


Introduction
The red panda (Ailurus fulgens) is a globally threatened species with an estimated >40% population decline over the past 50 years in the wild. It occurs in the wild in India, Nepal, Myanmar and China, where it is mainly distributed in Sichuan, Yunnan, Guizhou and Tibet, with numbers estimated to be 3000–7000 (Wei et al., Reference Wei, Feng, Wang and Hu1999). The International Union for Conservation of Nature upgraded the status of the species from vulnerable to endangered in 2015 (Goździewska-Harłajczuk et al., Reference Goździewska-Harłajczuk, Hamouzová, Klećkowska-Nawrot and Čížek2021). The red panda was also included in a national key protected wildlife list in China by the State Forestry and Grassland Administration and the Ministry of Agriculture and Rural Affairs in 2021 (www.forestry.gov.cn/main/5461/20210205/122418860831352.html).
Disease is one of the most important factors to consider when managing endangered species (Sharma and Achhami, Reference Sharma and Achhami2022). Parasitic diseases are particularly serious given that they can influence the reproductive rate and population abundance in captive wild animals (Albon et al., Reference Albon, Stien, Irvine, Langvatn, Ropstad and Halvorsen2002). Ogmocotyle spp. are intestinal trematodes of various mammalian species and belong to the family Notocotylidae, suborder Pronocephalata, order Plagiorchiida. Ogmocotyle trematodes have been reported in a variety of definitive hosts, including ruminants (Ma et al., Reference Ma, He, Liu, Blair, Liu, Liu and Zhu2016), primates (Coil, Reference Coil1966; Yoshimura et al., Reference Yoshimura, Hishinuma and Sato1969; Iwaki et al., Reference Iwaki, Okada, Seki, Izawa and Sakurai2012), waterfowl (Xu et al., Reference Xu, Zhu, Zhu, Ma, Li and Li2021), panda (Li et al., Reference Li, Karim, Li, Zhang and Zhang2020) and red panda (Pice, Reference Pice1954). The flukes mainly inhabit the small intestine, with large numbers of flukes resulting in ogmocotylosis, which manifests clinically as diarrhoea, mucosal haemorrhage, haemorrhagic enteritis, malnutrition and even death (Ma et al., Reference Ma, He, Liu, Blair, Liu, Liu and Zhu2016). Trematodes of this genus are mainly distributed in Asian countries, including China, Korea, Japan and India (Coil, Reference Coil1966; Eom et al., Reference Eom, Rim and Jang1984; Iwaki et al., Reference Iwaki, Okada, Seki, Izawa and Sakurai2012; Ma et al., Reference Ma, He, Liu, Blair, Liu, Liu and Zhu2016).
Morphological descriptions have had a key role in identifying and differentiating parasites, with some synonym species within the genus Ogmocotyle from various hosts being reported. Sey and Graber (Reference Sey and Graber1991) conducted a systematic morphological description of Ogmocotyle trematodes known at that time, and identified 6 valid species in the genus: Ogmocotyle africanum, Ogmocotyle ratti, Ogmocotyle fujianensis, Ogmocotyle indica (synonym Ogmocotyle capricori), Ogmocotyle sikae (synonym Ogmocotyle pygargi) and Ogmocotyle ailuri (synonym Ogmocotyle macacae) (Sey and Graber, Reference Sey and Graber1991; Yang and Zhang, Reference Yang and Zhang2013). Two species, O. indica and O. ailuri, were reported to infest red pandas. The main morphological identification characteristics distinguishing the 2 species were the location of the cirrus pouch and testis shape. In O. indica, the cirrus pouch occurs longitudinally in one-third of the body, and the testis is either kidney shaped or oval with a smooth surface; by contrast, in O. ailuri, the cirrus pouch lies on the front half of the body in an arc shape, and the testis is lobular (Sey and Graber, Reference Sey and Graber1991).
The emergence of molecular data has resolved many of the limitations associated with morphological approaches for comparing related species and any further in-depth analyses. For example, genus Orientobilharzia was originally called Schistosoma, and then re-named Ornithobilharzia based upon the presence of numerous testes. Subsequently, it transferred to the new genus Orientobilharzia to accommodate blood flukes whose definitive hosts were mammals (Wang et al., Reference Wang, Chen, Zhao, Chen, Zhai, Li and Zhu2009). Aldhoun and Littlewood (Reference Aldhoun and Littlewood2012) then proposed on molecular grounds that the genus Orientobilharzia was in fact a junior synonym of Schistosoma (Aldhoun and Littlewood, Reference Aldhoun and Littlewood2012). In addition, controversy has surrounded whether the hookworms Bunostomum trigonocephalum and Bunostomum phlebotomum represent different species or strains, given significant similarities in morphology. However, molecular data were used to show that B. trigonocephalum and B. phlebotomum represent distinct but closely related species (Gao et al., Reference Gao, Zhao, Liu, Zhang, Zhang, Wang, Chang, Wang and Zhu2014). Thus, more molecular data are needed to reassess traditional morphological classifications.
