



Introduction
An island can be defined as a suitable habitat for species that is isolated and surrounded by an unsuitable matrix. While the traditional ‘island’ term is used to define a land surrounded by water, biologists use this term as well for mountain peaks (Brown, Reference Brown1971), fragmented forest (Harris, Reference Harris1984), lakes (Harris et al. Reference Harris, Woolnouh and Zanatta2011) or caves (Culver, Reference Culver1970). These different ecosystems have been widely studied but an island-like ecosystem, oases, has been somehow neglected. One of the reasons may be that oases are not natural but semi-natural continental islands. In fact, the structure, functioning and size of the oases, that range from few tens to thousands hectares (Kassah, Reference Kassah1996), depend on environmental, historical and socioeconomic factors, since they are directly dependent on the availability of water and on human activities for irrigation and maintenance (Selmi and Boulinier, Reference Selmi, Boulinier, Gillespie and Clague2009). To date, the dynamics and maintenance of biodiversity associated with oases have been poorly investigated. For instance, to our knowledge, only one study examined the classic species–area relationship (Tjørve and Tjørve, Reference Tjørve and Tjørve2008; Triantis et al. Reference Triantis, Guilhaumon and Whittaker2012) and found that the number of breeding bird species in a given oasis was positively related to its area (Selmi et al. Reference Selmi, Boulinier and Barbault2002). In addition, metapopulation processes (i.e. dispersal and colonization/extinction dynamics) seem to play important roles in shaping bird distribution and abundance in southern Tunisian oases. In fact, because oases are organized into distinct geographic clusters, bird exchanges occurred more frequently within rather than between oasis clusters (Selmi et al. Reference Selmi, Boulinier and Barbault2002, Reference Selmi, Boulinier and Faivre2003).
An appealing objective for the evolutionary parasitology field is to understand how parasites circulate in wildlife inhabiting such insular-like systems. To date, studies in oases have looked only at parasites infecting humans (Fasciola hepatica: Hammami et al. Reference Hammami, Hamed and Ayadi2007) and animal exposure to viruses (West Nile virus: Hammouda et al. Reference Hammouda, Lecollinet, Hamza, Nasri, Neb and Selmi2015; Usutu virus: Ben Hassine et al. Reference Ben Hassine, De Massis, Calistri, Savini, Bel Haj Mohamed, Ranen, Di Gennaro, Sghaier and Hammami2014). To our knowledge, no study has yet explored the diversity and dynamics of parasites infecting wild animals that live in this atypical ecosystem. To do so, we used the avian blood parasites that are an appropriate model system for diverse reasons (Lapointe et al. Reference Lapointe, Atkinson and Samuel2012; Sehgal, Reference Sehgal2015). First, haemosporidian parasites have repeatedly proven to be particularly useful when investigating the roles of host ecology and habitat features in shaping the spatial distribution, prevalence and host specificity of parasites in various ecosystems, such as tropical forests (e.g. Chasar et al. Reference Chasar, Loiseau, Valkiūnas, Iezhova, Smith and Sehgal2009; Laurance et al. Reference Laurance, Jones, Westcott, Mckeown, Harrington and Hilbert2013), arctic forests (e.g. Loiseau et al. Reference Loiseau, Harrigan, Cornel, Guers, Dodge, Marzec, Carlson, Seppi and Sehgal2012) or urban landscapes (e.g. Carbó-Ramírez et al. Reference Carbó-Ramírez, Zuria, Schaefer and Santiago-Alarcon2017). Furthermore, during the past decade, the increasing number of studies using this parasite system created an invaluable amount of knowledge on the parasite diversity worldwide that has been gathered into the online database MalAvi (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009), which comprises now more than 2700 parasite lineages from 350 publications.
