


Introduction
Under normal conditions, the adipose tissue is able to fine-tune a series of neuroendocrine signals to precisely adapt the balance between TAG synthesis (lipogenesis) and breakdown (lipolysis) to meet physiological needs. In higher eukaryotes adipocyte TAG depots represent the major energy reserve of the organism as a result of the constant flux between lipolysis and re-esterification( Reference Frayn 1 – Reference Girousse and Langin 5 ). During energy surplus adipocytes accomodate the excess fuel as TAG for retrieval during periods of negative energy balance such as fasting, starvation or long-term exercise. The hydrolysis of TAG produces NEFA and glycerol that are released into the vasculature for use as energy substrates by other organs. Since TAG are not able to pass through biological membranes they need to be cleaved by TAG hydrolases, also termed lipases, before entering or exiting cells( Reference Zechner, Zimmermann and Eichmann 6 , Reference Young and Zechner 7 ). The ability to rapidly mobilise lipid reserves as NEFA to subvene energy demands represents a highly adapted metabolic response. In addition, the balance between the lipogenic drive and the lipolytic rate prevents an exaggerated elevation of plasma NEFA, which is considered a key aetiological factor in the development of insulin resistance( Reference Guilherme, Virbasius and Puri 8 , Reference Girousse, Tavernier and Valle 9 ). Thus, the fat-storing ability of adipocytes prevents the appearance of lipotoxicity (lipid-induced dysfunction) and lipoapoptosis (lipid-induced programmed cell death) in other tissues (especially skeletal muscle and liver)( Reference Virtue and Vidal-Puig 10 – Reference Unger, Scherer and Holland 12 ). While the metabolic importance of lipolysis remains unchanged, established models of adipose tissue lipolysis have undergone substantial revision lately. Notably, adipocyte lipid droplets are now considered dynamic organelles critical for the handling of lipid stores, containing specific structural proteins and lipid-metabolising enzymes involved in the modulation of both basal and hormone-regulated lipolysis( Reference Gross, Miyoshi and Hosaka 13 – Reference Walther and Farese 17 ). Current knowledge in this field is reviewed from the broader perspective of providing an overview of the classic lipolytic factors as well as by focusing on the recently identified influence of the subcellular compartmentalisation of lipases, the relevance of lipid droplet proteins and lipid-binding proteins, as well as the activation of the different signalling pathways together with their regulation.
Control of lipolysis
Lipolysis constitutes the catabolic process leading to the breakdown of TAG into glycerol and NEFA in the adipose tissue( Reference Arner 2 ). Basal lipolytic activity of adipocytes is conditioned by sex, age, physical activity, fat depot location, species and genetic variance, whereas stimulated adipocyte lipolysis is regulated by multiple factors, which are depicted in Fig. 1 ( Reference Langin, Lucas and Lafontan 18 , Reference Frühbeck and Gómez-Ambrosi 19 ). Interestingly, fat cell lipolysis exhibits species-unique characteristics based on the predominance of specific receptors and their relative density and expression( Reference Bousquet-Melou, Galitzky and Lafontan 20 , Reference Langin, Portillo and Saulnier Blache 21 ). A decreased lipolytic rate is observed both in the early years of life and the elderly in relation to the action of catecholamines and insulin( Reference Blaak 22 – Reference Herrera 25 ). For the same BMI, women exhibit higher NEFA circulating concentrations than men due to their constitutively larger fat depots and subcutaneous adipocytes( Reference Blaak 26 ). Regional differences in the sensitivity to catecholamine-stimulated and insulin-inhibited lipolysis further underlie these sex-specific characteristics, which will be described more extensively below. An increased basal lipolysis together with an enhanced lipolytic sensitivity to catecholamines take place during situations of negative energy balance such as fasting, starvation or semi-starvation, contributing to the increased mobilisation of NEFA from adipocytes and the subsequent fat mass loss when maintained over time( Reference Arner 2 ). As in situations of energy deprivation, during prolonged exercise plasma NEFA increase in response to the elevated release of catecholamines and decreased production of insulin( Reference Lange 27 ). Both short- and long-term endurance training make adipocytes more sensitive to catecholamine stimulation via adrenoceptor signal transduction changes( Reference Mauriege, Prud'Homme and Marcotte 28 – Reference De Glisezinski, Marion-Latard and Crampes 31 ).
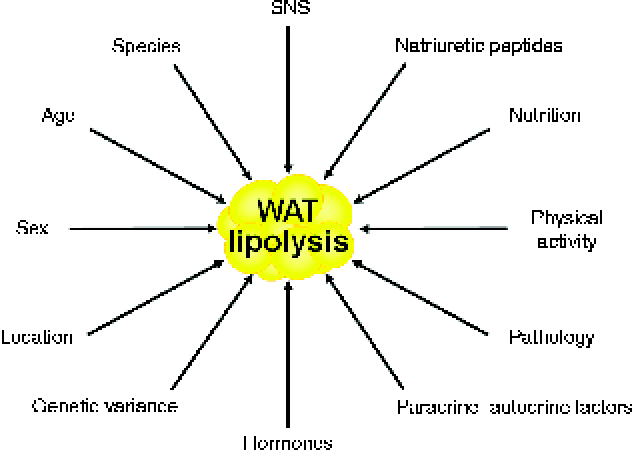
Fig. 1 Main factors influencing adipocyte lipolysis. SNS, sympathetic nervous system; WAT, white adipose tissue. (A colour version of this figure can be found online at http://www.journals.cambridge.org/nrr)
Some dietary compounds also have the capacity to exert a direct impact on lipolysis regulation. The well-known lipolytic effect of caffeine and other methylxanthines occurs by elevating the cyclic AMP (cAMP) intracellular levels by two mechanisms. On the one hand, this is through A1-adenosine receptor antagonism, leading to a reduction of adenylyl cyclase activity and subsequent increased lipolysis. On the other hand, methylxanthines further prevent the breakdown of cAMP by inhibiting phosphodiesterase activity( Reference Duncan, Ahmadian and Jaworski 3 ). Thus, coffee consumption increases lipid turnover and raises plasma NEFA, while a high intake of methylxanthines may also contribute to weight loss and maintenance through an enhanced fat oxidation and thermogenesis( Reference Westerterp-Plantenga, Lejeune and Kovacs 32 , Reference Murosaki, Lee and Muroyama 33 ). Another dietary compound influencing adipocyte lipolysis is Ca, with high intakes being associated with decreased adiposity and a reduced risk of obesity in diverse epidemiological studies( Reference Duncan, Ahmadian and Jaworski 3 ). Ca supplementation reportedly favours weight loss in both obese mice and human subjects undergoing energy-restricted diets, stimulating lipolysis via inhibition of the secretion of parathyroid hormone (PTH)( Reference McCarty and Thomas 34 ) and the subsequent activation of 25-hydroxycholecalciferol to 1,25-dihydroxycalciferol( Reference Shi, Dirienzo and Zemel 35 – Reference Major, Chaput and Ledoux 38 ). While acute ethanol intake exerts an anti-lipolytic effect, chronic ethanol consumption suppresses the β-adrenergic receptor-mediated lipolytic action via an increased activation of phosphodiesterase, resulting in a decreased protein kinase A (PKA) stimulation and a diminished activating phosphorylation of perilipin-1 and hormone-sensitive lipase (HSL)( Reference Kang and Nagy 39 ).
Genetic variance also plays a role in determining lipolytic rate( Reference Girousse and Langin 5 , Reference Langin, Lucas and Lafontan 18 , Reference Arner 40 ). Variations in adrenoceptors have been intensely analysed for their putative functional effects on lipolysis and association with the development of obesity. The most studied are the polymorphisms in codon 64 of the β3-adrenergic receptor and in codons 16, 27 and 164 of the β2-adrenoceptor. The Trp64Arg missense mutation of the β3-adrenergic receptor gene was reportedly associated with decreased lipolysis induced by β3-adrenoceptor agonists( Reference Umekawa, Yoshida and Sakane 41 ). However, other studies have failed to show any phenotypic effect of this polymorphism, so its true pathophysiological contribution to fat metabolism and energy homeostasis in humans remains controversial( Reference Langin, Lucas and Lafontan 18 ). Noteworthy, variations in non-coding regions of calpain 10 lead to a decreased β3-adrenergic receptor function. In the β2-adrenergic receptor gene the Arg16Gly mutation has been shown to be associated with altered β2-adrenergic receptor function, with carriers of this mutation showing a five-fold increased agonist sensitivity( Reference Langin, Lucas and Lafontan 18 ). The Gln27Glu substitution was found to be twice as common in obese than in non-obese subjects in some populations, with homozygotes exhibiting an average excess fat mass of 20 kg and about 50 % larger fat cells( Reference Large, Hellstrom and Reynisdottir 42 ). On the contrary, the rare Thr164Ile substitution in the β2-adrenergic receptor gene has not been consistently observed in obese individuals. Polymorphisms in the G-β3 gene, encoding for a specific G-coupling protein that links α- as well as β-adrenergic receptors to adenylate cyclase, alter catecholamine-induced lipolysis in human fat cells, improving the lipolytic function of β-adrenoceptors at the same time as enhancing the anti-lipolytic activity of α2-adrenoceptors. Furthermore, variations in intronic dinucleotide repeats of the HSL gene are accompanied by a decreased function of the lipase with a reduced lipolytic effect of catecholamines( Reference Klannemark, Orho and Langin 43 , Reference Magre, Laurell and Fizames 44 ).
Classic factors
In humans the main elements controlling lipolysis are the activity of the autonomic nervous system and the endocrine influence derived from the release of insulin( Reference Arner 2 , Reference Langin, Lucas and Lafontan 18 , Reference Arner 45 ). Adipose tissue is richly innervated by both the sympathetic and parasympathetic nervous systems with nerve terminals running along blood vessels and a certain number of adipocytes in direct contact with nerve varicosities. Thus, electrical stimulation of sympathetic nervous system nerve endings results in an increase in lipolytic activity, while surgical sympathectomy reportedly reduces lipolysis in the denervated adipose depot( Reference Dodt, Lonnroth and Fehm 46 – Reference Bartness and Song 49 ). Although the parasympathetic nervous system has been shown to also innervate white adipose tissue and decrease lipolysis, stimulating an increase in insulin sensitivity( Reference Kreier, Fliers and Voshol 50 , Reference Bartness 51 ), its true functional role has been subsequently questioned( Reference Giordano, Song and Bowers 52 ).