As a result of maternal inheritance, an apparent lack of recombination, rapid evolutionary rates and comparatively conserved genomic structures, mitochondrial (mt) genomes are highly suitable for trematode taxonomy, population genetics and systematics studies (Ichikawa-Seki et al., Reference Ichikawa-Seki, Peng, Hayashi, Shoriki, Mohanta, Shibahara and Itagaki2017; Le et al., Reference Le, Pham, Doan, Le, Saijuntha, Rajapakse and Lawton2020; Valentyne et al., Reference Valentyne, Spratt, Aghazadeh, Jones and Šlapeta2020). Although 6 valid species have been recognized in the genus Ogmocotyle, only the O. sikae mt genome is currently available in GenBank. The paucity of molecular data is a limitation for population genetic and phylogenetic studies of these parasites.
Therefore, the objectives of the current study were to determine the complete O. ailuri mt genome, compare the mtDNA genome with those previously reported for Pronocephalata trematodes, and reconstruct the phylogenetic relationships with other trematodes based on a dataset comprising the concatenation of 12 protein-coding gene (PCG) nucleotide sequences. The results provide a molecular resource for further studies of Pronocephalata taxonomy, population genetics and systematics.
Materials and methods
Parasites and total genomic DNA isolation
An autopsy was performed to determine the cause of death in red pandas in Longsha Zoological and Botanical Garden, Heilongjiang Province, China. Trematodes were collected from the small intestine of red panda following Wildlife Protection Law protocols of the People's Republic of China (Fang et al., Reference Fang, Liu, Wu, Wei and Wang2022). The trematodes were identified to the species level based on previously described morphological keys (Sey and Graber, Reference Sey and Graber1991) (Fig. S1). They were then fixed in 75% ethanol and stored at −20°C until use. Total genomic DNA was isolated from a single fluke using a sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification using Genomic DNA Purification System (Promega, Madison, Wisconsin, USA) according to the manufacturer's protocol.
Sequencing and mt genome assembly
Illumina paired-end shotgun libraries were prepared using the standard protocol of the Nextera™ DNA Sample Prep Kit (Epicentre, Madison, Wisconsin, USA) and sequenced using an Illumina NovaSeq sequencing platform (Personal Biotechnology Co, Ltd, Shanghai, China) using 2 × 100 cycles. Raw sequence data were deposited into the Short Read Archive database (www.ncbi.nlm.nih.gov/sra/) under accession number PRJNA896356. Clean data without sequencing adapters were assembled using NOVOPlasty software (Dierckxsens et al., Reference Dierckxsens, Mardulyn and Smits2017) with the parameters of the genome range (13 000–15 000) and k-mer 39. The completeness of the mt genome assembly was further verified by polymerase chain reaction and Sanger sequencing using 4 pairs of primers designed on the basis of conserved regions (Table S1; Fig. S2).
Sequence analysis and gene annotation
Mt genome sequences were used for a BLAST search of the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast) (Altschul et al., Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997). Twelve PCGs were initially identified using ‘ORF Finder’ through NCBI and the MITOS Web Server (Rombel et al., Reference Rombel, Sykes, Rayner and Johnston2002) to specify the mt genetic code of invertebrates. The MITOS Web Server was then used to calculate the potential stem-loop secondary structures within these tRNA gene sequences (Bernt et al., Reference Bernt, Donath, Jühling, Externbrink, Florentz, Fritzsch, Pütz, Middendorf and Stadler2013). The codon usage of the 12 PCGs was analysed using the invertebrate genetic code and the Codon Usage web server (www.bioinformatics.org/sms2/codon_usage.html). An analysis of compositional skews was conducted using equations (1) and (2) (Perna and Kocher, Reference Perna and Kocher1995):


The relative synonymous codon usage of the 12 PCGs was calculated using the Sequence Manipulation Suite (www.detaibio.com/sms2/codon_usage.html) (Stothard, Reference Stothard2000). A sliding window analysis [window length = 300 base pairs (bp), step size = 10 bp] was conducted using DnaSP v.5 (Librado and Rozas, Reference Librado and Rozas2009) to assess nucleotide diversity Pi (π) between 12 PCGs from 14 Pronocephalata mt genomes. Nucleotide diversity was plotted against the mid-point positions of each window, and gene boundaries were identified. The differences between nucleotide and amino acid sequences were calculated using MEGA 11.0 and MegAlign (Burland, Reference Burland2000; Tamura et al., Reference Tamura, Stecher and Kumar2021). A gene map of the mt genome was constructed using the online mitochondrial visualization tool OrganellarGenomeDRAW (Lohse et al., Reference Lohse, Drechsel, Kahlau and Bock2013). To determine the occurrence of Plagiorchiida spp. gene recombination, the gene arrangement of the O. ailuri mt genome was compared with those of 47 Plagiorchiida species currently available. The circular mt genomes were linearized at the 5′ end of their cox3 genes in the H-strand direction, as previously reported (Gao et al., Reference Gao, Mao, Li, Sun, Gao, Zhang, Jin, An, Zhang, Zhang, Wei, Lan and Wang2021).