Here, our objective was to explore how the diversity, prevalence and host specificity of avian haemosporidian parasites in southern Tunisian oases vary among hosts and according to habitat features. The avifauna in southern Tunisia has been well described (Selmi, Reference Selmi2000; Selmi and Boulinier, Reference Selmi and Boulinier2003, Reference Selmi and Boulinier2004) and among the list of breeding bird species recorded in this area, only a few are found in the entire oases system (Selmi and Boulinier, Reference Selmi and Boulinier2003, Reference Selmi and Boulinier2004). Among them, we decided to focus on the hybrid sparrow Passer domesticus × hispaniolensis and the laughing dove Spilopelia senegalensis that share some ecological characteristics, as they are both abundant sedentary breeders with anthropophilic behaviours. We also focused on the most common avian parasites, Plasmodium and Haemoproteus, respectively, transmitted by mosquitoes (Culicidae) and biting midges (Ceratopogonidae) or louse flies (Hippoboscidae; Valkiūnas, Reference Valkiūnas2004). First, we molecularly characterized the lineage diversity of haemosporidian parasites infecting doves and sparrows and compared the lineage diversity within and between oases. We used the database MalAvi to: (i) identify lineages that may have been found before or that are restricted to the oasis system and (ii) estimate the host specificity of the lineages (Poulin and Mouillot, Reference Poulin and Mouillot2003; Hellgren et al. Reference Hellgren, Pérez-Tris and Bensch2009). Second, we statistically tested for each species how the prevalence of avian parasites varied according to the host age and also to two main oasis characteristics, namely vegetation structure and distance to the coast.
We predicted that each bird species should harbour distinct parasite lineages, but within each host species parasite, diversity should not significantly vary among oases because bird dispersal and exchanges between local populations may be rather common. However, we expected parasite prevalence to vary among oases according to habitat conditions and their suitability to vectors. Inland oases that are more exposed to the effects of Sahara are drier than coastal oases (Kassah, Reference Kassah1996), and this may lead to a reduced vector presence and abundance and hence a reduced bird exposure to parasites. Moreover, densely vegetated oases are expected to offer more suitable habitat conditions for vectors, thus increasing bird exposure to parasites, compared with the less-vegetated areas.
Material and methods
Study sites
Data were collected in six study locations in southern Tunisia (Fig. 1; Table 1): three coastal sites (Kettana, Nakta and Zrig), with distance to sea ranging from 2 to 4·5 km, and three inland sites (Gafsa, El-Manachi and Oum-Errous), with distance to sea ranging from 115 to 168 km. Kettana and Gafsa sites are densely vegetated areas, as they correspond to traditional oases where the vegetation is composed of a mixture of cultivated and natural plants organized into