Catecholamine-induced regulation
Catecholamines, adrenaline and noradrenaline, exert their impact on lipolysis upon binding to the diverse adrenergic receptor subtypes located on the plasma membrane of adipocytes( Reference Arner 2 , Reference Arner 45 , Reference Jocken and Blaak 53 ). These receptors are linked to G-proteins, with G-protein receptor complexes regulating adenylate cyclase in the cell membrane. In mammals at least four adrenoceptors exert their action with marked species characteristics( Reference Lafontan and Langin 4 ). In humans β1- and β2-adrenoceptors are the most active lipolytic elements, while the contribution of β3-adrenergic receptors remains to be better established. The presence of β3-adrenoceptors in human white adipocytes has been clearly proven with tissue and subcellular distribution as well as response to stimulators being consistent with participation in lipolysis( Reference De Matteis, Arch and Petroni 54 ). However, the failure of β3-adrenoceptor agonists to elicit clear-cut lipolytic and weight-loss effects in obese patients casted doubts on the true physiological relevance of this β-adrenoceptor subtype in humans( Reference Buemann, Toubro and Astrup 55 , Reference Redman, de Jonge and Fang 56 ). Contrarily, β3-adrenoceptors are abundantly expressed in adipocytes of rodents( Reference Gómez-Ambrosi, Frühbeck and Aguado 57 ). Upon binding to their ligand, β-adrenergic receptors initiate the activation of the lipolytic cascade through the stimulation of cAMP production and subsequent activation of the cAMP-dependent PKA, which is followed by the phosphorylation of perilipin and HSL, ultimately leading to lipolysis stimulation (Fig. 2). Another peculiarity of human adipocytes resides in the presence of abundant α2-adrenoceptors, which are coupled to G-inhibitory proteins (Gi), thereby inhibiting cAMP production and, thus, lipolysis( Reference Lafontan and Berlan 58 , Reference Langin 59 ). Therefore, the balance between the lipolytic effect of β-adrenergic receptors and the opposing anti-lipolytic activity of α2-adrenoceptors also determines the net outcome of catecholamine-induced fat mobilisation in humans. The identification of brown adipose tissue in human adults beyond the vestigial amounts originally acknowledged and its association with BMI and adiposity has triggered a re-focusing of attention to the true relevance of β3-adrenoceptors in lipid metabolism and energy homeostasis( Reference Frühbeck, Becerril and Sáinz 60 , Reference Frühbeck, Sesma and Burrell 61 ).

Fig. 2 Principal regulators and major pathways involved in adipocyte lipolysis. A1R, A1 adenosine receptor; AC, adenylyl cyclase; ADRP, adipophilin/adipocyte differentiation-related protein; AMPK, AMP-activated protein kinase; AQP7, aquaporin 7; AR, adrenoreceptor; ATGL, adipocyte TAG lipase; cAMP, cyclic AMP; CGI-58, comparative gene identification-58; cGMP, cyclic GMP; CIDEA, cell death-inducing DFFA (DNA fragmentation factor-α)-like effector A; CL, calcitonin receptor-like; EP-3R, PGE receptor 3; FABP4, fatty acid binding protein 4; FSP27, fat-specific protein 27; GC, guanylyl cyclase; Gi, inhibitory GTP-binding proteins; Gs, stimulatory GTP-binding proteins; HSL, hormone-sensitive lipase; IRS-1, insulin receptor substrate-1; JNK, Jun kinase; NOS, NO synthase; NPR, natriuretic peptide receptor; NPY, neuropeptide Y; NPY-R1, neuropeptide Y receptor 1; PDE3B, phosphodiesterase 3B; PEDF, pigment epithelium-derived factor; PI3K, phospatidylinositol-3 kinase; PKA, protein kinase A; PKB, protein kinase B; PKG, protein kinase G; RAMP2, receptor activity modifying protein-2; TIP47, tail-interacting protein of 47 kDa; TNF-α-R, TNF-α receptor; ZAG, zinc-α2-glycoprotein. (A colour version of this figure can be found online at http://www.journals.cambridge.org/nrr)
Hormone-mediated control
A number of hormones are known to participate in the regulation of lipolysis. Among all endocrine factors, insulin is quantitatively and qualitatively the most relevant one. The impact of growth hormone (GH), adrenocorticotropic hormone, cortisol, thyroid hormones, PTH and glucagon is comparatively much more reduced than that of insulin. The mechanisms of action of all are briefly discussed below.
Hormone-mediated control: insulin
Insulin is a key regulator of white adipose tissue biology, controlling not only lipogenesis but also the rate of lipolysis and NEFA efflux. Insulin regulates glucose uptake by adipocytes and triggers fatty acid transport protein translocation as well as fatty acid uptake by fat cells( Reference Lafontan 62 ). Binding of insulin to its specific cell-surface receptor produces tyrosine phosphorylation and activation of the insulin receptor, which leads to the interaction with the insulin receptor substrates (IRS-1 and IRS-2), in turn activating the phosphatidyl inositol 3-kinase (PI3K) complex( Reference Arner 2 ). Insulin powerfully inhibits basal and catecholamine-induced lipolysis through phosphorylation (via a PKB/Akt-dependent action) and activation of phosphodiesterase-3B (PDE-3B). The phosphodiesterase catalyses the breakdown of cAMP to its inactive form, thereby decreasing cAMP levels, which in turn reduces PKA activation and, therefore, also translates into preventing HSL stimulation. Insulin may also suppress lipolysis through phosphorylation of the regulatory subunit of protein phosphatase-1 (PP-1), which once activated rapidly dephosphorylates and deactivates HSL, thus decreasing the lipolytic rate( Reference Jaworski, Sarkadi-Nagy and Duncan 63 ). The anti-lipolytic effect of insulin is observed already minutes upon binding of the hormone to its receptors.
Hormone-mediated control: growth hormone
While insulin repesents the primary anabolic hormone exerting the main influence periprandially, GH operates directly and through stimulation of insulin growth factor-1, insulin and NEFA during stress and fasting( Reference Møller and Jørgensen 64 ). Thus, GH represents a less potent though critically important regulator of lipolysis, which influences body composition, stimulating muscle mass accretion at the same time as reducing adiposity by a direct lipolytic effect using cAMP- and PKA-dependent pathways. GH-deficient individuals can experience up to a 40 % reduction in plasma NEFA and lipolysis that are returned to normal values by GH replacement therapy. Interestingly, GH activates adenylyl cyclase by selectively shifting the Giα2 subunit and removing cAMP production inhibition( Reference Yip and Goodman 65 ). Exogenous GH administration produces an increase in NEFA after 2–3 h, thus reflecting a delayed lipolytic effect when compared with that of catecholamines. In this context, small physiological GH pulses reportedly increase interstitial glycerol levels in abdominal and femoral fat( Reference Gravholt, Schmitz and Simonsen 66 ). In addition, suppression of the normal nocturnal rise in GH is followed by a reduction in subsequent lipolysis in subcutaneous adipose tissue( Reference Samra, Clark and Humphreys 67 ). Endogenous GH has been shown to play a limited metabolic role during the daily fed–fast cycle, whereas it is essential for the increased lipolytic rate observed with more prolonged fasting( Reference Sakharova, Horowitz and Surya 68 ). Recently, adipocyte-specific disruption of JAK2 (JAK2A) in mice has been shown to result in GH resistance in adipocytes, with reduced lipolysis and increased body fat, thereby offering complementary mechanistic insights into the well-recognised effects of GH on lipid flux( Reference Nordstrom, Tran and Sos 69 ).
Hormone-mediated control: other hormones
Cortisol also exerts a lipolytic effect, which is less potent than that of catecholamines at the same time as being delayed (minutes in the case of adrenaline v. hours for cortisol)( Reference Lafontan 62 , Reference Djurhuus, Gravholt and Nielsen 70 ). Importantly, the in vivo lipolysis stimulation is counteracted by the corticoid-induced insulin release( Reference Samra, Clark and Humphreys 71 , Reference Ottosson, Lonnroth and Björntorp 72 ), whereby the net outcome of a short-term treatment with a standard dose of corticosteroids is an increase in abdominal adipose tissue lipolysis, without changes in GH concentrations, hyperglucagonaemia and insulin resistance. While a stimulation of lipolysis in human adipose tissue has been also ascribed to PTH( Reference Bousquet-Melou, Galitzky and Lafontan 20 , Reference Taniguchi, Kataoka and Kono 73 ), it has also been suggested that a PTH excess may promote weight gain by impeding catecholamine-induced lipolysis( Reference McCarty and Thomas 34 ). Whereas in rodents testosterone up-regulates catecholamine-induced lipolysis( Reference Xu, De Pergola and Bjorntorp 74 ), in humans testosterone in physiological concentrations causes a depot-specific reduction of catecholamine-stimulated lipolysis in subcutaneous fat cells, probably due to reduced protein expression of β2-adrenoceptors and HSL( Reference Dicker, Ryden and Naslund 75 – Reference Zang, Ryden and Wahlen 77 ). The relevance of androgen signalling in lipolysis regulation became evident from the observation that late-onset obesity development in androgen receptor-null male mice was caused in part by a decreased lipolytic activity( Reference Fan, Yanase and Nomura 78 ). The direct molecular mechanism accounting for the hypertrophic adipocytes and expanded white adipose tissue of these mice depends on an altered lipid homeostasis characterised by a decreased lipolysis but not an increased lipogenesis. Interestingly, transcripts for HSL were strikingly decreased, whereas those for lipogenic genes were unchanged or decreased. Androgens slightly decrease lipoprotein lipase (LPL) activity in human adipose tissue organ cultures, but markedly inhibit adipogenesis in primary preadipocyte cultures obtained from subcutaneous and omental depots of both sexes( Reference Blouin, Nadeau and Perreault 79 ). Thus, the androgenic effects on adipose tissue in men as opposed to women may differ more in terms of the magnitude of their negative impact on adipogenesis and lipid synthesis rather than in the direction of the lipolytic action.
