranda-Saavedra, Malas, Mourier, Naghra, Nair, Otto, Rawlings, Rivailler, Sanchez-Flores, Sanders, Subramanian, Tay, Woo, Wu, Barrell, Dear, Doerig, Gruber, Ivens, Parkinson, Rajandream, Shirley, Wan, Berriman, Tomley and Pain2014 #517). Both families have signal peptides and GPI anchor addition sites. Furthermore, the individual domain sequences in each family both tend to contain six cysteines and are of similar lengths (200–300 amino acids). However, these common properties appear to be convergent, as the families have distinct origins. While Toxoplasma srs genes have been proposed to derive from metazoan ephrins by lateral transfer (Arredondo et al. Reference Arredondo, Cai, Takayama, MacDonald, Anderson, Aravind, Clore and Miller2012). Eimeria sag genes meanwhile are thought to be related to CAP domains (Reid, Reference Reid, Blake, Ansari, Billington, Browne, Bryant, Dunn, Hung, Kawahara, Miranda-Saavedra, Malas, Mourier, Naghra, Nair, Otto, Rawlings, Rivailler, Sanchez-Flores, Sanders, Subramanian, Tay, Woo, Wu, Barrell, Dear, Doerig, Gruber, Ivens, Parkinson, Rajandream, Shirley, Wan, Berriman, Tomley and Pain2014 #517). CAP domains are widely used across the tree of life.
Theileria parasites invade and transform host leucocytes (reviewed in Dobbelaere and Kuenzi, Reference Dobbelaere and Kuenzi2004). It is thought that they transform the host cell using families of secreted proteins. On such example is the SVSP family, which encode a signal peptide and a Frequently Associated IN Theileria (FAINT) domain. FAINT domains are common to a range of Theileria subtelomeric genes from different families, all predicted to be secreted (Pain et al. Reference Pain, Renauld, Berriman, Murphy, Yeats, Weir, Kerhornou, Aslett, Bishop, Bouchier, Cochet, Coulson, Cronin, de Villiers, Fraser, Fosker, Gardner, Goble, Griffiths-Jones, Harris, Katzer, Larke, Lord, Maser, McKellar, Mooney, Morton, Nene, O'Neil and Price2005). It has been speculated that this low-complexity glutamine- and proline-rich domain may be difficult for vertebrate immune systems to recognize and therefore could provide some anonymity for the parasite (Gardner et al. Reference Gardner, Bishop, Shah, de Villiers, Carlton, Hall, Ren, Paulsen, Pain, Berriman, Wilson, Sato, Ralph, Mann, Xiong, Shallom, Weidman, Jiang, Lynn, Weaver, Shoaibi, Domingo, Wasawo, Crabtree, Wortman, Haas, Angiuoli, Creasy, Lu and Suh2005).
Domain structures in these families suggest a diverse array of structures and of phylogenetic origins for surface proteins and other families. This is likely to be a result of the requirement for constant innovation in the parasites’ battle with the host but also of differing requirements related to the niche of the parasite and its strategy for transmission.
EXPRESSION PATTERNS AND PROTEIN LOCALIZATION
Gene families involved in antigenic variation sensu stricto are expressed monoallelically, with only one family member localized to the parasite or host cell surface at a time (Borst, Reference Borst1991). Although this is observed for var and VESA families, it is not what has been observed for most apicomplexan large gene families. Frequently, where measurements have been made, multiple members are expressed. Unlike switching of vsg in trypanosomes, switching to different genes in apicomplexans does not involve genomic rearrangement. Although many contingency gene families encode protein localized to the parasite or host cell surface, some families localize to the host cytosol or nucleus (e.g. ropk) and some families have different members with distinct localizations (e.g. rif; Fig. 2).