three main layers (palm trees, fruit trees and herbaceous plants). Seventeen bird species (three non-passerine and 14 passerine species) are known to breed regularly in these oases (Selmi, Reference Selmi2001). The remaining sites are characterized by a simpler vegetation structure. In Oum-Errous and El-Manachi, the vegetation is exclusively composed of palm trees arranged in regular and spaced rows. Nakta is surrounded by low-density olive fields and Zrig was located inside the University of Gabès where the vegetation is minimal, with trees surrounded by buildings. These less-vegetated areas support fewer than ten breeding bird species (Selmi, Reference Selmi2001). In addition, all sites are frequented by humans, as they contain human dwellings and/or cattle stables.
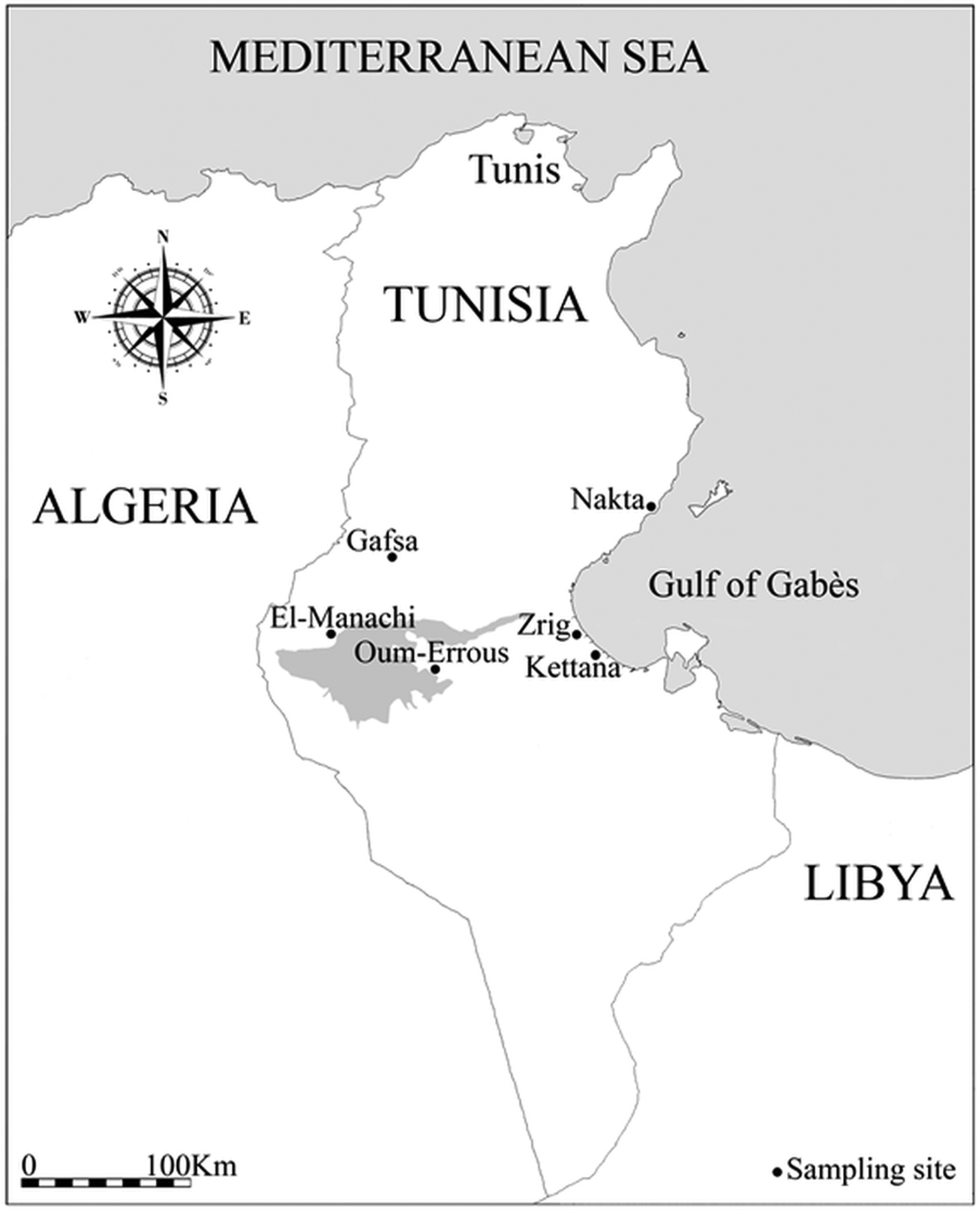
Fig. 1. Map of Tunisia showing the location of the six sampled sites. The shading represents a large endorheic salt lake named Chott El-Jerid.
Table 1. Avian haemosporidian parasite diversity and prevalence from southern Tunisian oases. For each parasite genus and for each site (In El-Manachi, we captured only doves, and in Zrig, only sparrows), we report here the number of infected individuals (N inf) out of the number of sampled birds (N tot), with the prevalence in brackets. Numbers and names of lineages are also given for each parasite genus and site. Lineages recorded in juveniles are marked with asterisks. (A) Spilopelia senegalensis, (B) Passer domesticus × hispaniolensis

Host species and data collection
The Laughing dove S. senegalensis is a Columbiform native to most of sub-Saharan Africa, Middle-East and Indian subcontinent (Baptista et al. Reference Baptista, Trail, Horblit, del Hoyo, Elliott and Sargatal1997). This species has largely expanded its geographic range in North Africa, occupying dry scrub, farmland habitats and areas close to human settlements (Selmi, Reference Selmi2000; Isenmann et al. Reference Isenmann, Gaultier, El Hili, Azafzaf, Dlensi and Smart2005). This dove colonized southern Tunisia late in the 19th century and is nowadays a common sedentary breeder in the oasis habitat (Selmi, Reference Selmi2000; Isenmann et al. Reference Isenmann, Gaultier, El Hili, Azafzaf, Dlensi and Smart2005; Boukhriss and Selmi, Reference Boukhriss and Selmi2009).
The hybrid sparrow is a result of hybridization between the Spanish sparrow Passer hispaniolensis and house sparrow P. domesticus (Johnston, Reference Johnston1969), with a male plumage intermediate to males of the Spanish and house sparrows. In southern Tunisia, hybrid sparrows are common sedentary birds that show anthropophilic behaviour and lifestyle. They nest on human settlements, stables, electric poles and large palm trees (Selmi, Reference Selmi2000; Isenmann et al. Reference Isenmann, Gaultier, El Hili, Azafzaf, Dlensi and Smart2005). House sparrows are totally absent from this area and Spanish sparrows do not breed inside the oasis habitat, therefore when we captured young sparrows, no confusion was possible with the house or Spanish sparrows.