Although commonly acting in rodent fat cells as lipolytic agents via stimulatory GTP-binding protein (Gs protein)-coupled receptors, thyrotropin-stimulating hormone, adrenocorticotropic hormone and α-melanocyte-stimulating hormone are either ineffective or very weak stimulators of lipolysis in human adipocytes( Reference Lafontan 62 ). Neither glucagon nor glucagon-like peptide-1 (GLP-1) has been clearly shown to stimulate lipolysis in vitro. Likewise, no significant effects of glucagon or GLP-1 on lipolytic rate or adipose tissue blood flow following local or experimental intravenous normo- and hyperglucagonaemia have been observed( Reference Bertin, Arner and Bolinder 80 , Reference Gravholt, Moller and Jensen 81 ). However, during the present decade the role of the GLP-1/GLP-1 receptor system in lipolysis has experienced renewed interest( Reference Sancho, Trigo and Martin-Duce 82 ). A dose-dependent lipolytic effect of GLP-1 in 3T3-L1 adipocytes in a receptor-dependent manner involving downstream adenylate cyclase/cAMP signalling has been shown( Reference Vendrell, El Bekay and Peral 83 ).
Cytokines and other ‘newcomers’
Over the past years adipose tissue has been recognised as an extraordinarily active endocrine organ with the ability to secrete numerous products of diverse nature such as hormones, cytokines, enzymes, complement factors, vasoactive peptides and growth factors, among others( Reference Frühbeck, Gómez-Ambrosi and Muruzábal 84 – Reference Sun, Kusminski and Scherer 87 ). All these adipose-derived factors, collectively termed adipokines, are involved in a pleiad of physiological functions ranging from energy homeostasis to reproduction, including inflammation and immunity as well as angiogenesis and bone metabolism, among others( Reference Fortuño, Rodríguez and Gómez-Ambrosi 88 – Reference Hefetz-Sela and Scherer 94 ). The dynamic cross-talk of adipokines with other non-metabolic biological processes extends to the cardiovascular( Reference Gómez-Ambrosi and Frühbeck 95 – Reference Poulain-Godefroy, Lecoeur and Pattou 99 ), gastrointestinal( Reference Muruzábal, Frühbeck and Gómez-Ambrosi 100 – Reference Frühbeck 103 ), respiratory( Reference Campo, Frühbeck and Zulueta 104 – Reference Weng, Raher and Leyton 106 ) and muscular( Reference Sáinz, Rodríguez and Catalán 107 – Reference Sáinz, Rodríguez and Catalán 110 ) systems. In addition to their participation in plentiful diverse physiological functions, many of the recently identified hormones and adipokines have also been shown to be able to directly affect lipolysis.
Cytokine regulation of lipolysis
Cytokine release by both adipocytes and stromovascular cells underlies the participation of adipose tissue in a dynamic cross-talk and potent feedback signalling with key neuroendocrine organs involved in the regulation of food intake, lipid metabolism, glucose disposal, energy expenditure and the stress response( Reference Gannagé-Yared, Yaghi and Habre 111 , Reference Trayhurn, Drevon and Eckel 112 ). The complex secretory activities of adipose tissue also contribute to the development of insulin resistance and atherogenic processes( Reference Berg and Scherer 113 – Reference Arner, Bernard and Salehpour 115 ). The release of cytokines further exerts important local autocrine and paracrine effects, mainly involved in adipose tissue remodelling, adipogenesis, angiogenesis, inflammation and immunity. Noteworthy, cytokines, like TNF-α, as well as some interleukins and adipokines, are important regulators of spontaneous lipolysis.
Cytokine regulation of lipolysis: TNF-α
TNF-α is produced in large amounts by adipocytes and other cell types within adipose tissue( Reference Frühbeck, Gómez-Ambrosi and Muruzábal 84 , Reference Arner, Rydén and Arner 116 ). In humans, contrarily to rodents, TNF-α is not released from adipose tissue into the circulation but rather acts predominantly as a local factor( Reference Langin and Arner 117 – Reference Cawthorn and Sethi 119 ). As with other lipolytic agents, important species differences have also been observed as regards TNF-α action. TNF-α is able to stimulate lipolysis by at least three separate mechanisms( Reference Langin and Arner 117 , Reference Ryden and Arner 120 , Reference González-Yanes and Sánchez-Margalet 121 ). First, it inhibits insulin receptor signalling, thereby counteracting the anti-lipolytic effect of the hormone. In this respect, TNF-α operates via the inactivation of IRS-1. This can be brought about by the inhibition of tyrosine phosphorylation and by a reduction in the amount of IRS-1 in adipocytes. In fact, TNF-α counteracts tyrosine phosphorylation by promoting serine phosphorylation of IRS-1. The most important TNF-α effect on adipocyte IRS-1 is mediated through the p42/44 mitogen-activated protein (MAP) kinase (Fig. 2). Second, TNF-α is able to stimulate lipolysis by inhibiting the Gi-protein-coupled adenosine receptor signalling to counteract the anti-lipolytic effect of adenosine. TNF-α markedly decreases the protein content of all three Giα subtypes in rodent fat cells, without changing the amount of Gs protein or β-subunit of the G-protein complex. This decrease in Gi protein mitigates the anti-lipolytic effect of adenosine. Interestingly, TNF-α decreases Gi-protein content through an induction of protein degradation by the proteasomal pathway( Reference Botion, Brasier and Tian 122 ). However, the TNF-α–Gi interaction appears to be specific for rodents because it has not been observed in human fat cells. The third way by which TNF-α induces lipolysis is via direct stimulation of basal lipolysis through interactions with the lipid-binding protein perilipin. Only TNF-α receptor 1 and MAP kinases promote lipolytic effects in fat cells leading to phosphorylation and decreased production of perilipin, the adipose lipid droplet coating protein that protects it from being hydrolysed by HSL( Reference Langin and Arner 117 , Reference Xu and Hotamisligil 123 , Reference Xu, Hirosumi and Uysal 124 ). Three MAP kinases, namely p44/42, Jun kinase (JNK) and p38, are activated by TNF-α in fat cells but only the first two have been linked to lipolysis so far.
Mechanistically, TNF-α can stimulate lipolysis in the absence of insulin, thus providing evidence that it does not simply antagonise the anti-lipolytic effects of insulin. Moreover, extracellular glucose is required for the TNF-α-induced lipolytic effect, suggesting that a certain nutritional state or substrate availability is required( Reference Cawthorn and Sethi 119 ). The downstream signals of the TNF-α receptor 1-dependent pathway involve the activation of extracellular signal-related kinases (ERK1/2), JNK, AMP-activated protein kinase (AMPK), inhibitor of κB kinase (IKK) and PKA( Reference Cawthorn and Sethi 119 , Reference Zhang, Halbleib and Ahmad 125 , Reference Souza, Palmer and Kang 126 ). However, in fat cells the TNF-α-induced activation of ERK1/2, JNK and IKK is rapid and transient, while TNF-α-induced lipolysis takes more than 6 h, suggesting the existence of more distant events that are likely to be controlled by transcriptional regulation( Reference Cawthorn and Sethi 119 , Reference Jager, Gremeaux and Gonzalez 127 ).
Cytokine regulation of lipolysis: IL-6 and IL-15
The IL-6 receptor and glycoprotein 130, key elements of the cytokine pathway, are expressed in human adipocytes, pointing to a direct autocrine/paracrine action of IL-6 on fat cells( Reference Lafontan 62 ). Infusions of recombinant human IL-6 have been reported to increase plasma NEFA and glycerol concentrations, leading the authors to conclude that IL-6 represents a novel lipolytic factor that operates as a potent stimulator of lipolysis( Reference van Hall, Steensberg and Sacchetti 128 , Reference Yang, Ju and Zhang 129 ). Interestingly, IL-6 infusions were accompanied by parallel increases in plasma cortisol and adrenaline levels, whereas the potential effect on GH concentrations was not analysed. In this regard, it is difficult to establish whether the increased lipolysis depends on the direct action of IL-6 or rather reflects the effects of other lipolytic factors such as GH, cortisol and noradrenaline( Reference Jensen 130 ). A more recent study has shown that higher circulating IL-6 concentrations are associated with an increased isoproterenol-stimulated lipolysis especially in omental adipocytes in women( Reference Morisset, Huot and Legare 131 ). In any case, the reported effect on lipolysis of IL-6 is relatively modest compared with that elicited by catecholamines and insulin. The potential involvement of IL-6 during the practice of exercise or other situations related to severe illness, where a clear need for an elevated lipid fuel takes place, has been set forward( Reference Holmes, Watt and Febbraio 132 , Reference Hiscock, Fischer and Sacchetti 133 ).
Another member of the interleukin family has been proposed to participate in the modulation of lipolysis. The administration of IL-15 has been shown to produce a significant reduction in white adipose tissue via both a decreased rate of lipogenesis and a reduction in LPL activity, without a concomitant decrease in food intake( Reference Carbo, Lopez-Soriano and Costelli 134 ). Comparative studies with other cytokines revealed that IL-15 is apparently more potent in its acute stimulation of lipolysis than IL-6 and TNF-α( Reference Ajuwon and Spurlock 135 ). Noteworthy, when specific inhibitors of PKA or Janus kinase were present an attenuation of the lipolytic effect of IL-15 was observed. IL-15 is known to be highly expressed in skeletal muscle, exerting a potent anabolic effect on muscle protein accretion while decreasing fat depots in adipose tissue( Reference Quinn, Strait-Bodey and Anderson 136 ). Taking these observations together, it can be speculated that IL-15 may operate as a homeorhectic factor that mobilises and directs energy away from the adipocyte to other cells during the acute phase of the inflammatory response.
Interestingly, IL-1β and TNF-α have been shown to activate MAP3K8, also called Tpl2, which is expressed in adipocytes and is implicated in cytokine-induced lipolysis( Reference Jager, Gremeaux and Gonzalez 127 ). Pharmacological inhibition or silencing of Tpl2 was able to prevent MAP kinase kinas/ERK1/2 activation by these cytokines but not by insulin, thereby providing evidence of its involvement in ERK1/2 activation particularly in response to inflammatory stimuli( Reference Jager, Gremeaux and Gonzalez 127 ).