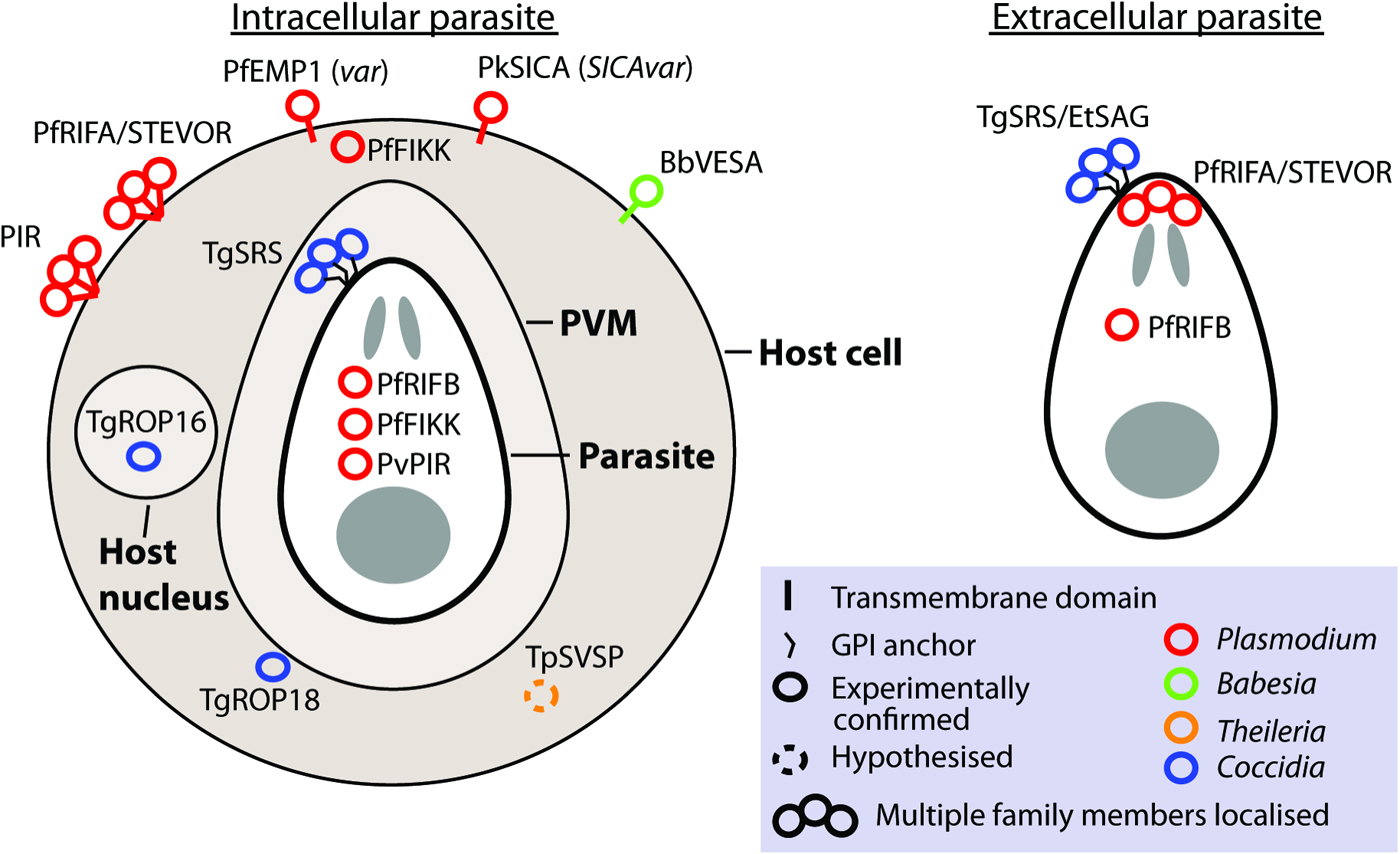
Fig. 2. Localization of gene family products. The localization of gene products is shown for intracellular and extracellular parasites, where known or hypothesized. Where multiple copies have been localized this is indicated by a bundle of circles. Abbreviations: PVM, parasitophorus vacuole membrane. n.b. The PVM is not present in Theileria infected leucocytes and is destroyed quickly after invasion by Babesia (Asada, 2012 #518). Evidence: TgROP18 localizes to host cytosol/PVM (Taylor et al. Reference Taylor, Barragan, Su, Fux, Fentress, Tang, Beatty, Hajj, Jerome, Behnke, White, Wootton and Sibley2006), TgROP16 localizes to host nucleus (Saeij et al. Reference Saeij, Coller, Boyle, Jerome, White and Boothroyd2007), TpSVSP predicted to localize to host cytosol (Weir et al. Reference Weir, Karagenc, Baird, Tait and Shiels2010), PfRIF/STEVOR localize to parasite apex (Petter et al. Reference Petter, Haeggstrom, Khattab, Fernandez, Klinkert and Wahlgren2007; Blythe et al. Reference Blythe, Yam, Kuss, Bozdech, Holder, Marsh, Langhorne and Preiser2008) and RBC surface (Kyes et al. Reference Kyes, Rowe, Kriek and Newbold1999), PvVIR14 and PvVIR10 are exported to the membrane of iRBC whereas PvVIR17 remains inside the parasite (Bernabeu et al. Reference Bernabeu, Lopez, Ferrer, Martin-Jaular, Razaname, Corradin, Maier, Del Portillo and Fernandez-Becerra2012), rodent PIR family localize on or close to the surface of the RBC (Cunningham et al. Reference Cunningham, Fonager, Jarra, Carret, Preiser and Langhorne2009), TgSRS multiple gene products have been localized to the parasite surface (Tomavo, Reference Tomavo1996), EtSAG multiple gene products localized to the parasite surface (Tabares et al. Reference Tabares, Ferguson, Clark, Soon, Wan and Tomley2004), PfFIKKs localize to Maurer's cleft/host membrane cytoskeleton and within parasite (Nunes et al. Reference Nunes, Okada, Scheidig-Benatar, Cooke and Scherf2010), PfEMP1 (var family) localizes to the host cell surface (Kyes et al. Reference Kyes, Horrocks and Newbold2001), PkSICA (SICAvar family) localizes to the host cell surface (Howard et al. Reference Howard, Barnwell and Kao1983), BbVESA (ves1 family) localizes to the host cell surface (O'Connor et al. Reference O'Connor, Lane, Stroup and Allred1997).