Phylogenetic analysis
Phylogenetic relationships among the 48 representative members of Plagiorchiida available in GenBank (Table S2) were determined based on concatenated amino acid sequences of 12 PCGs, using Postharmostomum commutatum (NC_044643) as the outgroup. The PCG amino acid sequences were aligned using MAFFT v7.471 (Katoh and Standley, Reference Katoh and Standley2013), and the alignment was processed for elimination of poorly aligned sites and divergent regions using the GBlocks Server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html).
Phylogenetic trees were reconstructed using Bayesian inference (BI) methods. The best-fit substitution model for phylogenetic analysis of the amino acid alignment was determined using jModeltest under Akaike information criterion, and identified as the SYM + I + G model (Darriba et al., Reference Darriba, Taboada, Doallo and Posada2012). MrBayes v3.2.6 (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001) was used to conduct BI analysis. This analysis was performed for 10 000 000 generations, in 2 simultaneous runs, with 4 chains (3 heated and 1 cold), to catalyse swapping among the Markov-chain Monte Carlo chains. Trees were sampled every 1000 generations. Tracer v1.6 software (http://tree.bio.ed.ac.uk/software/tracer/) was used to investigate the convergence of sampled parameters and potential autocorrelation (effective sample size for all parameters >200). In addition, the average standard deviations of the split frequencies were checked between both runs (<0.01). Bayesian posterior probabilities were obtained from the 50% majority-rule consensus of the post-burn-in trees sampled at stationarity after removing the first 25% of trees as a ‘burn-in’ stage. The final phylogenetic tree was graphically visualized and edited using FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
General features of the O. ailuri mt genome
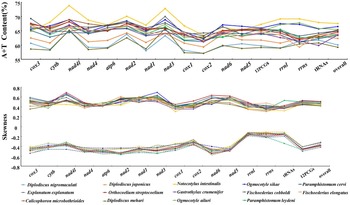
The O. ailuri mt genome (GenBank accession number OP414758) was a typical closed circular molecule, 14 642 bp in size. It contained 12 PCGs (cox1–3, nad1–6, nad4L, cytb and atp6), 22 tRNA genes (1 for each amino acid and 2 each for leucine and serine), 2 rRNA genes (rrnL and rrnS) and 2 non-coding regions [NCRs; long (LNCR) and short (SNCR)], but lacked atp8 (Fig. 1; Table 1). All the genes were transcribed in the same direction. There were 23 intergenic spacers, ranging from 1 to 37 bp, and 2 locations with gene overlaps, ranging from 1 to 40 bp (Table 1). The nucleotide composition of the entire mt genome of O. ailuri was biased towards A and T, with an overall AT content of 65.51% (Fig. 2A).

Figure 1. Gene map of the mt genome of Ogmocotyle ailuri. All 22 tRNAs are designated by the 1-letter code for the corresponding amino acid, with numerals differentiating each of the 2 leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families UCN and AGN, respectively), LNCR and SNCR refer to long and short non-coding region. All genes are transcribed in the anticlockwise direction.

Figure 2. A + T content and nucleotide skew of genes, individual elements and the complete mt genome of 14 Pronocephalata trematodes. Lines colour and point represent a specific Pronocephalata species.