Cytokine regulation of lipolysis: leptin
More than a decade ago the identification of functional leptin receptors (OB-R) in white adipose tissue suggested the involvement of leptin in the direct peripheral regulation of adipocyte metabolism( Reference Frühbeck, Jebb and Prentice 137 – Reference Frühbeck 139 ). In fact, leptin was shown to directly participate in lipid metabolism control through the inhibition of lipogenesis and the stimulation of lipolysis. Leptin reportedly exerts an autocrine–paracrine lipolytic effect on isolated white adipocytes both in vitro and ex vivo ( Reference Frühbeck, Aguado and Martínez 140 – Reference Frühbeck, Gómez Ambrosi and Salvador 143 ).
Adenosine A1 receptors have been shown to be markedly expressed in adipocytes and influence fat cell metabolism via the regulation of adenylyl cyclase and, therefore, participate in lipolysis control via the inhibitory guanosine 5′-triphosphate (GTP) binding proteins, Gi ( Reference Honnor, Dhillon and Londos 144 , Reference Honnor, Dhillon and Londos 145 ). The adenosinergic system increases leptin secretion by directly activating adenosine A1 in white adipose tissue( Reference Rice, Fain and Rivkees 146 ). In this respect, a defective leptin-induced stimulation of lipolysis that opposes the adenosine-mediated tonic inhibition was identified( Reference Frühbeck, Gómez Ambrosi and Salvador 143 ). Interestingly, the lipolytic effect of leptin is located at the adenylyl cyclase-inhibitory G protein step (Fig. 2), providing an explanation for the defective stimulation of adipocyte adenylate cyclase and the blunted lipolysis observed in leptin-deficient and OB-R-lacking rodents as well as in morbidly obese humans( Reference Greenberg, Taylor and Londos 147 – Reference Martin, Klim and Vannucci 149 ). Moreover, storage of surplus energy in white adipose tissue and the development of diet-induced obesity require the blockade of a latent leptin-stimulated energy sump in white adipocytes( Reference Wang, Orci and Ravazzola 150 ). In this regard, the pleiotropic effects of leptin in other metabolically relevant organs like brown adipose tissue, skeletal muscle, pancreas, liver and heart need to be considered( Reference Sáinz, Rodríguez and Catalán 108 , Reference Gómez-Ambrosi, Frühbeck and Martínez 151 – Reference Huynh, Neumann and Wang 157 ).
Cytokine regulation of lipolysis: adiponectin
Adiponectin, also known as Acrp30, AdipoQ, apM1 or GBP28, is a hydrophilic 30-kDa protein highly expressed and secreted by adipocytes( Reference Fortuño, Rodríguez and Gómez-Ambrosi 88 , Reference Ahima and Lazar 90 ). The three-dimensional structure of the C-terminal globular domain of adiponectin shows a high structural homology with TNF-α, another well-known lipolytic cytokine( Reference Shapiro and Scherer 158 ). Interestingly, HSL activity has been shown to be positively correlated to adiponectin expression, with percentage body fat and adiponectin mRNA arising as the only independent predictors of adipose tissue HSL activity explaining 26 % of its variability( Reference Bullo, Salas-Salvado and Garcia-Lorda 159 ). Increased adipose tissue mass has been suggested to explain the association between low adiponectin and reduced NEFA tolerance( Reference Lavoie, Frisch and Brassard 160 ). Adiponectin has been shown to inhibit spontaneous and catecholamine-induced lipolysis in human adipocytes of non-obese subjects through AMPK-dependent mechanisms( Reference Wedellova, Dietrich and Siklova-Vitkova 161 ). In contrast to most adipokines, which are markedly up-regulated in obesity, adipose tissue expression and circulating concentrations of adiponectin are decreased in both overweight and obesity, thereby implying a plausibly decreased impact on overall lipolysis. Adiponectin gene knockout mice and primary adipocytes obtained from these mice exhibit an increased lipolysis( Reference Qiao, Kinney and Schaack 162 ). Moreover, adiponectin was shown to suppress HSL activation without modifying adipocyte TAG lipase (ATGL) and comparative gene identification-58 (CGI-58) expression in adipocytes. In addition, adiponectin reportedly reduced the type 2 regulatory subunit RIIα protein levels of PKA by reducing its protein stability, with ectopic expression of RIIα abolishing the inhibitory effects of adiponectin on lipolysis in adipocytes( Reference Qiao, Kinney and Schaack 162 ). The proportion of secreted high-molecular-weight v. total adiponectin has been shown to be higher in visceral than in subcutaneous adipose tissue explants in non-obese individuals, while no differences were observed in obese individuals( Reference Kovacova, Tencerova and Roussel 163 ). More recently, full-length adiponectin was shown to exert an anti-lipolytic effect in non-obese subcutaneous adipose tissue, while the globular and trimeric isoforms exhibited anti-lipolytic activity in obese subcutaneous and visceral adipose tissue, respectively( Reference Wedellova, Kovacova and Tencerova 164 ).
Other elements involved in lipolysis
Analysis of the involvement of other factors in the control of lipolytic pathways is unravelling a huge number of potential modulators, which vary greatly not only in their biochemical structure but also in their main physiological effect and the signalling cascade activated.
Other elements involved in lipolysis: nitric oxide
NO or related redox species have been described to act as regulators of lipolysis both in rodent and human adipocytes( Reference Gaudiot, Jaubert and Charbonnier 165 – Reference Engeli, Boschmann and Adams 170 ). Inhibition of NO release increased lipolysis independently of local blood flow changes. While chemical NO donors stimulate basal lipolysis, they block the characteristic isoproterenol-induced lipolytic activity via the inhibition of adenylyl cyclase and PKA. Inducible NO synthase has emerged as a negative modulator of lipolysis via an oxidative signalling pathway upstream of cAMP production( Reference Penfornis and Marette 169 ).
A functional relationship between leptin and NO has been established in several physiological processes( Reference Frühbeck 139 , Reference Frühbeck 171 – Reference Becerril, Rodríguez and Catalán 175 ). Given the co-localisation of both factors in fat cells and their involvement in lipolysis, a potential role of NO in the leptin-induced lipolytic effect seemed plausible. In fact, 1 h after exogenous leptin administration a dose-dependent increase in both serum NO concentrations and basal adipose tissue lipolytic rate was observed( Reference Frühbeck, Gómez Ambrosi and Salvador 143 ). Up to 27 % of the variability taking place in lipolysis was attributable to the changes in NO concentrations. The leptin-induced NO production in white adipocytes was shown to be mediated through PKA and MAP kinase activation( Reference Mehebik, Jaubert and Sabourault 176 ). Inhibition of NO synthesis by N ω-nitro-l-arginine methyl ester (l-NAME) pretreatment was followed by a reduction in the leptin-mediated lipolysis stimulation compared with leptin-treated control animals. Contrarily, in adipocytes obtained from rats under acute ganglionic blockade, the leptin-induced lipolytic effect did not show differences with the lipolytic rate achieved by leptin in control rats. The NO donor S-nitroso-N-acetyl-penicillamine (SNAP) was able to exert a significant inhibitory effect on isoproterenol-stimulated lipolysis. Thus, NO has emerged as a potentially relevant autocrine–paracrine physiological signal to fine-tune lipolysis by facilitating leptin-induced lipolysis and, at the same time, being able to inhibit catecholamine-induced lipolysis( Reference Frühbeck and Gómez-Ambrosi 173 ).
Other elements involved in lipolysis: natriuretic peptides
Until recently, human fat cell lipolysis was thought to be mediated essentially by a cAMP-dependent PKA-regulated pathway under the control of catecholamines and insulin. However, Lafontan et al. ( Reference Lafontan, Moro and Berlan 177 ) provided evidence that natriuretic peptides also have the ability to potently stimulate lipolysis in human adipocytes to the same degree as a non-selective β-adrenoceptor agonist. This lipolytic effect is mediated mainly by natriuretic peptide receptor type A through a cyclic GMP-dependent PKG (cGK-I) signalling pathway (Fig. 2) that does not involve PDE-3B inhibition or cAMP production and PKA activity( Reference Sengenes, Berlan and De Glisezinski 178 – Reference Moro, Galitzky and Sengenes 182 ). Noteworthy, in vitro studies have shown that HSL can also be phosphorylated by the cyclic GMP-dependent signalling cascade. In fact, cGK-I phosphorylates perilipin and HSL. Increases in plasma atrial natriuretic peptide levels by physiological (exercise) or pharmacological stimuli are followed by an enhanced lipid mobilisation( Reference Moro, Pillard and de Glisezinski 183 , Reference Moro, Pasarica and Elkind-Hirsch 184 ). In humans atrial natriuretic peptide also reportedly induces postprandial lipid oxidation, energy expenditure, and concomitantly arterial blood pressure( Reference Birkenfeld, Budziarek and Boschmann 185 , Reference Moro and Lafontan 186 ). Taken together, this pathway that participates in lipid mobilisation and energy homeostasis becomes especially important during chronic treatment with β-adrenoceptor antagonists, which inhibit catecholamine-induced lipolysis but enhance cardiac atrial natriuretic peptide release.
Other elements involved in lipolysis: endocannabinoid system
Our understanding of the participation of the endocannabinoid system in energy homeostasis has progressed enormously over the past years( Reference Horvath 187 – Reference Moreno-Navarrete, Catalán and Whyte 189 ). In particular, the observation of the presence of G protein-coupled cannabinoid receptor (CB) CB1 receptors in adipocytes provided a clue for the involvement of endocannabinoids in the peripheral control of lipid metabolism( Reference Cota, Marsicano and Tschop 190 – Reference Matias, Gonthier and Orlando 193 ). Selective CB1 antagonism was shown to coordinately induce key genes of the fatty acid catabolic pathway, thereby favouring lipolysis and reducing fat storage in adipose tissue( Reference Jbilo, Ravinet-Trillou and Arnone 191 ). Interestingly, the selective antagonism of CB1 receptors reportedly induced β3-adrenoceptors and GH receptors at the same time as repressing the expression of catechol-O-methyltransferase, an enzyme involved in the degradation of catecholamines. The reduced expression of this methyltransferase along with the induction of the receptors of two well-known hormones with lipolytic effects further supports the molecular basis for the participation of endocannabinoids in the modulation of lipolysis.