Correct expression of P. falciparum var genes involves the suppression of all but one gene and switching between individual genes; control occurs at the level of transcription initiation (Scherf et al. Reference Scherf, Lopez-Rubio and Riviere2008). Expression is the highest early after cell invasion, presumably because the protein must quickly be expressed on the surface of the invaded red cell. Control of transcription is thought to require genetic elements such as the intron (Deitsch et al. Reference Deitsch, Calderwood and Wellems2001) and the ups promoter region (Voss et al. Reference Voss, Healer, Marty, Duffy, Thompson, Beeson, Reeder, Crabb and Cowman2006). An ApiAP2 transcription factor is known to bind to the intron, which acts as a bidirectional promoter and from which non-coding RNA is transcribed (Zhang et al. Reference Zhang, Huang, Zhang, Fang, Claes, Duchateau, Namane, Lopez-Rubio, Pan and Scherf2011). Plasmodium falciparum subtelomeres are characteristically marked by the heterochromatic epigenetic mark H3K9me3 (Salcedo-Amaya et al. Reference Salcedo-Amaya, van Driel, Alako, Trelle, van den Elzen, Cohen, Janssen-Megens, van de Vegte-Bolmer, Selzer, Iniguez, Green, Sauerwein, Jensen and Stunnenberg2009). During transcription the active var gene is marked by more highly acetylated histone H4 and also H3K4me3 and H3K4me2 (Freitas-Junior et al. Reference Freitas-Junior, Hernandez-Rivas, Ralph, Montiel-Condado, Ruvalcaba-Salazar, Rojas-Meza, Mancio-Silva, Leal-Silvestre, Gontijo, Shorte and Scherf2005; Lopez-Rubio et al. Reference Lopez-Rubio, Gontijo, Nunes, Issar, Hernandez Rivas and Scherf2007). When not expressed it loses H3K4me3, but retains H3K4me2. This is thought to act as memory so that the same var gene is activated in the next invaded red cell (Lopez-Rubio et al. Reference Lopez-Rubio, Gontijo, Nunes, Issar, Hernandez Rivas and Scherf2007). Conversely, methylation of H3K36 represses var gene expression (Jiang et al. Reference Jiang, Mu, Zhang, Ni, Srinivasan, Rayavara, Yang, Turner, Lavstsen, Theander, Peng, Wei, Jing, Wakabayashi, Bansal, Luo, Ribeiro, Scherf, Aravind, Zhu, Zhao and Miller2013).
The mechanism for controlling monoallelic expression of Babesia ves1 genes is less well understood. Expressed genes are found in a tandem configuration and are expressed from LATs by a bidirectional promoter. It is thought that chromatin remodelling is required for transcription suggesting there may be some commonalities with control of var expression (Huang et al. Reference Huang, Xiao and Allred2013). However, it is not clear that chromatin remodelling plays a direct role in monoallelic expression.