Table 1. Features of the mt genome of Ogmocotyle ailuri

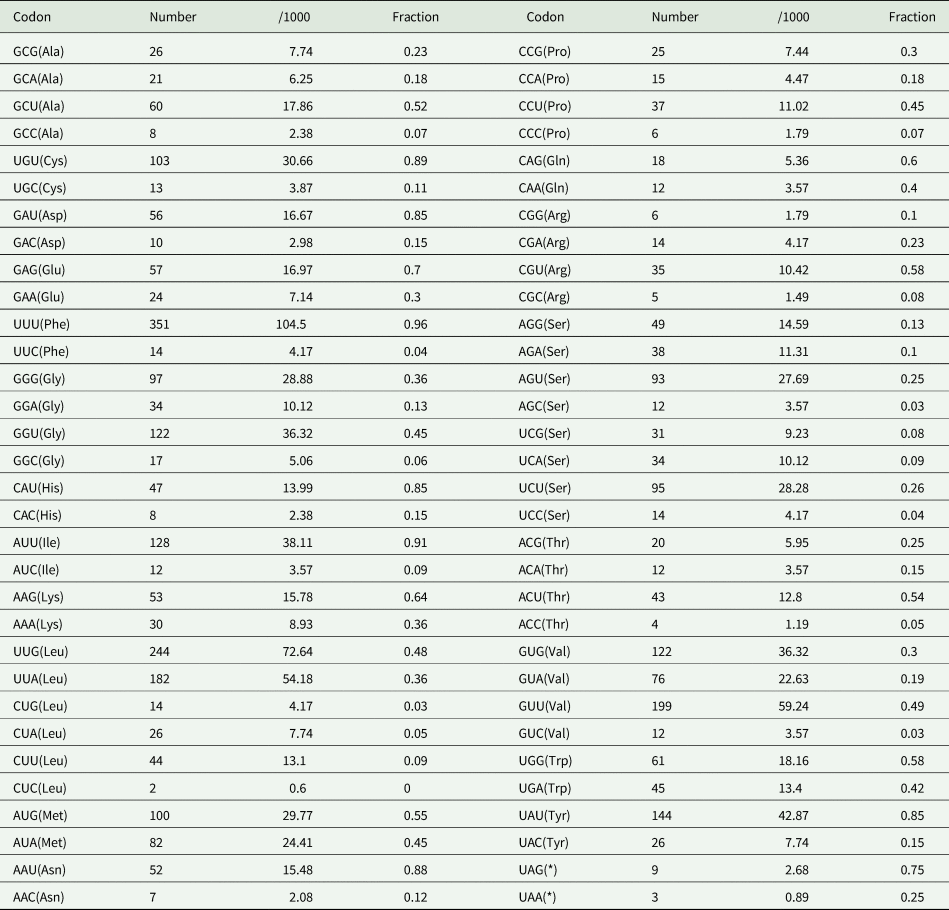
In the O. ailuri mt genome, the 12 PCGs accounted for 10 098 bp and encoded 3354 amino acids, excluding the termination codons. The average AT content of the 12 PCGs was 64.8% (Fig. 2A). The initiation and termination codons of the PCGs in the O. ailuri mt genome are listed in Table 1. The most common initiation codon for O. ailuri was ATG (9 of 12 PCGs), followed by ATA (2 of 12 PCGs) and GTG (1 of 12 PCGs). Two types of termination codon, TAG (9) and TAA (3), occurred in the O. ailuri mt genome. The codon usage analyses of the 12 PCGs in the mt genome are summarized in Table 2. The most frequent amino acid was Phe (TTT, 10.46%), followed by Leu (TTG, 9.25%), Val (GTT, 5.94%) and Leu (TTA, 5.44%). The least frequently used were Leu (CTC, 0.06%), Thr (ACC, 0.12%) and Arg (CGC, 0.15%) (Table 2).
Table 2. Codon usage analysis of 12 PCGs in the mitochondrial genome of Ogmocotyle ailuri
The 22 tRNA genes identified in the O. ailuri mt genome ranged from 60 to 70 bp. The total length of the 22 tRNA genes was 1457 bp, and the AT content was 65.80%. RrnL was located between trnT and trnC, and rrnS between trnC and cox2. rrnL and rrnS were 995 and 748 bp in O. ailuri, respectively, and had an AT content of 67.14 and 67.78%, respectively. The mt genome of O. ailuri contained 2 NCRs, LNCR located between trnG and trnE, and SNCR located between trnE and cox3. LNCR and SNCR were 677 and 487 bp in O. ailuri, respectively, and had an AT content of 69.40 and 66.47%, respectively.