Amides of fatty acids with ethanolamine (FAE) are biologically active lipids participating in a variety of physiological effects, including appetite regulation. While the polyunsaturated FAE anandamide (arachidonoylethanolamide) increases food intake by activating G protein-coupled cannabinoid receptors, the monounsaturated FAE oleoylethanolamide (OEA) reduces feeding as well as body-weight gain and stimulates lipolysis by activating the nuclear receptor PPAR-α( Reference Guzman, Lo Verme and Fu 194 , Reference Martinez de Ubago, Garcia-Oya and Perez-Perez 195 ).
Other elements involved in lipolysis: ghrelin
Beyond its strong orexigenic effect, the gastrointestinal twenty-eight-amino acid octanoylated peptide ghrelin exerts a wide spectrum of actions including the inhibition of isoproterenol-induced lipolysis in rodent adipocytes( Reference Muccioli, Pons and Ghe 196 ). Both ghrelin and des-acyl ghrelin have been shown to antagonise the catecholamine-stimulated lipolysis via a non-type 1A GH secretagogue receptor. Moreover, acylated and unacylated ghrelin have been also shown to attenuate isoproterenol-induced lipolysis in isolated rat visceral adipocytes through activation of phosphoinositide 3-kinase γ and PDE-3B( Reference Baragli, Ghe and Arnoletti 197 ). However, ghrelin infusion in human subjects was observed to induce acute insulin resistance and lipolysis independent of GH signalling( Reference Vestergaard, Gormsen and Jessen 198 ). All of the elements of the ghrelin system have been identified in human adipocytes, including receptors and isoforms as well as the ghrelin-O-acyltransferase or GOAT enzyme( Reference Rodríguez, Gómez-Ambrosi and Catalán 199 , Reference Rodríguez, Gómez-Ambrosi and Catalán 200 ). Interestingly, in differentiating omental adipocytes, incubation with both acylated and desacyl ghrelin increased PPAR-γ and sterol regulatory element-binding protein-1 mRNA levels, as well as fat storage-related proteins, like acetyl-CoA carboxylase, fatty acid synthase, LPL and perilipin( Reference Rodríguez, Gómez-Ambrosi and Catalán 199 ). Consequently, both ghrelin forms stimulate intracytoplasmatic lipid accumulation at the same time as exhibiting an anti-lipolytic effect.
Other elements involved in lipolysis: other miscellaneous agents
The potent anti-lipolytic effect of nicotinic acid together with its specific binding to adipose tissue was firmly established more than half a century ago( Reference Carlson and Oro 201 , Reference Carlson and Hanngren 202 ). However, the mechanistic basis for this action on lipolysis control has been provided only more recently( Reference Karpe and Frayn 203 ). Activation of the nicotinic acid receptor triggers an inhibitory G-protein signal, which decreases cAMP concentrations in adipocytes, thereby inhibiting lipolysis. Continuous 24 h nicotinic acid infusion in rats reportedly alters gene expression and basal lipolysis in adipose tissue, producing a NEFA rebound and insulin resistance( Reference Oh, Oh and Choi 204 ) that are consistent with clinical observations following treatment with this compound.
Other agents originating from either adipocytes or surrounding cells are known to negatively control adenylyl cyclase activity and inhibit lipolysis via their interaction with plasma membrane receptors belonging to the seven-transmembrane domain receptor family. Autacoid agents, as already mentioned including adenosine, prostaglandins and their metabolites, exert a clear anti-lipolytic effect. Whereas adenosine and neuropeptide Y reportedly inhibit lipolysis, for PGE2 a biphasic effect has been put forward with nanomolar concentrations suppressing lipolysis, but micromolar levels resulting in lipolysis stimulation( Reference Jaworski, Sarkadi-Nagy and Duncan 63 ). On the contrary, PGI2 showed no effect or exerted also a biphasic effect, whereby nanomolar concentrations stimulated lipolysis, whereas at micromolar levels lipolysis was suppressed.
Cachexia-inducing tumours produce a lipid-mobilising factor (LMF) that causes an immediate glycerol release when incubated with murine adipocytes, with the stimulation of lipolysis by LMF being associated with an elevation in intracellular cAMP concentrations( Reference Tisdale 205 – Reference Cabassi and Tedeschi 207 ). Zn-α2-glycoprotein (ZAG), a tumour-related LMF of 43 kDa, has been found to be expressed in 3T3-L1 cells as well as in the major fat depots of mice, being up-regulated in rodents with cancer cachexia( Reference Bing, Bao and Jenkins 208 ). Both ZAG expression and protein have been also detected in human adipocytes of visceral and subcutaneous origin. Remodelling of adipose tissue together with decreased lipid storage constitute a hallmark of cancer patients with cachexia. In addition to ATGL- and HSL-enhanced lipolysis, in cancer other factors such as ZAG have been shown to participate in TAG degradation leading to white adipose tissue atrophy. ZAG expression and release by adipose tissue are up-regulated in weight-losing cancer patients, suggesting that ZAG operates both locally and systemically to stimulate lipid mobilisation( Reference Bing, Mracek and Gao 206 ). However, ZAG did not display the thermogenic effects of the β-adrenoceptor agonist, nor did it increase β3-adrenoceptor or UCP1 (uncoupling protein 1) gene expression in brown adipose tissue, thereby implying that it does not behave as a typical β3/2-adrenoceptor agonist( Reference Wargent, O'Dowd and Zaibi 209 ). Thus, ZAG has emerged as a novel adipokine, being identified as an additional adipose tissue factor closely related to body weight loss not only via modulation of lipolysis in fat cells but also by activating AMPK in skeletal muscle cells( Reference Bing, Bao and Jenkins 208 , Reference Eckardt, Schober and Platzbecker 210 ).
The octapeptide angiotensin II (Ang II) is the active component of the renin–angiotensin system (RAS). A local RAS is present in adipose tissue, with all the elements of the system, including angiotensinogen, renin and angiotensin-converting enzyme, having been identified in adipocytes( Reference Sarzani, Salvi and Dessi-Fulgheri 211 ). Noteworthy, Ang II has been shown to decrease local blood flow in a dose-dependent manner and to inhibit lipolysis in adipose tissue with the effects being similar in both normal-weight and obese individuals( Reference Goossens, Blaak and Saris 212 ). In the last decade evidence has been provided that adipose tissue is a source of vasoactive peptides that further exert metabolic actions( Reference Frühbeck 213 ). Thus, endothelin-1 is a powerful vasoconstrictor primarily produced and secreted by endothelial cells to operate on the underlying vascular smooth muscle cell layer that can also act on adipocytes inducing lipolysis via the ERK pathway( Reference Juan, Chang and Lai 214 , Reference Juan, Chang and Huang 215 ). In human subjects endothelin-1 has been shown to selectively counteract insulin inhibition of visceral adipocyte lipolysis, decreasing the expression of insulin receptor, IRS-1 and PDE-3B and increasing the expression of the endothelin receptor-B (ETBR) in visceral but not subcutaneous adipocytes( Reference van Harmelen, Eriksson and Astrom 216 ). The ETBR-mediated effects were signalled via the PKC and calmodulin pathways. Subsequently, it was further observed that long-term incubation of human adipocytes with endothelin-1 increases lipolysis via the activation of ETAR( Reference Eriksson, van Harmelen and Stenson 217 ). Likewise, the fifty-two-amino acid vasoactive peptide adrenomedullin together with its receptor components (calcitonin receptor-like receptor and receptor activity modifying protein-2 (CRLR/RAMP2)) have been identified to be concomitantly expressed in adipose tissue (Fig. 2), exhibiting a tissue-specific up-regulation during the development of obesity( Reference Shichiri, Fukai and Ozawa 218 , Reference Fukai, Yoshimoto and Sugiyama 219 ). Interestingly, in adipose tissue adrenomedullin acts as an autocrine–paracrine factor to regulate lipid mobilisation, inhibiting lipolysis through NO-mediated β-adrenergic agonist oxidation( Reference Harmancey, Senard and Pathak 220 ). In this context, it has been proposed that adrenomedullin alone is devoid of lipolytic function and inhibits β-adrenergic-stimulated lipolysis by shifting the concentration–response curve for isoproterenol by a NO-dependent mechanism; specifically, adrenomedullin-induced NO modifies isoproterenol through an extracellular oxidative reaction to yield its aminochrome, isoprenochrome. However, other studies have provided evidence for adrenomedullin dose-dependently elevating cAMP levels and the lipolytic rate( Reference Iemura-Inaba, Nishikimi and Akimoto 221 ). In this case, adrenomedullin was shown to increase the phosphorylation of PKA, ERK and Akt and would reportedly exhibit additive effects on isoproterenol-induced lipolysis.
Apelin represents a further peptide with vasoactive characteristics that has been subsequently shown to be secreted by adipocytes of both humans and rodents, being up-regulated in states of obesity( Reference Boucher, Masri and Daviaud 222 ). The identification in adipocytes of apelin and the apelin receptor (APJ), a G-protein-coupled receptor, supported a plausible autocrine participation of this peptide in adipobiology. In this line, apelin was shown to dose-dependently stimulate AMPK phosphorylation in human adipose tissue, which was associated with increased glucose uptake( Reference Attane, Daviaud and Dray 223 ). Apelin reportedly decreased isoproterenol-induced NEFA and glycerol release in 3T3-L1 cells and isolated adipocytes abrogating the catecholamine-induced HSL phosphorylation via G-protein q polypeptide (Gq), Gi pathways and AMPK activation( Reference Yue, Jin and Xu 224 ). The apelin-induced inhibition of basal lipolysis was exerted through AMPK-dependent enhancement of perilipin expression by preventing lipid droplet fragmentation and hormone-stimulated acute lipolysis inhibition mediated by decreasing perilipin phosphorylation( Reference Than, Cheng and Foh 225 ). Moreover, apelin also suppressed adipogenesis through MAP kinase kinase/ERK signalling.