For all other apicomplexan families multiple members appear to be expressed at once. A small number of P. knowlesi SICAvars are expressed by the parasite at the same time (Barnwell et al. Reference Barnwell, Howard, Coon and Miller1983). Interestingly none are expressed in splenectomized monkeys, suggesting that they are regulated in response to the host immune system (Barnwell et al. Reference Barnwell, Howard, Coon and Miller1983). Such a response has also been observed for pir genes in several Plasmodium species. In P. chabaudi it was shown that parasites, which are serially passaged between mice, avoiding the mosquito and liver stages, express a reduced set of pirs and RMP-fam-as and that this is associated with an increase in virulence (Spence et al. Reference Spence, Jarra, Levy, Reid, Chappell, Brugat, Sanders, Berriman and Langhorne2013). Whereas perhaps 10–20% of the pir repertoire (20–40 genes) is expressed in serially blood-passaged parasites (Lawton et al. Reference Lawton, Brugat, Yan, Reid, Bohme, Otto, Pain, Jackson, Berriman, Cunningham, Preiser and Langhorne2012), mosquito transmitted parasites express more than half of their repertoire (>100 genes), quite the opposite of what we would expect from genes involved in antigenic variation sensu stricto (Spence et al. Reference Spence, Jarra, Levy, Reid, Chappell, Brugat, Sanders, Berriman and Langhorne2013). Similar results have been shown for Plasmodium yoelii, with B or T cell deficient mice showing differences in expression of these genes (Cunningham et al. Reference Cunningham, Jarra, Koernig, Fonager, Fernandez-Reyes, Blythe, Waller, Preiser and Langhorne2005).
Change in expression of the srs family of surface antigens in T. gondii is known to be important for evading the host immune response (Tomavo, Reference Tomavo1996). Around half its repertoire of srs genes is expressed during the rapidly growing tachyzoite stage and multiple protein products have been localized to the parasite cell surface (Tomavo, Reference Tomavo2001; Reid et al. Reference Reid, Vermont, Cotton, Harris, Hill-Cawthorne, Konen-Waisman, Latham, Mourier, Norton, Quail, Sanders, Shanmugam, Sohal, Wasmuth, Brunk, Grigg, Howard, Parkinson, Roos, Trees, Berriman, Pain and Wastling2012). During switch to the encysted, bradyzoite form, there is a switch to a largely non-overlapping set of srs genes (Jung et al. Reference Jung, Lee and Grigg2004). The regulatory mechanism controlling this is unknown.
Several families are known to localize within the host cell and have been shown to adapt the host cell to function as a better home for the parasite. Fikk kinases are expressed in the blood stage of P. falciparum, with different members expressed at different stages of development. Their products have been shown to locate to within the parasite itself, the Maurer's cleft in the host erythrocyte and to the inner side of the erythrocyte membrane (Nunes et al. Reference Nunes, Goldring, Doerig and Scherf2007). Transcriptional data indicates that unlike SfiI sub-telomeric genes, SVSP family genes of T. annulata are expressed in a stage-specific manner by the macroschizont and that the majority of the gene products contribute to the secretome (Weir et al. Reference Weir, Karagenc, Baird, Tait and Shiels2010). These are predicted to localize to the host cytosol. No members of large gene families in Theileria are thought to localize to the parasite or host membranes.
Genomic data can give us clues about whether gene products are secreted, exported, membrane-bound or GPI-anchored. However, experimental approaches are required to confirm this and provide greater detail. There is great difficulty in localizing multiple members of protein families due to the requirement for specific antibodies or genetic manipulation of multiple, similar, genes. Improvements in locating the product of each family member over time would allow greater insight into a family's function.
FUNCTIONAL ROLES
It is generally assumed that large variant gene families have a role in antigenic variation, this being the most straightforward explanation for maintaining such a large repertoire of related genes. However, it is clear that most large gene families in Apicomplexa are not involved in antigenic variation as it is often understood and many may not be involved in antigenic variation at all (Table 1). It has been proposed that gene families, which are involved in antigenic variation, might have a distinct primary function. The primary functions of vars for instance appear to be sequestration to avoid clearance by the spleen and rosetting to enhance efficiency of invasion (Rowe et al. Reference Rowe, Moulds, Newbold and Miller1997). They may vary greatly because they are exposed to the host immune system, not exposed to the host immune system simply in order to provide a variety of epitopes. Other Plasmodium species do not have var genes, or a family, which might obviously replace them, except perhaps for SICAvar in P. knowlesi. However P. chabaudi does sequester (Brugat et al. Reference Brugat, Cunningham, Sodenkamp, Coomes, Wilson, Spence, Jarra, Thompson, Scudamore and Langhorne2014) and shows antigenic variation (Brannan et al. Reference Brannan, Turner and Phillips1994; Phillips et al. Reference Phillips, Brannan, Balmer and Neuville1997), suggesting these functions can be achieved by genes with very different properties. It is possible that immune evasion could be mediated by expression of all or most of the repertoire of a gene family. Many similar proteins, expressed at relatively low levels, might prevent induction of an effective immune response, resulting in immune evasion without mutually exclusive expression (Cunningham et al. Reference Cunningham, Jarra, Koernig, Fonager, Fernandez-Reyes, Blythe, Waller, Preiser and Langhorne2005, Reference Cunningham, Fonager, Jarra, Carret, Preiser and Langhorne2009). Ves1 genes in Babesia bovis are implicated in antigenic variation and cytoadherence and, like Plasmodium, this species can sequester in the brain and can cause neurological damage (Sondgeroth et al. Reference Sondgeroth, McElwain, Allen, Chen and Lau2013).