Comparative analysis of O. ailuri mt genome within Pronocephalata trematodes
The complete mt genome of O. ailuri was relatively large among Pronocephalata species, although was shorter than that of Diplodiscus nigromaculati (14 697 bp) and Gastrothylax crumenifer (14 801 bp), but longer than that of other Pronocephalata (Table 3). The gene transcription direction of the complete mt genomes of O. ailuri was the same as all Pronocephalata mt genomes reported thus far. The rank order of the 12 PCGs of O. ailuri by length was as follows: nad5 > cox1 > nad4 > cytb > nad1 > nad2 > cox3 > cox2 > atp6 > nad6 > nad3 > nad4L.
Table 3. Comparison of 12 PCGs among Ogmocotyle ailuri and other suborder Pronocephalata trematodes

Note: Dn, Diplodiscus nigromaculati; Dm, Diplodiscus mehari; Dj, Diplodiscus japonicus; Ni, Notocotylus intestinalis; Ogs, Ogmocotyle sikae; Oga, Ogmocotyle ailuri; Pc, Paramphistomum cervi; Cm, Calicophoron microbothrioides; Ee, Explanatum explanatum; Ors, Orthocoelium streptocoelium; Gc, Gastrothylax crumenifer; Fc, Fischoederius cobboldi; Fe, Fischoederius elongatus; Pl, Paramphistomum leydeni.
The A + T content of O. ailuri mt genomes was lower than those of O. sikae and Notocotylus intestinalis, which was the lowest in the Notocotylidae reported thus far (Fig. 2A). The AT bias of the O. ailuri mt genome was consistent with other Pronocephalata. The O. ailuri AT-skew values were negative, and the GC-skew values were positive, ranging from −0.486 (nad6) to −0.144 (rrnL) and 0.375 (cytb) to 0.604 (nad1), respectively (Fig. 2B).
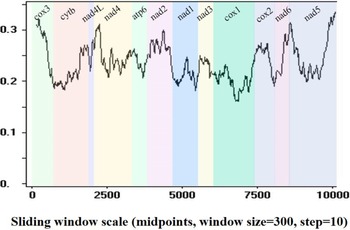
Sequence identities and sliding window analyses were used to determine the diversity and mutation rate of mt genes among 14 Pronocephalata. Sequence identities of the 12 PCGs from the 14 Pronocephalata species were 68.2–90.3% at the nucleotide level and 51.5–95.3% at the amino acid level. The complete mt genome nucleotide identities among the 14 trematodes ranged from 64.3 to 89.8% (Table 3). Among the 12 PCGs, cox3 had the fewest similarities among the 14 species, whereas cox1 had the highest similarity (Table 3). A sliding window analysis of the concatenated nucleotide sequences of 12 PCGs showed obvious differences in 12 PCGs from the 14 Pronocephalata trematodes. By computing the number of variable positions per unit length of gene, the curve indicated that cox1 (0.161) was the least variable gene, whereas cox3 (0.321) and nad5 (0.336) showed the highest sequence variation (Fig. 3).

Fig. 3. Sliding window analysis of the complete mt genome sequences of 14 Pronocephalata trematodes. A sliding window of 300 bp (in 10 bp overlapping steps) was used to estimate nucleotide diversity Pi (π) across the alignments. Nucleotide diversity was plotted against the mid-point positions of each window. Each gene boundary is identified.
Gene arrangements
The sequenced mt genome of the 48 Plagiorchiida showed 5 types of gene arrangement, with that of the O. ailuri mt genome being type I (Fig. 4). The gene arrangements of Plagiorchiida shared an identical arrangement based on 12 PCGs and 2 rRNA genes (the order: cox3 > cytb > nad4L > nad4 > atp6 > nad2 > nad1 > nad3 > cox1 > rrnL > rrnS > cox2 > nad6 > nad5), and the gene rearrangement events only occurred in transposed tRNAs.

Figure 4. Mt genome arrangement in Ogmocotyle ailuri compared with those in Plagiorchiida trematodes. The circular mt genomes were linearized at the 5′ end of cox3 gene for illustration purpose. Non-coding regions were not shown.
The rearrangement events mainly occurred in 2 gene blocks (trnN-trnP-trnI-trnK and trnG-trnE) of the mt genome in Plagiorchiida. Compared with type I rearrangements, trnG and trnE were interchanged within nad5 and cox3 in type II rearrangements. Compared with type II rearrangements, trnP and trnI were interchanged with nad1 and nad3 in type III rearrangements. Compared with type III rearrangements, trnE and trnG were interchanged with nad5 and cox3 in type IV rearrangements. trnE between nad5 and cox3 in type IV rearrangements was translocated between nad1 and nad3 in type V rearrangements. Additionally, trnY, which is between nad6 and nad5 in type IV rearrangements, was translocated between nad5 and cox3 in type V rearrangements.