Pigment epithelium-derived factor (PEDF) is a 50-kDa protein of the non-inhibitory serpin family of serine protease inhibitors originally identified as a regulator of hepatic TAG metabolism involved in the development of insulin resistance in obesity( Reference Zechner, Zimmermann and Eichmann 6 , Reference Crowe, Wu and Economou 226 , Reference Sabater, Moreno-Navarrete and Ortega 227 ). Subsequently it was tested whether this adipocyte-secreted factor also exhibits autocrine–paracrine lipolytic effects. PEDF was shown to stimulate TAG hydrolysis in adipose tissue, muscle and liver via ATGL( Reference Borg, Andrews and Duh 228 ). The exact mechanisms underlying the participation of PEDF in insulin resistance, obesity and non-alcoholic fatty liver disease still need to be fully elucidated( Reference Yamagishi, Amano and Inagaki 229 – Reference Moreno-Navarrete, Touskova and Sabater 231 ). The potential role of other recently identified adipose-related factors on lipolysis such as serum amyloid A, osteopontin, osteocalcin, osteoprotegerin, obestatin, lipocalin 2, visfatin, nerve growth factor-inducible derived peptides, omentin, mammalian chitinase-like protein YKL40, chemerin, vitamin D and tenascin C, among others, beyond their originally reported effects merits to be specifically investigated( Reference Gannagé-Yared, Yaghi and Habre 111 , Reference Sabater, Moreno-Navarrete and Ortega 227 , Reference Gómez-Ambrosi, Salvador and Rotellar 232 – Reference Fernández-Real, Izquierdo and Ortega 245 ).
Influence of subcellular compartmentalisation of lipases
Multicellular organisms ranging from insects to mammals have evolved specialised systems to store surplus lipid energy for subsequent mobilisation in times of need. In mammals the storage and mobilisation of lipids are fundamental functions of adipocytes. About 80 % of the total adipose tissue weight is due to the fat content, with over 90 % of lipids being stored as TAG( Reference Bays, González-Campoy and Bray 246 ). The major secretory products of adipose tissue are NEFA( Reference Wood and Trayhurn 247 ), which are derived from the lipolysis of stored TAG in a process involving three main steps and requiring, at least, three different lipases, which are regulated by both adipocyte and non-adipocyte factors( Reference Young and Zechner 7 ). Thus, the classic lipolytic pathway encompasses the three following consecutive steps: (i) TAG hydrolysation by ATGL to generate fatty acids and diacylglyerol (DAG)( Reference Haemmerle, Lass and Zimmermann 248 ); (ii) subsequently, HSL catalyses the hydrolysis of DAG to monoacylglycerol (MAG) and fatty acids( Reference Reynisdottir, Dauzats and Thorne 249 , Reference Osuga, Ishibashi and Oka 250 ); (iii) monoacylglycerol lipase (MGL) is required to complete the hydrolysis of MAG into one fatty acid and glycerol( Reference Fredrikson, Tornqvist and Belfrage 251 ). HSL and ATGL are quantitatively the most important lipases based on the blunted isoprenaline-induced lipolysis observed in adipocytes of Atgl- and Hsl-knockout mice( Reference Haemmerle, Lass and Zimmermann 248 , Reference Osuga, Ishibashi and Oka 252 ).
TAG hydrolysis
Only a decade ago the initiation of TAG hydrolysis was thought to be exclusively controlled by HSL( Reference Arner 2 – Reference Young and Zechner 7 , Reference Shi and Burn 253 – Reference Bezaire and Langin 255 ). However, the generation of Hsl-knockout mice revealed the existence of residual HSL-independent TAG lipase activity, pointing to the existence of previously unidentified adipose tissue lipases. Currently, ATGL is well recognised to be the lipase responsible for initiating TAG breakdown to yield DAG( Reference Girousse and Langin 5 , Reference Zechner, Zimmermann and Eichmann 6 ). ATGL is a 54-kDa TAG hydrolase, also named phospholipase A2ξ or desnutrin, belonging to the family of patatin-like phospholipase domain-containing proteins (PNPLA) with specificity for TAG as a substrate( Reference Zechner, Zimmermann and Eichmann 6 , Reference Haemmerle, Lass and Zimmermann 248 , Reference Zimmermann, Strauss and Haemmerle 256 , Reference Zechner, Kienesberger and Haemmerle 257 ). Atgl-knockout mice and knockdown studies in adipocytes provided evidence for the involvement of ATGL in TAG but not DAG hydrolysis. Atgl-null mice exhibited a blunted lipolysis, producing a more than 75 % reduction in NEFA release and a significant TAG accumulation in adipocytes leading to obesity( Reference Haemmerle, Lass and Zimmermann 248 , Reference Schweiger, Schreiber and Haemmerle 258 ). The co-activator of ATGL, CGI-58, also known as α/β-hydrolase domain-containing protein 5 (ABHD5), was shown to stimulate TAG hydrolase activity in wild-type and Hsl-deficient but not Atgl-deficient mice. ATGL and HSL are responsible for 95 % of TAG lipase activity, thereby suggesting a complementary relationship between the two lipases( Reference Zechner, Kienesberger and Haemmerle 257 – Reference Thompson, Lobo and Bernlohr 259 ).
ATGL is highly expressed in adipose tissue, with its expression being profoundly elevated during adipocyte differentiation. Two phosphorylation sites (Ser404 and Ser428) have been identified within the C-terminal region of ATGL. Furthermore, the enzymic activity and its interaction with CGI-58 are dependent on the C-terminal region( Reference Schweiger, Schoiswohl and Lass 260 ). Overexpression of Atgl elevates TAG hydrolysis as well as basal and catecholamine-stimulated lipolysis, while Atgl silencing decreases TAG hydrolase activity, TAG storage and lipid droplet size( Reference Zechner, Kienesberger and Haemmerle 257 ). Alterations of Atgl expression resulted in dramatic changes in whole-cell lipolysis. Conversely, silencing of Atgl or CGI-58 significantly reduced basal lipolysis and essentially abolished forskolin-stimulated lipolysis. Taken together, these findings suggest that in humans the ATGL–CGI-58 complex acts independently of HSL and precedes its action in the sequential hydrolysis of TAG.
Fasting, glucocorticoids and PPAR agonists increase Atgl mRNA expression, whereas food intake and insulin decrease it( Reference Lass, Zimmermann and Oberer 261 , Reference Chakrabarti, Kim and Singh 262 ). Cellular TAG lipolysis by ATGL produces essential mediators involved in lipid ligand generation for PPAR activation, with Atgl deficiency in mice reducing mRNA levels of PPAR-α and PPAR-δ target genes( Reference Haemmerle, Moustafa and Woelkart 263 ). While mammalian target of rapamycin (mTOR)-dependent signalling has been observed to decrease Atgl mRNA expression, FoxO1 activation by SIRT1-mediated deacetylation elevated it( Reference Chakrabarti, Kim and Singh 262 , Reference Chakrabarti and Kandror 264 – Reference Chakrabarti, English and Karki 266 ). However, the role of AMPK in lipolysis control remains controversial( Reference Yin, Mu and Birnbaum 267 – Reference Gaidhu, Bikopoulos and Ceddia 271 ). In this sense, the precise mechanisms of ATGL regulation need to be fully established. Recently, a protein encoded by the G0/G1 switch gene 2 (G0S2) has been identified as a selective regulator of ATGL by attenuating its action both in vitro and in vivo ( Reference Yang, Lu and Lombès 272 , Reference Schweiger, Paar and Eder 273 ). G0S2 is highly expressed in adipose tissue and differentiated adipocytes interacting specifically with ATGL to inhibit its TAG hydrolase activity. While knockdown of endogenous G0S2 enhances both basal and stimulated lipolysis in adipocytes, overexpression of G0S2 decreases the lipolytic rate of adipocytes and adipose tissue explants. G0S2 has been further shown to regulate human lipolysis influencing ATGL activity and intracellular localisation by anchoring the lipase to lipid droplets (Fig. 3) independently of the C-terminal lipid-binding domain of ATGL( Reference Schweiger, Paar and Eder 273 ). Moreover, G0S2 expression has been observed to be diminished in poorly controlled type 2 diabetes, thereby establishing a potential link between adipose tissue G0S2 down-regulation and insulin resistance. Given that the above-mentioned characteristics reveal ATGL as an attractive therapeutic target, the development and characterisation of a selective small-molecule inhibitor of ATGL, atglistatin, may prove of interest for the pharmacological treatment of dyslipidaemic and metabolic disorders( Reference Mayer, Schweiger and Romauch 274 ).