Bird sampling took place between October 2013 and June 2015. Hybrid sparrows were sampled in five out of the six above cited sites (Table 1) during three sampling periods (October 2013, October 2014 and June 2015). All laughing dove samples were collected during June 2015 in five sites (Table 1). Birds were trapped by means of mist nets placed around highly frequented feeding sites (cowsheds and grain storage garages). Once captured, birds were aged based on their plumage, following Svensson (Reference Svensson1984) for sparrows and Baptista et al. (Reference Baptista, Trail, Horblit, del Hoyo, Elliott and Sargatal1997) for laughing doves. A blood sample was taken from the jugular or the brachial veins using heparinized capillary tubes or syringes, and stored in lysis buffer (10 mM Tris–HCL pH 8·0, 100 mm EDTA, 2% SDS) at −20 °C until molecular analyses could be performed. Before being released, birds were colour-ringed to avoid resampling.
Molecular analysis and lineage identification
DNA was extracted from whole blood from birds with a DNeasy kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. For Plasmodium and Haemoproteus detection, we used a nested polymerase chain reaction (PCR) to amplify a fragment of cytochrome b with the primers HAEMF/HAEMR2 – HAEMNF/HAEMNR2 following Waldenström et al. (Reference Waldenström, Bensch, Hasselquist and Ostman2004). We used REDTaq®ReadyMix™ PCR Reaction Mix manufactured by the Sigma-Aldrich Co. LLC (St. Louis, MO, USA) that includes 20 mm Tris–HCl, 100 mm KCl, 3 mm MgCl2, 0·002% gelatin, 0·4 mm dNTP mix and 0·06 unit µL−1 of Taq DNA Polymerase. Primers were mixed with purified water and added to the PCR tubes to make a total volume of 20 µL including the DNA template. We included positive controls using samples with known infections as well as negative controls using purified water in place of DNA template.
The PCR products were run out on a 2% agarose gel, and visualized by an ethidium bromide stain under ultraviolet light to check for positive infections. We identified lineages by sequencing the fragments; positive PCR products were sent out for bi-directional sequencing to Eurofins Genomics, Ebersberg, Germany. The sequences were edited and aligned using the program Geneious 7 (Kearse et al. Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012) and we compared the lineages with all sequences from haemosporidian parasites already deposited in Genbank and MalAvi database (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009; Version 2.2.8, accessed on 30 May 2017). Parasite sequences that differed by a single base pair were treated as distinct lineages (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009). When a new haplotype was obtained, we verified that this haplotype was present in at least two individuals in our populations before giving it a new name.
Data analyses
First, we performed phylogenetic analyses using 38 mitochondrial cytochrome b sequences of avian Haemoproteus spp. from passerines and Columbidae species recovered from MalAvi (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009) and Križanauskienė et al. (Reference Križanauskienė, Iezhova, Sehgal, Carlson, Palinauskas, Bensch and Valkiūnas2013). The GenBank accession numbers of these sequences are given in Fig. 2. The sequences were aligned with the two new detected lineages using Geneious 7 (Kearse et al. Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012). All individual sequences were grouped into a consensus that was 509 bp long with two lineages of Plasmodium relictum used as outgroup (Fig. 2). The phylogenetic tree was constructed using the Bayesian phylogenetics as implemented in MrBayes 3.2.2 (Ronquist et al. Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2011) after finding an appropriate model of sequence evolution using the software jModelTest 2 (Guindon and Gascuel, Reference Guindon and Gascuel2003; Darriba et al. Reference Darriba, Taboada, Doallo and Posada2012). A general time-reversible model with gamma-distributed rate variation among sites (GTR + G) was used. Two Markov Chain Monte Carlo simulations were run simultaneously for 2 million generations with sampling every 200 generations, generating 10 000 trees. Of these trees, 25% were discarded as burn-in material. The remaining 7500 trees were used to construct a majority consensus tree (Fig. 2).