Phylogenetic analysis
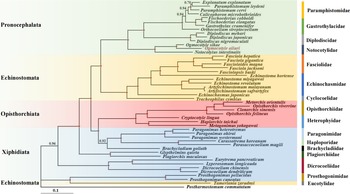
BI approaches were used to estimate the phylogenetic position of O. ailuri within 47 other Plagiorchiida trematodes based on the concatenated amino acid sequences of 12 PCGs (Fig. 5). The result showed that the Pronocephalata and Opisthorchiata suborders form independent monophyletic groups in the phylogenetic tree. Xiphidiata trematodes have a complicated taxonomic relationship, although each family clustered together in 1 branch; however, Xiphidiata trematodes formed 4 lineages rather than a monophyletic group. Prosthogonimus pellucidus and Prosthogonimus cuneatus clustered together with Tamerlania zarudnyi to form a group on the outermost branch, which was next to other Plagiorchiida trematodes. Except for T. zarudnyi (Eucotylidae), 12 Echinostomata trematodes clustered together to form a group.

Figure 5. Phylogenetic relationships of Ogmocotyle ailuri with other 48 Plagiorchiida trematodes based on concatenated amino acid sequences of 12 protein-coding genes analysed by BI using Postharmostomum commutatum as the outgroup. Posterior probability values are indicated. Suborders and families are highlighted by individual colours. Circles indicate BI = 1.0, other values are given above the nodes.
The branch of Pronocephalata includes the clades Paramphistomidae, Gastrothylacidae, Diplodiscidae and Notocotylidae. Paramphistomidae, Gastrothylacidae and Diplodiscidae clustered together to form a group, with Notocotylidae forming another group. In the Notocotylidae branch, O. ailuri isolated from this study was closer to O. sikae than to N. intestinalis.
Discussion
In this study, the entire mt genome of O. ailuri was sequenced for the first time. The mt genome of O. ailuri was the classical structure of trematodes, contained 12 PCGs, 22 tRNA genes, 2 rRNA genes and 2 non-coding regions. There were a 40 bp gene overlap between nad4L and nad4 in O. ailuri mt genome, the result was consistent with most Plagiorchiida, except Dicrocoelium dendriticum (8 bp), Dicrocoelium chinensis (8 bp), Lyperosomum longicauda (37 bp), Paragonimus westermani (14 bp) and N. intestinalis (0 bp) (Biswal et al., Reference Biswal, Chatterjee, Bhattacharya and Tandon2014; Liu et al., Reference Liu, Yan, Otranto, Wang, Zhao, Jia and Zhu2014; Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang, Zhu and Ma2020; Xu et al., Reference Xu, Zhu, Zhu, Ma, Li and Li2021).
A common feature of the mt genome in most metazoans is a bias towards a higher representation of A and T nucleotides, which leads to subsequent biases in the corresponding encoded amino acids (Hu et al., Reference Hu, Zhang, Sun and Bu2020). The AT content of the entire mt genome and the 12 PCGs was 65.51 and 64.8%, respectively, and the result was consistent with other Plagiorchiida trematodes biased towards A and T (Atopkin et al., Reference Atopkin, Semenchenko, Solodovnik, Ivashko and Vinnikov2021; Ivashko et al., Reference Ivashko, Semenchenko, Solodovnik and Atopkin2022). The negative AT-skew indicated a higher incidence of T compared with A nucleotides, and the positive GC-skew indicated that G was more abundant than C (Le et al., Reference Le, Nguyen, Nguyen, Doan, Agatsuma and Blair2019). The AT-skew values were negative, and the GC-skew values were positive in O. ailuri mt genome; these trends in AT- and GC-skew were similar to those of other Pronocephalata (Yan et al., Reference Yan, Wang, Lou, Li, Blair, Yin, Cai, Dai, Lei, Zhu, Cai and Jia2013; Xu et al., Reference Xu, Zhu, Zhu, Ma, Li and Li2021).