Pir is the only large gene family conserved across all Plasmodium species examined. It has been shown that pir and RMP-fam-a expression (Table 1) is modulated in response to the host immune system (Cunningham et al. Reference Cunningham, Jarra, Koernig, Fonager, Fernandez-Reyes, Blythe, Waller, Preiser and Langhorne2005; Spence et al. Reference Spence, Jarra, Levy, Reid, Chappell, Brugat, Sanders, Berriman and Langhorne2013). In particular, expression of a smaller repertoire of P. chabaudi pirs is associated with higher parasitemia and more damage to the host. These genes may have a role in attracting the immune system to a sufficient degree that it is prevented from doing excessive harm to the host.
A-type RIFINs and STEVORs have been shown to locate to the apical end of the invading merozoite stage of P. falciparum. It has been suggested that they provide a shield from immune attack against the more conserved proteins involved in invasion, through binding to erythrocyte surface molecules (Petter et al. Reference Petter, Haeggstrom, Khattab, Fernandez, Klinkert and Wahlgren2007; Blythe et al. Reference Blythe, Yam, Kuss, Bozdech, Holder, Marsh, Langhorne and Preiser2008). There is evidence for molecular mimicry amongst the related pir family in P. knowlesi (Pain et al. Reference Pain, Bohme, Berry, Mungall, Finn, Jackson, Mourier, Mistry, Pasini, Aslett, Balasubrammaniam, Borgwardt, Brooks, Carret, Carver, Cherevach, Chillingworth, Clark, Galinski, Hall, Harper, Harris, Hauser, Ivens, Janssen, Keane, Larke, Lapp, Marti and Moule2008). Seven P. knowlesi pirs contain regions of up to 36 amino acids, which perfectly match the immune system protein CD99 from the natural host Macaca mulatta. This would suggest that these pirs may compete with host CD99 for binding and could interfere with the host immune response.
Within large variant gene families individual members are able to mutate quite freely due to redundancy of function and reduced selective pressure. They may therefore have a great deal of opportunity to develop new functions. Despite the great differences in var gene repertoire, even between different strains of P. falciparum, a small number are have been under strong positive selection to maintain their structure and function (Rowe et al. Reference Rowe, Kyes, Rogerson, Babiker and Raza2002). Var2csa is able to bind chondroitin sulphate A on the placenta and its expression is associated with placental malaria in women pregnant for the first time (Salanti et al. Reference Salanti, Dahlback, Turner, Nielsen, Barfod, Magistrado, Jensen, Lavstsen, Ofori, Marsh, Hviid and Theander2004).
DBLα domains are found in var genes and in genes involved in invasion in P. falciparum. The srs gene family takes a similar multi-functional role in Toxoplasma. Host cell invasion by apicomplexans is preceded by attachment and reorientation (Carruthers and Boothroyd, Reference Carruthers and Boothroyd2007). In Toxoplasma, the same gene family involved in immune evasion is also involved in the first of these processes (Jung et al. Reference Jung, Lee and Grigg2004). SRS57 has been shown to act in binding host cells (Dzierszinski et al. Reference Dzierszinski, Mortuaire, Cesbron-Delauw and Tomavo2000), while SRS34A has a role in attracting an immune response. SRS29B, SRS34A and SRS29C are specific to the rapidly growing tachyzoite stage and are strongly antigenic (Lekutis et al. Reference Lekutis, Ferguson, Grigg, Camps and Boothroyd2001). Along with many other srs genes these are turned off during the switch to bradyzoites. The tachyzoite may attract the immune system, or the immune system may prompt the parasite to switch forms (Bohne et al. Reference Bohne, Heesemann and Gross1993). This may have a role in limiting parasite proliferation and also distracting attention from the bradyzoite, which can then hide in tissue and await transmission. Perhaps this family might have started out with a cell adhesion function, which showed antigenic variation because it was exposed. It could then have evolved a function in immune evasion switching which allowed the quiescent form to hide in tissues.