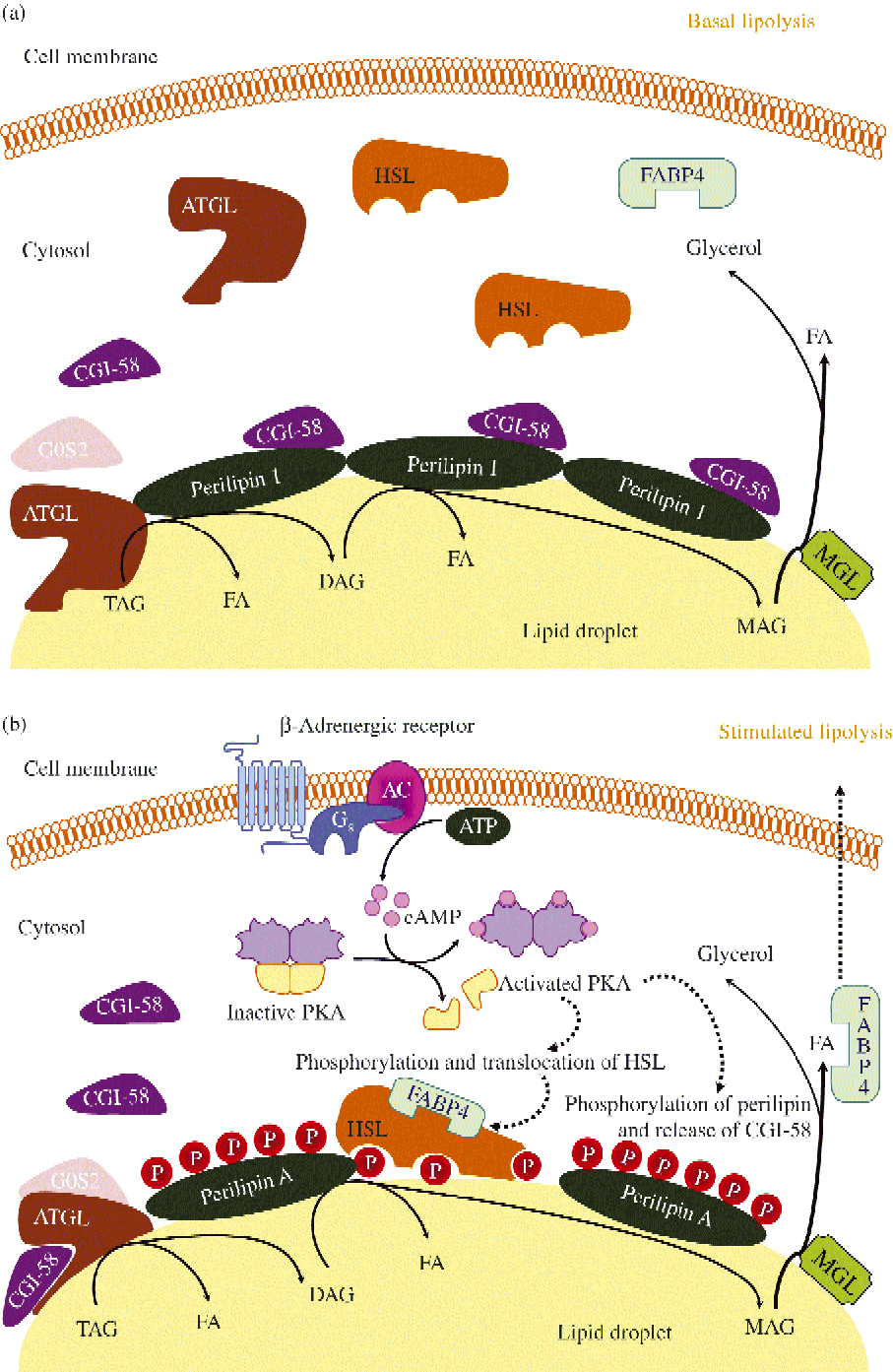
Fig. 3 Schematic representation of basal (a) and stimulated (b) lipolysis, the catabolic pathway by which TAG are hydrolysed into fatty acids (FA). AC, adenylyl cyclase; ATGL, adipocyte TAG lipase; cAMP, cyclic AMP; CGI-58, comparative gene identification-58; DAG, diacylglycerol; FABP4, fatty acid binding protein 4; G0S2, G0/G1 switch gene 2; Gs, stimulatory GTP-binding proteins; HSL, hormone-sensitive lipase; MAG, monoacylglycerol; MGL, monoacylglycerol lipase; P, phosphate; PKA, protein kinase A. (A colour version of this figure can be found online at http://www.journals.cambridge.org/nrr)
Diacylglycerol hydrolysis
HSL, an 84-kDa cytoplasmic protein with demonstrated activity for a wide range of substrates including TAG, DAG, cholesteryl esters and retinyl esters, was presumed to be the rate-limiting enzyme in the initial steps of the lipolytic process. However, several important findings challenged this view of the unique regulatory and rate-limiting role of HSL on lipolysis, pointing to the existence of alternative lipases targeting TAG molecules to counterbalance the strong affinity of HSL for DAG( Reference Lafontan and Langin 4 , Reference Girousse and Langin 5 , Reference Young and Zechner 7 , Reference Osuga, Ishibashi and Oka 250 , Reference Zechner, Kienesberger and Haemmerle 257 , Reference Okazaki, Osuga and Tamura 275 ): (i) PKA-dependent HSL phosphorylation led only to a 2- to 3-fold increase in TAG hydrolase activity, while whole-cell lipolysis resulted in a 100-fold increase; (ii) Hsl-null mice exhibited a normal body weight with decreased adiposity; (iii) these mutants further showed DAG adipocyte accumulation; (iv) the existence of residual TAG hydrolase activity and lipolysis despite HSL silencing or specific pharmacological inhibition; and (v) failure of HSL overexpression to promote whole-cell lipolysis. As mentioned previously, the identification of ATGL provided explanations for these findings( Reference Osuga, Ishibashi and Oka 250 , Reference Granneman and Moore 254 , Reference Haemmerle, Zimmermann and Strauss 276 ).
Fig. 3 illustrates ATGL and HSL regulation in basal and stimulated conditions. ATGL and HSL have the capacity to hydrolyse in vitro the first ester bond of TAG. ATGL exhibits 10-fold higher substrate specificity for TAG than DAG, selectively enabling the first step in TAG hydrolysis, leading to the formation of DAG and fatty acid. An important step in lipolysis activation comprises the translocation of HSL from a cytosolic compartment to the surface of the lipid droplet. Upon lipolytic stimulation, HSL moves from the cytosol to the surface of lipid droplets where it interacts with perilipin-1 and neutral lipids. Noteworthy, adipocytes lacking perilipin-1 are incapable of translocating HSL to the lipid droplet after increases in cAMP( Reference Holm 277 , Reference Peyot, Nolan and Soni 278 ). Perilipin-1 operates as a dynamic scaffold to coordinate the access of enzymes to the lipid droplet in a way that is responsive to the metabolic state of the adipocyte( Reference Tansey, Sztalryd and Gruia-Gray 279 , Reference Tansey, Sztalryd and Hlavin 280 ). Thus, in basal conditions (Fig. 3(a)) perilipin-1 limits lipase access to the lipid droplet( Reference Sztalryd, Xu and Dorward 281 ). Lipolysis stimulation is followed by HSL translocation from the cytosol to lipid droplets and redistribution of ATGL, resulting in enriched colocalisation of the two lipases. Interestingly, the ATGL–CGI-58 complex acts independently of HSL and precedes its action in the sequential hydrolysis of TAG in humans. The increased number of ATGL–CGI-58 complexes formed following perilipin-1 phosphorylation (which releases CCI-58) and docked on small lipid droplets govern PKA-stimulated lipolysis (Fig. 3(b)). The association between fatty acid binding protein 4 (FABP4) and HSL represents a further regulatory step. Fatty acid binding to FABP4 and HSL phosphorylation precede the association of FABP4 and HSL. FABP4 also participates in the trafficking of fatty acids from the site of hydrolysis (i.e. the lipid droplet) to the plasma membrane. In addition to supporting fatty acid trafficking to the plasma membrane in a reaction that is independent of its physical association with HSL, FABP4 bound to fatty acids associates with activated, phosphorylated HSL on the surface of lipid droplets. The sequential effect of ATGL-accentuated TAG hydrolysis, phosphorylated HSL and MGL activity yields massive increases in NEFA release in response to PKA activation.
The expression profile of HSL basically mirrors that of ATGL, given that both enzymes coordinatedly hydrolyse TAG and, therefore, share some regulatory characteristics but differ in the mechanisms of enzyme control( Reference Zechner, Zimmermann and Eichmann 6 ). Whereas β-adrenergic stimulation exerts ATGL regulation mainly via CGI-58 recruitment, HSL constitutes the main target for PKA-catalysed phosphorylation( Reference Belfrage, Fredrikson and Olsson 282 ). Adipocyte HSL encompasses an N-terminal domain (that interacts with FABP4) and a C-terminal catalytic domain (that contains the active site as well as a regulatory module with all the known phosphorylation sites of HSL)( Reference Lafontan and Langin 4 , Reference Bezaire and Langin 255 , Reference Ray, Beylot and Arner 283 ). Phosphorylation of HSL at Ser563, Ser659 and Ser660 by PKA and at Ser660 via the ERK pathway activate lipolysis( Reference Greenberg, Shen and Muliro 284 ). The PKA-dependent lipolytic effect is exerted increasing HSL's intrinsic activity and promoting its access to TAG molecules within the adipocyte. Conversely, AMPK exerts an anti-lipolytic effect, blocking the translocation of HSL to the lipid droplets by its phosphorylation at Ser565( Reference Lass, Zimmermann and Oberer 261 ). Deactivation of lipolysis mediated by insulin is associated with down-regulation of HSL and ATGL expression( Reference Kralisch, Klein and Lossner 285 , Reference Kershaw, Hamm and Verhagen 286 ). Moreover, insulin signalling phosphorylates and activates PDE isoforms via PKB, cAMP hydrolysis and PKA inhibition, resulting in the prevention of HSL and perilipin-1 phosphorylation, HSL activation and translocation as well as CGI-58-mediated ATGL activation. The peripheral control of insulin is accompanied by a central mechanism via the sympathetic nervous system that reduces the activitiy of both HSL and ATGL( Reference Scherer, O'Hare and Diggs-Andrews 287 ).
Monoacylglycerol hydrolysis
The final step of lipolysis is catalysed by MGL, which is constitutively expressed in adipose tissue and has no affinity for DAG, TAG or cholesteryl esters( Reference Bezaire and Langin 255 ). The enzymic activity of MGL is required in the final hydrolysis of the 2-monoacylglycerols produced by HSL activation. Site-directed mutagenesis has shown the relevance of Ser122, Asp239 and His269 in the lipase and esterase activities of MGL( Reference Bezaire and Langin 255 , Reference Taschler, Radner and Heier 288 ).
Other lipases
The contribution of alternative lipases to ATGL and HSL to the overall lipolytic capacity and maintenance of the highly dynamic TAG turnover has yet to be completely discerned. Potential TAG hydrolases have been identified within members of the carboxylesterase/lipase and the patatin homology domain families( Reference Zechner, Zimmermann and Eichmann 6 ). Carboxylesterase-3/TAG hydrolase-1 is supposedly involved in HSL-independent lipolysis in adipocytes and participates in the assembly and secretion of VLDL in the liver ( Reference Soni, Lehner and Metalnikov 289 , Reference Wei, Ben Ali and Lyon 290 ). Among the patatin homology domain family, PNPLA4 and PNPLA5 have been observed to exhibit TAG hydrolase, DAG transacylase and retinylester hydrolase activity in vitro, which needs to be confirmed in vivo ( Reference Kienesberger, Oberer and Lass 291 ). Noteworthy, the member with the highest ATGL homology is PNPLA3 or adiponutrin( Reference Polson and Thompson 292 – Reference Basantani, Sitnick and Cai 295 ).
Lipid droplet proteins
Cytoplasmic lipid droplets are organelles in which cells store neutral lipids for use as an energy source in times of need, but they also play important roles in the regulation of key metabolic processes, with excess accumulation of intracellular lipids being associated with obesity, type 2 diabetes and atherosclerosis. Fat droplets may constitute up to 95 % of the total adipocyte volume, being mainly composed by TAG. Intracellular TAG storage droplets have emerged as extraordinarily dynamic organelles, with signalling events underlying lipid mobilisation by shuttling protein trafficking to a specialised subset of these droplets( Reference Greenberg, Coleman and Kraemer 15 ). Thus, lipid droplet scaffold proteins are key elements in organising and directing the lipolytic signalling cascade( Reference Greenberg, Coleman and Kraemer 15 , Reference Bays, González-Campoy and Bray 246 ).