Three types of initiation codon (ATG, ATA and GTG) and 2 types of termination codon (TAG and TAA) occurred in the O. ailuri mt genome; these codons were commonly used in Plagiorchiida trematodes. The result was consistent with most Plagiorchiida trematodes, but different from some Plagiorchiida trematodes, such as Eurytrema pancreaticum, L. longicauda and Plagiorchis maculosus that utilized incomplete stop codon T as termination codon (Chang et al., Reference Chang, Liu, Gao, Zheng, Zhang, Duan, Yue, Fu, Su, Gao and Wang2016; Suleman et al., Reference Suleman, Ma, Khan, Tkach, Muhammad, Zhang and Zhu2019, Reference Suleman, Khan, Tkach, Muhammad, Zhang, Zhu and Ma2020). Two NCRs were identified in the O. ailuri mt genome, which is consistent with other related Plagiorchiida, such as Fischoederius elongatus, G. crumenifer and Paragonimus ohirai (Yang et al., Reference Yang, Zhao, Wang, Feng, Tan, Lei, Zhao, Hu and Fang2015, Reference Yang, Wang, Chen, Feng, Shen, Hu and Fang2016; Le et al., Reference Le, Pham, Doan, Le, Saijuntha, Rajapakse and Lawton2020). However, this result is inconsistent with other related Plagiorchiida, such as Diplodiscus japonicus and Tracheophilus cymbius, in which only 1 NCR has been identified in the mt genome (Li et al., Reference Li, Ma, Lv, Hu, Qiu, Chang and Wang2019; An et al., Reference An, Qiu, Lou, Jiang, Qiu, Zhang, Li, Zhang, Wei, Chen, Gao and Wang2022).
The gene cox1 was considered to be a useful barcode for metazoans, and widely used for trematode studies (Mioduchowska et al., Reference Mioduchowska, Czyż, Gołdyn, Kur and Sell2018). The mitochondrial genes cox1 have also been used to study the population genetic structure of trematode on a local and global scale (Djuikwo-Teukeng et al., Reference Djuikwo-Teukeng, Kouam Simo, Allienne, Rey, Njayou Ngapagna, Tchuem-Tchuente and Boissier2019; Shumenko and Tatonova, Reference Shumenko and Tatonova2022; Wang et al., Reference Wang, Zhang, Zhang, Li, Shang, Ning, Ji, Qiao, Meng and Cai2022). In the present study, the sequence identities and sliding window analysis indicated cox1 was the most conserved gene among 12 PCGs in 14 Pronocephalata species; this result was similar to those from a study of L. longicauda, O. sikae, N. intestinalis, Paramphistomum leydeni and T. zarudnyi (Ma et al., Reference Ma, He, Liu, Zhou, Liu, Liu and Zhu2015, Reference Ma, He, Liu, Blair, Liu, Liu and Zhu2016; Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang, Zhu and Ma2020, Reference Suleman, Muhammad, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021; Xu et al., Reference Xu, Zhu, Zhu, Ma, Li and Li2021).
Gene arrangement in mt genomes provides a source of information for phylogenetic inference (Le et al., Reference Le, Humair, Blair, Agatsuma, Littlewood and McManus2001). Wey-Fabrizius et al. (Reference Wey-Fabrizius, Podsiadlowski, Herlyn and Hankeln2013) reviewed how mt genome data contribute to the phylogeny debates within Platyzoan, which showed that the gene order of PCGs and rRNA genes within Platyhelminthes were highly conserved with exceptions of partial Monogenea and Seriata trematodes. For majority trematodes, the gene order is cox3 > cytb > nad4L > nad4 > atp6 > nad2 > nad1 > nad3 > cox1 > rrnL > rrnS > cox2 > nad6 > nad5 (Wey-Fabrizius et al., Reference Wey-Fabrizius, Podsiadlowski, Herlyn and Hankeln2013), which was well supported by the present study. Solà et al. (Reference Solà, Álvarez-Presas, Frías-López, Littlewood, Rozas and Riutort2015) reported the mt genomes and gene order of 2 free-living flatworm species (Crenobia alpina and Obama sp.), and compared with other relevant helminthes, it was found that the gene order among free-living flatworms differs considerably from the parasitic flatworms. However, the gene order of both free-living and parasitic flatworms was highly conservative with only a few exceptions in each kind of flatworm (Solà et al., Reference Solà, Álvarez-Presas, Frías-López, Littlewood, Rozas and Riutort2015). The phenomenon was also found in Plagiorchiida trematodes in the present study; 48 Plagiorchiida trematodes available in GenBank shared an identical gene order based on PCGs and rRNA genes.