The intracellular stage of the bovine parasite Theileria, equivalent to the malaria blood stage, is not known to express any molecules on the surface of the invaded cell, a leucocyte (Boulter and Hall, Reference Boulter and Hall1999). The largest gene family in Theileria, Tar/Tpr, is predicted to encode mostly integral membrane proteins; however, no known function has been attributed to it (Weir et al. Reference Weir, Karagenc, Baird, Tait and Shiels2010). The next two largest variant gene families of Theileria appear to encode largely secreted proteins (Pain et al. Reference Pain, Renauld, Berriman, Murphy, Yeats, Weir, Kerhornou, Aslett, Bishop, Bouchier, Cochet, Coulson, Cronin, de Villiers, Fraser, Fosker, Gardner, Goble, Griffiths-Jones, Harris, Katzer, Larke, Lord, Maser, McKellar, Mooney, Morton, Nene, O'Neil and Price2005). It has been proposed that SVSPs have a role in transforming the host cell. They possess nuclear localization signals and it is thought they might localize to the host nucleus much like products of the smaller TashAT family (Shiels et al. Reference Shiels, McKellar, Katzer, Lyons, Kinnaird, Ward, Wastling and Swan2004). Alternatively they might provide an immunological smokescreen effect by distracting immune cells from more conserved surface-bound antigens (Weir et al. Reference Weir, Karagenc, Baird, Tait and Shiels2010).
There are large kinase gene families from Plasmodium and Coccidia, which are also thought to be involved in modifying the host cell. Plasmodium falciparum has a unique expansion of the Apicomplexan-specific FIKK family of kinases (Nunes et al. Reference Nunes, Goldring, Doerig and Scherf2007). There is evidence that some members are involved in phosphorylation of host membrane skeleton proteins and alter the mechanical properties of the red blood cell (Nunes et al. Reference Nunes, Okada, Scheidig-Benatar, Cooke and Scherf2010). ROP kinases and inactivated pseudokinases are stored in club-shaped organelles known as rhoptries, which introduce their cargo to the host cell upon invasion. In T. gondii, ROP16 localizes to host nucleus where it activates the JAK-STAT pathway and modulates host gene expression (Saeij et al. Reference Saeij, Coller, Boyle, Jerome, White and Boothroyd2007). TgROP18 localizes to the PVM (Taylor et al. Reference Taylor, Barragan, Su, Fux, Fentress, Tang, Beatty, Hajj, Jerome, Behnke, White, Wootton and Sibley2006) and is able to prevent immune-related GTPases (IRGs) from attacking this membrane and destroying the parasite (Fentress et al. Reference Fentress, Behnke, Dunay, Mashayekhi, Rommereim, Fox, Bzik, Taylor, Turk, Lichti, Townsend, Qiu, Hui, Beatty and Sibley2010). This family seems to provide a set of exquisite tools for modifying the host cell.
It has been proposed that Cryptosporidium species do not display any form of antigenic variation (Singh et al. Reference Singh, Theodos and Tzipori2005). Interestingly, they also have a paucity of large variant gene families. Only the mucin-like glycoproteins show a large expansion. These have been proposed to function in tethering the sporozoite stage to the oocyst wall. However, it is not clear why 30 genes would be required to perform this task. Why Cryptosporidium should be able to survive without the wealth of large gene families present in other apicomplexans is currently unclear.
Thus, a variety of roles have already been uncovered for large apicomplexan gene families. Most families however have no known function and await characterization (Table 1). Small research communities are assembling around some of these families, but many remain essentially unstudied.
CONCLUSIONS
The P. falciparum var gene family is the most intensely studied large gene family in the Apicomplexa. It is involved in the best-understood mechanisms of antigenic variation and sequestration in this phylum. While other species exhibit antigenic variation and sequestration they use gene families with different properties and evolutionary histories. Many gene families are concerned with manipulating the host immune response or modifying the host cell rather than antigenic variation or sequestration. This review has highlighted some of the common themes and important contrasts that exist amongst large variant gene families in apicomplexan species. These may be useful in directing research to understand how apicomplexans interact with their hosts.
ACKNOWLEDGEMENTS
I would like to thank Chris Newbold and Magdalena Zarowiecki for critical reading of the manuscript.
FINANCIAL SUPPORT
This work was supported by the Wellcome Trust.




 Open access
Open access