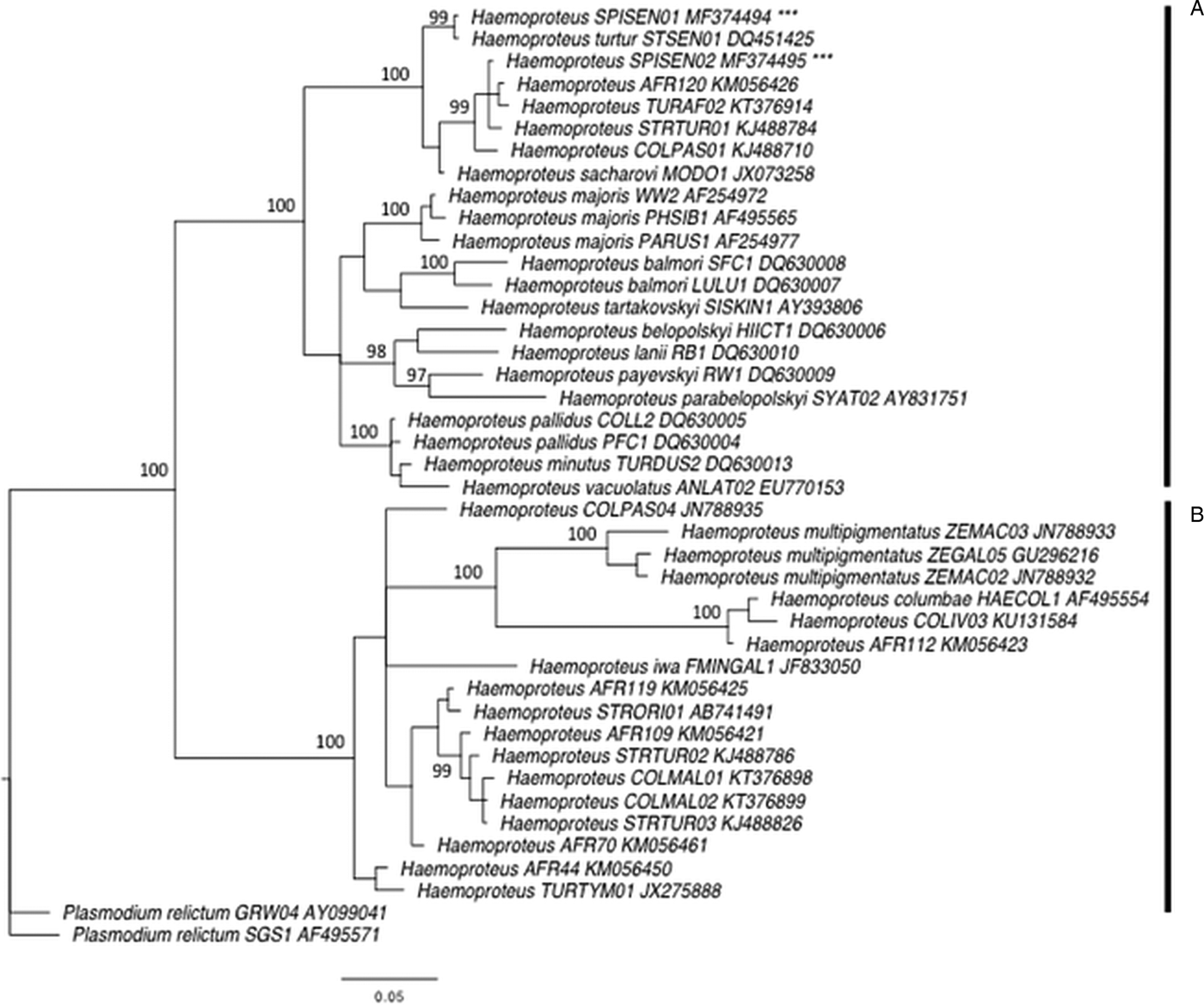
Fig. 2. Bayesian phylogeny of 40 mitochondrial cytochrome b lineages of Haemoproteus spp. and two lineages of Plasmodium relictum used as an outgroup. Posterior probabilities are indicated near the nodes. GenBank accession numbers are given for each lineage. Vertical bar A indicates a clade representing species of Parahaemoproteus subgenus transmitted by biting midges (Ceratopogonidae), and the vertical bar B represents a clade of species of Haemoproteus subgenus transmitted by hippoboscid flies. *** indicates the new lineages SPISEN01 and SPISEN02.
To calculate host specificity indices we used data from available online databases. Host specificity of each parasite lineage from our study was estimated using the modified version of the host specificity index (STD*; Poulin and Mouillot, Reference Poulin and Mouillot2003; Hellgren et al. Reference Hellgren, Pérez-Tris and Bensch2009). This index accounts not only for the number of host species that a parasite lineage can infect, but also for the mean and the variance of taxonomic distances among them.
Finally, the prevalence of blood parasites was investigated for each bird species and parasite genus separately. We conducted Generalized Linear Mixed Models (GLMM), with a logit link-function and binary distribution, to assess the relevance of the geographic position (coastal vs inland), vegetation structure (simple vs stratified), bird age (young vs adult) and the interaction geographic position × vegetation structure, as predictors of the occurrence probability of blood parasites. In these models, we included the sampling site as a random factor. Analyses were conducted using the GLIMMIX procedure in SAS software (SAS Institute, 2008).