The gene rearrangement showed 5 types among 48 Plagiorchiida trematodes, and only occurred in transposed tRNAs. This was different from Schistosoma trematodes, the gene arrangement of which was radically altered by PCGs (Le et al., Reference Le, Humair, Blair, Agatsuma, Littlewood and McManus2001; Zhang et al., Reference Zhang, Li, Zou, Wu, Li, Jakovlić, Zhang, Chen and Wang2019). In addition, the rearrangement events mainly occurred in nad1-N-P-I-K-nad3 and nad5-G-E-cox3 gene blocks of the mt genome in Plagiorchiida. Zhang et al. reported that type I (trnG-trnE) and type II (trnE-trnG) rearrangements were generated by the gene recombination of ancestral gene rearrangements in Trematoda, and hypothesized that type II evolutionarily pre-dates type I gene rearrangements (Zhang et al., Reference Zhang, Li, Zou, Wu, Li, Jakovlić, Zhang, Chen and Wang2019). Thus, these gene rearrangements might be an important molecular marker of Plagiorchiida trematode evolution.
Previous research reported the phylogenetic position of O. sikae based on the complete mt genome (Ma et al., Reference Ma, He, Liu, Blair, Liu, Liu and Zhu2016). However, molecular data were rare at that time, and only morphological-based approaches could not fully reflect the phylogenetic relationship within Plagiorchiida trematodes. Thus, the phylogenetic relationships with other Pronocephalata trematodes previously reported were reconstructed based on the concatenation of 12 PCGs. In the phylogenetic tree, most of trematodes were consistent with the current taxonomic status of Plagiorchiida (Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003). Notably, except for T. zarudnyi (Eucotylidae), 12 Echinostomata trematodes clustered together to form a group. However, T. zarudnyi clustered together with Prosthogonimidae flukes, a result consistent with previous phylogenetic assessments (Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022). Thus, the current results showed that suborder Echinostomata is polyphyletic. Eucotylidae was considered to belong to Cyclocoeloidea (Strigeida) by Kanev et al., while Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) showed that Eucotylidae should belong to the suborder Xiphidiata as a long-branched clade based on nuclear ribosomal DNA (Kanev et al., Reference Kanev, Radev, Fried, Gibson, Jones and Bray2002; Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003). This hypothesis was also supported by the present study based on the complete mt genome of O. ailuri.
In the branch of Pronocephalata, 3 family trematodes (Paramphistomidae, Gastrothylacidae, Diplodiscidae) clustered together to form a group, and 1 family trematode (Notocotylidae) forming another group. A similar result was also reported in previous studies using the mtDNA genome (Atopkin et al., Reference Atopkin, Semenchenko, Solodovnik, Ivashko and Vinnikov2021; An et al., Reference An, Qiu, Lou, Jiang, Qiu, Zhang, Li, Zhang, Wei, Chen, Gao and Wang2022; Ivashko et al., Reference Ivashko, Semenchenko, Solodovnik and Atopkin2022). However, this was not consistent with those of a previous study using ITS2 for phylogenies, which demonstrated a close genetic affinity of Notocotylidae with Bucephalidae (of the order Strigeidida), rather than with Paramphistomidae and Gastrothylacidae (Xu et al., Reference Xu, Zhu, Zhu, Ma, Li and Li2021).
In the Notocotylidae branch, O. ailuri was closer to O. sikae than to N. intestinalis, which is consistent with the current taxonomic status of Notocotylidae. These data provide novel and useful genetic markers for studying systematics and population genetics, but only 3 complete mt genomes have been published for Notocotylidae species in GenBank. Therefore, more mt genomes should be sequenced to re-examine the phylogenetic relationships of Pronocephalata and other trematodes.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182023000379.
Data availability
All data generated or used during the study appear in the submitted article.
Acknowledgements
We thank Longsha Zoological and Botanical Garden for collecting the trematode specimens, and we also thank International Science Editing (http://www.international-scienceediting.com) for editing this manuscript.
Author's contribution
The research was designed by J.-F. Gao and C.-R. Wang. Material preparation and data collection were performed by W. Wei, B. Jia, J. Zhang and B. Li. Y.-Y. Chen, Y.-Y. Sun and M.-R. Hou analysed the data. The first draft of the manuscript was written by A.-H. Zhang. X.-W. Liu, J.-W. Wang and X.-H. Zhang critically revised the manuscript.
Financial support
This work was supported by the National Natural Science Foundation of China (32172886), Heilongjiang Provincial Natural Science Foundation of China (LH2021C071), Heilongjiang Province Postdoctoral Science Foundation (LBH−Z19191), Heilongjiang Bayi Agricultural University support Program for San Heng San Zong (ZRCQC202204) and Personnel Foundation of Heilongjiang Bayi Agricultural University (XYB202108).
Conflict of interest
None.
Ethical standards
The autopsy of red panda and collection of trematode specimens following the standards and procedures by the Animal Ethics Committee of Heilongjiang Bayi Agricultural University (Approval ID: WKJXY-2022020).








 Open access
Open access