Results
Parasite diversity and host specificity
In total, 138 laughing doves (49 adults and 89 young birds) and 235 sparrows (206 adults and 29 young birds) were sampled. Six Haemoproteus lineages were identified (Table 2). Laughing doves were exclusively infected by two new Haemoproteus lineages, hereafter called SPISEN01 and SPISEN02 (Fig. 2). Phylogenetic analyses revealed that these two new Haemoproteus lineages are closely related to lineages recorded in S. senegalensis in Israel, and others from dove species from Africa. Both lineages SPISEN01 and SPISEN02 fall into the clade of Haemoproteus that is transmitted by biting midges (Ceratopogonidae). SPISEN01 was detected in four sites (Kettana, Oum-Errous, El-Manachi and Gafsa) and SPISEN02 was detected in three sites (Nakta, Oum-Errous and El-Manachi). Thus, in doves, 1 to 2 lineages were detected per site (Table 1). The Haemoproteus SPISEN01 lineage was also detected in sparrows from Nakta (N = 3) and Oum-Errous (N = 1). The other Haemoproteus lineages detected in sparrows (4 lineages) were previously described in the literature (Table 2). The number of Haemoproteus lineages detected in sparrows in a site varied from 1 (Gafsa) to 5 (Oum-Errous). With regard to Plasmodium, no infection was recorded in laughing doves. However, 5 Plasmodium lineages were detected in sparrows; 2 to 3 lineages per site. These lineages were previously described in the literature (Table 2).
Table 2. Host specificity index STD* are given for each parasite lineage recovered in southern Tunisia. Number of orders (O), family (F), genus (G) and species (Sp) in which each lineage has been found in the past are given, as well as the number of localities (including Tunisia), with A = Africa, E = Europe, As = Asia, Am = America, O = Oceania and the total number of countries in brackets

The host specificity index of Plasmodium lineages ranged from 3·0 (PAGRI02 restricted to the genus Passer in North Africa) to 90·3 (SGS1 globally distributed; Table 2). Haemoproteus lineages showed reduced host range (STD* from 0 to 5·2; Table 2) but also exhibited a large geographic distribution (Europe and Africa).
Parasite prevalence
Among the 138 sampled laughing doves, 58 (42%) were infected by Haemoproteus (Table 1). Haemoproteus and Plasmodium infection cases were also detected in 76 and 50 sparrows respectively out of the 235 sampled, giving an overall prevalence of 32 and 21%, respectively (Table 1).
Using GLMMs, we found that the occurrence probability of detecting Haemoproteus in laughing doves varied significantly according to geographic position and vegetation structure, while no significant effect was detected for dove age (Table 3). Haemoproteus occurrence probability was higher in inland sites compared with coastal sites, and higher in less-vegetated sites compared with densely vegetated sites (Fig. 3). With regard to sparrows, none of the investigated variables provided a significant predictor of Haemoproteus occurrence probability. However, Plasmodium prevalence was significantly related to vegetation structure, but in a direction opposite to that observed in Haemoproteus infecting laughing doves, with more individuals infected with Plasmodium in densely vegetated sites (Table 4; Fig. 4).

Fig. 3. Plot of estimated Haemoproteus occurrence probability in laughing doves as a function of site geographic position and vegetation structure. Estimated probabilities are the least-squares mean (±s.e.) derived from a GLMM accounting for vegetation structure, geographic position and dove age as fixed effects and site identity as a random factor.

Fig. 4. Plot of Plasmodium occurrence probability in hybrid sparrows as a function of vegetation structure. The estimates are the least-squares mean (±s.e.) derived from a GLMM accounting for vegetation structure, geographic position and sparrow age as fixed effects and site identity as a random factor.
Table 3. Results of GLMM of Haemoproteus occurrence probability in laughing doves as a function of vegetation structure (two classes), geographic position (two classes) and bird age (two classes), accounting for sampling site as a random factor. Sample size = 138. Model generalized χ 2/d.f. = 1·03

Table 4. Results of GLMMs of the occurrence probabilities of Plasmodium and Haemoproteus in hybrid sparrows as functions of vegetation structure (two classes), geographic position (two classes) and bird age (two classes), accounting for site and sampling period as random factors. Sample size = 235. Model generalized χ2/d.f. = 1·01 and 0·99 for Plasmodium and Haemoproteus respectively

Discussion
To our knowledge, this is the first study that explored the diversity, host specificity and prevalence of avian haemosporidian parasites in a desert oasis system. Overall, parasite diversity was low, with only two new Haemoproteus lineages detected in the laughing dove. However, parasite prevalence was relatively high and showed a remarkable spatial variation, in particular, according to the vegetation structure.
Diversity and host specificity
Overall, haemosporidian parasites were found across all sites and the diversity per site in the two host species varied between 4 and 9 lineages. Some lineages were present in all sites (SPISEN01, SGS1, PAHIS1), which was expected given the great potential for bird dispersal and exchange between oases. Indeed, metapopulation dynamics processes are thought to play an important role in determining the distribution of birds in the oasis system (Selmi et al. Reference Selmi, Boulinier and Barbault2002; Selmi et al. Reference Selmi, Boulinier and Faivre2003; Selmi and Boulinier, Reference Selmi, Boulinier, Gillespie and Clague2009). Thus, doves and sparrows have the ability to disperse between sites and therefore to carry parasites from one site to the other.
In doves, we did not detect any Plasmodium infection and the diversity of Haemoproteus was relatively low but not surprising. The MalAvi database and several studies on Columbiformes revealed, in general, a low diversity of Haemoproteus (from 1 to 3 lineages per species), with few exceptions such as the Galapagos dove (Santiago-Alarcon et al. Reference Santiago-Alarcon, Outlaw, Ricklefs and Parker2010), the Mourning dove on Socorro Island (Carlson et al. Reference Carlson, Martínez-Gómez, Valkiūnas, Loiseau, Bell and Sehgal2013) or the Woodpigeons in the UK (Dunn et al. Reference Dunn, Stockdale, Bradford, McCubbin, Morris, Grice, Goodman and Hamer2017). According to the current taxonomy, Haemoproteus is the largest genus of avian haemosporidian parasites (Valkiūnas, Reference Valkiūnas2004) and is categorized into subgenera Haemoproteus and Parahaemoproteus. Species of these subgenera are transmitted by hippoboscid flies (Hippoboscidae) and biting midges (Ceratopogonidae), respectively. The subgenus Parahaemoproteus was considered to not infect doves and pigeons (Columbiformes) until a study investigates the phylogenetic relationships of various Haemoproteus lineages (Križanauskienė et al. Reference Križanauskienė, Iezhova, Sehgal, Carlson, Palinauskas, Bensch and Valkiūnas2013). The authors found that two Haemoproteus species infecting doves H. sacharovi and H. turtur were clustering with many other species of Parahaemoproteus, which were proved experimentally to be transmitted by biting midges, i.e. H. balmorali, H. lanii, H. tartakovskyi and H. parabelopolskyi (Valkiūnas, Reference Valkiūnas2004). Our phylogenetic analyses revealed that the two new lineages found in our survey, SPISEN01 and SPISEN02, were also part of the clade of Parahaemoproteus species (Fig. 2). Our finding corroborates the results of Križanauskienė et al. (Reference Križanauskienė, Iezhova, Sehgal, Carlson, Palinauskas, Bensch and Valkiūnas2013) that doves could be infected by the subgenera Parahaemoproteus transmitted by biting midges. Blood smears would be ideally necessary for further morphological identification.
In sparrows, we detected higher parasite diversity, with five lineages of Plasmodium and five lineages of Haemoproteus. This result is similar to those found in other sparrow populations in another part of the world (Marzal et al. Reference Marzal, Ricklefs, Valkiūnas, Albayrak, Arriero, Bonneaud, Czirják, Ewen, Hellgren, Hořáková, Iezhova, Jensen, Križanauskienė, Lima, de Lope, Magnussen, Martin, Møller, Palinauskas, Pap, Pérez-Tris, Sehgal, Soler, Szöllősi, Westerdahl, Zetindjiev and Bensch2011; Birget and Larcombe, Reference Birget and Larcombe2015; Coon et al. Reference Coon, Garcia-Longoria, Martin, Magallanes, Lope and Marzal2016). Haemoproteus lineages were rather specific to the genus Passer, while Plasmodium lineages were highly generalist, which confirms the general pattern of host specificity of these two genera, Haemoproteus found repeatedly to be more specialist than Plasmodium (Clark and Clegg, Reference Clark and Clegg2017; Loiseau et al. Reference Loiseau, Melo, Lobato, Beadell, Fleischer, Reis, Doutrelant and Covas2017). Identifying factors that influence host specificity and parasite assemblages carried by hosts remains challenging (Gervasi et al. Reference Gervasi, Civitello, Kilvitis and Martin2015), and we believe that more in-depth analyses using higher numbers of host species and sites need to be performed in our study system. These analyses should disentangle the relative effects of host phylogeny and habitat on host specificity, as it has been proposed recently (Clark and Clegg, Reference Clark and Clegg2017).
Parasite prevalence
We found an intriguing pattern for Haemoproteus and Plasmodium prevalence that most likely reveals differences in their respective vectors’ ecology. Haemoproteus prevalence was higher in sites with simple vegetation whereas Plasmodium prevalence was higher in sites harbouring complex vegetation structure. Biting midges and mosquitoes do not have the same ecological requirements, and this has been proposed in several studies to explain the prevalence variation between distinct habitats (Chasar et al. Reference Chasar, Loiseau, Valkiūnas, Iezhova, Smith and Sehgal2009; Sehgal, Reference Sehgal2015).
In fact, a considerable number of biotic and abiotic factors, such as habitat types and aquatic vegetation, seasonal variation and physicochemical parameters of water, affect the abundance and distribution of immature mosquitoes (Gardner et al. Reference Gardner, Anderson, Hamer, Johnson, Varela, Walker and Ruiz2013; Ma et al. Reference Ma, Huang and Leng2016). In addition, different mosquito species choose various types of habitats for diurnal rest, either having a solid background or within vegetation (Clements, Reference Clements1999). As an example, mosquitoes of the genus Culex, which is one of the main genus that transmits avian malaria, are known to rest in the vegetation, and in arid habitats they seem to be concentrated in a limited micro-habitat, at a distance of one meter only from the water (Schlein and Müller, Reference Schlein and Müller2012). These conditions are more satisfied in the two traditional oases included in our study sample, namely Kettana and Gafsa, compared to the remaining less-vegetated sites. Indeed, within these traditional oases, the agricultural production system is based on the use of groundwater for the irrigation of a great diversity of crops that are organized into three main layers: palm trees, fruit trees and herbaceous plants. Complex open irrigation and drainage systems, as well as water collection basins are thus available, which offer plenty of suitable water surfaces for mosquito larval development. Moreover, the high density and stratification of plants create a relatively wet micro-climate that is favourable for mosquitoes during the hottest hours of the day. These areas are also used as resting sites by birds during the night, which may increase the probability of contact between birds and mosquitoes, thus favouring the transmission of Plasmodium.
The ecology of biting midges has been less studied than that of mosquitoes but one study showed that the abundance of midges increased with temperature and decreased with wind speed (Garvin and Greiner, Reference Garvin and Greiner2003). Thus, one possible explanation could be that the stronger winds on the coast would impact the life cycle of biting midges and lead to a lower occurrence of Haemoproteus compared with the inland sites. However, to properly evaluate correlations between climate factors, prevalence and ecology and behaviour of mosquitoes and midges inhabiting the oasis habitat, we would need to sample vectors in a greater number of sites at different distances from the coast.
To conclude, our study was a first exploration of avian haemosporidian parasites in a largely unexplored area and a poorly known habitat system. Although our results identified some factors that may affect parasite prevalence in this area, we believe that a complete study gathering data on a larger number of oases and bird species and taking into account of the habitat requirements and behaviour of vectors is needed to better understand the transmission dynamics of avian parasites in the oasis system. This would also permit a test of whether the island biogeography theory (MacArthur and Wilson, Reference MacArthur and Wilson1967) applies to avian parasites in such island-like systems.
Acknowledgements
We are thankful to Samuel Perret for his help in the field. Authorizations for bird sampling were obtained from the Forest Service of the Tunisian Ministry of Agriculture (permit numbers: 947/17-04-2014 and 1035/04-05-2015). We thank the SMGE platform of CEFE for support for the analyses and Claire Doutrelant via the Languedoc Roussillon French Region Program.
Financial support
This work was conducted as a part of the OISEAU-FCAIMED project financed by the DGRST-CNRS collaboration programme (grant number 14/R0901) between France (T.B.) and Tunisia (S.S). This work was funded by National Funds through FCT Portugal – Foundation for Science and Technology under the IF/00744/2014/CP1256/CT0001 Exploratory Research Project to C.L.