The function of lipid droplets is regulated by their coating proteins, collectively termed PAT proteins after perilipin, adipophilin/adipocyte differentiation-related protein (ADRP), and tail-interacting protein of 47 kDa (TIP47)( Reference Lafontan and Langin 4 , Reference Brasaemle 296 , Reference Brasaemle, Subramanian and Garcia 297 ). Further members of the family are S3-12, oxidative tissue-enriched PAT protein (OXPAT), myocardial lipid droplet protein (MLDP) and lipid storage droplet protein 5 (LSDP5)( Reference Robenek, Robenek and Buers 298 , Reference Robenek, Robenek and Troyer 299 ). The members of this family share varying levels of sequence similarity, lipid droplet association and functions in stabilising lipid droplets.
Lipid droplet proteins: perilipin
Lipid droplets in most tissues are coated by two or more members of the perilipin family, which are now numbered according to the order of discovery( Reference Kienesberger, Oberer and Lass 291 ). Expression of perilipin-1 is mainly restricted to white and brown adipocytes and, to a lesser extent, steroidogenic cells of adrenal cortex, testes and ovaries. Perilipin-2 (formerly adipophilin or ADRP) and perilipin-3 (formerly TIP47) are ubiquitously expressed and, therefore, lipid droplet components of most tissues. While perilipin-4 (formerly S3-12) is primarily expressed in white adipocytes, perilipin-5 (formerly OXPAT, MLDP, or LSDP5) is expressed in brown adipocytes as well as myocytes of skeletal muscle and heart, all of which rely on lipolysis to provide fatty acids to mitochondria for β-oxidation to drive either ATP production or heat generation. Thus, the perilipin composition of lipid droplets within a specific tissue constitutes an important component of lipolysis regulation.
Perilipin is the best-known member of the PAT family, with perilipin-1 being the predominant isoform found in mature adipocytes, the most abundant protein on the lipid droplet surface and the major substrate for cAMP-dependent PKA in lipolytically stimulated adipocytes( Reference Brasaemle, Subramanian and Garcia 297 , Reference Londos, Brasaemle and Schultz 300 – Reference Yang, Heckmann and Zhang 308 ). Perilipin limits the access of cytosolic lipases to lipid droplets, thereby facilitating TAG storage under basal conditions (Fig. 3(a)). When energy is needed, perilipin is phosphorylated by PKA, facilitating maximal lipolysis by ATGL and HSL (Fig. 3(b)). Thus, perilipin expression and its phosphorylation state are key in lipolysis control, with phosphorylation of Ser492 producing a lipid droplet remodelling, widely increasing the surface area for lipase binding, while Ser517 is essential for ATGL-dependent lipolysis in stimulated conditions( Reference Lafontan and Langin 4 ). Perilipin-1 is also phosphorylated by the cyclic GMP-dependent PKG.
Perilipin ablation confers resistance to genetic or diet-induced obesity, producing a lean phenotype with smaller adipocytes, increased basal lipolysis and attenuated stimulated lipolysis( Reference Martínez-Botas, Anderson and Tessier 301 ). Recently, perilipin-1 has been shown to move between the fat droplet and the endoplasmic reticulum( Reference Skinner, Harris and Shew 309 ), which is physiologically reasonable given that lipid droplets are largely derived from the endoplasmic reticulum. In this regard, perilipin-mediated lipid droplet formation in adipocytes was demonstrated to promote sterol regulatory element-binding protein-1 (SREBP-1) processing and TAG accumulation, suggesting an interplay between lipid droplet formation and SREBP-1 activation via a positive feedback loop( Reference Takahashi, Shinoda and Furuya 310 ). Therefore, the lysosomal protein degradation machinery of perilipin may constitute a target mechanism for enhancing adipocyte lipolysis. Interestingly, a genome-wide RNA interference (RNAi) screen in Drosophila S2 cells highlighted the relevance of elements of the vesicle-transport systems in lipolysis regulation through the identification of the vesicle-mediated coat protein complex I (COPI) as an evolutionary-conserved regulator of PAT protein composition at the lipid droplet surface( Reference Beller, Sztalryd and Southall 311 , Reference Guo, Walther and Rao 312 ). In addition to regulating PAT protein composition, COPI promotes the association of ATGL with the lipid droplet surface to mediate lipolysis. These genes are conserved in mammalian cells, thus suggesting that a similar complex might be operative in adipocytes. Although COPI-mediated transport reportedly participates in delivery of ATGL to the lipid droplet surface, depletion of β-COP (a subunit of the COPI coat complex) does not affect association of ATGL with lipid droplets or ATGL-mediated lipolysis, pointing to the possibility of alternative transport mechanisms implicated in the regulation of lipid homeostasis( Reference Takashima, Saitoh and Hirose 313 ).
Lipid droplet proteins: coactivator comparative gene identification-58 (CGI-58) or α/β-hydrolase domain-containing protein 5 (ABHD5)
CGI-58 lacks lipase activity in itself but potently and selectively stimulates lipolysis by activating ATGL. As mentioned above, in basal unstimulated conditions CGI-58 binds tightly to lipid droplets by interacting with perilipin-1 and is unable to activate ATGL( Reference Lafontan and Langin 4 ). However, following β-adrenoceptor stimulation CGI-58 is quickly dispersed to the cytosol, favouring ATGL co-localisation and migration to small lipid droplets. Thus, under stimulated conditions, the intracellular cAMP elevation and PKA activation promote perilipin-1 phosphorylation, which is followed by the dissociation from perilipin of CGI-58, which subsequently interacts with ATGL and activates TAG hydrolysis (Fig. 3(b)). In addition to ATGL activation, a further physiological function for CGI-58 in phospholipid synthesis with lysophosphatidic acid acyltransferase activity has been observed( Reference Lafontan and Langin 4 ).
Lipid droplet proteins: Cide domain-containing proteins
A further family of lipid droplet-associated proteins encompasses the cell death-inducing DFFA (DNA fragmentation factor-α)-like effectors (Cide), which includes three members (Cidea, Cideb and Cidec/Fsp27) with tissue-specific expression( Reference Girousse and Langin 5 ). In spite of Cidea and Cideb not being expressed in white adipose tissue, their deletion yielded rodents with lower body weight and improved insulin sensitivity as well as resistant to diet-induced obesity( Reference Zhou, Yon Toh and Chen 314 , Reference Li, Ye and Xue 315 ). In the Cidea knockout model the elevated energy expenditure was attributable to brown adipose tissue via enhanced AMPK activity leading to increased fatty acid oxidation( Reference Qi, Gong and Zhao 316 ). The Cideb mutants exhibited a decreased hepatic VLDL secretion and de novo fatty acid oxidation related to enhanced hepatic oxidative activity( Reference Ye, Li and Liu 317 , Reference Tiwari, Siddiqi and Siddiqi 318 ). Cidea is also involved in human adipocyte lipolysis, TAG deposition and fatty acid oxidation via cross-talk with TNF-α, which inhibits the transcription of the gene( Reference Nordstrom, Ryden and Backlund 319 – Reference Christianson, Boutet and Puri 321 ). Cidea co-localises with perilipin around lipid droplets in fat cells. An increased lipolysis is observed in Cidea-depleted human adipocytes. Contrarily, ectopical expression of Cidea in preadipocytes markedly enhances lipid droplet size, promoting lipid accumulation( Reference Puri, Ranjit and Konda 322 ). Noteworthy, Cidea expression is elevated in human cancer cachexia, exhibiting a correlation with elevated NEFA concentrations and weight loss( Reference Laurencikiene, Stenson and Arvidsson Nordstrom 323 ). In humans Cidec, also referred to as fat-specific protein 27, FSP27, is predominantly expressed in subcutaneous adipocytes, being down-regulated in response to a reduced energy intake( Reference Magnusson, Gummesson and Glad 324 ). Small interfering RNA-mediated knockdown of Cidec translated into an increased basal release of NEFA, and decreased responsiveness to adrenergic lipolysis stimulation( Reference Lafontan and Langin 4 , Reference Ranjit, Boutet and Gandhi 325 ). The interaction between the diverse lipases is also starting to be unfolded. FSP27 and perilipin-1 interaction promotes the formation of large lipid droplets in human adipocytes( Reference Kim, Cho and Yun 326 – Reference Nian, Sun and Yu 329 ). Recently, the unilocular to multilocular transformation that takes place during ‘browning’ of white adipose tissue has been related to Cide-triggered dynamic changes in lipid droplet-associated proteins( Reference Barneda, Frontini and Cinti 330 ).
Lipid droplet proteins: other proteins (GPIHBP1 and Rab)
Glycosylphosphatidylinositol-anchored HDL-binding protein (GPIHBP1) is a 28-kDa glycosylphosphatidylinositol-anchored glycoprotein located on the luminal surface of endothelial cells in tissues where lipolysis takes place such as adipose tissue, skeletal muscle and heart( Reference Young and Zechner 7 , Reference Muller, Wied and Walz 331 ). The expression of GPIHBP1 in mice is modulated by fasting and refeeding as well as by PPAR-γ agonists. GPIHBP1 knockout mice exhibit chylomicronaemia, even on a low-fat diet, with highly elevated plasma TAG concentrations( Reference Muller, Jung and Wied 332 – Reference Adeyo, Goulbourne and Bensadoun 334 ). GPIHBP1 is highly expressed in the same tissues that express high levels of LPL, namely, heart, adipose tissue, and skeletal muscle where it binds both LPL and chylomicrons, suggesting that GPIHBP1 functions as a platform for LPL-dependent lipolytic processing of TAG-rich lipoproteins, stabilising LPL without activating it.
Rab GTPases, which are key regulators of membrane trafficking, have emerged as particularly relevant molecules in the highly dynamic cellular interactions involved in lipid mobilisation. In this sense, proteomic analyses have consistently identified the small GTPase Rab18 as a component of the lipid droplet coat( Reference Peinado, Pardo and de la Rosa 335 ). Thus, Rab18 provides an excellent marker to follow the dynamics of lipid droplets in living cells as well as to gain insight into the complex regulatory mechanisms involved in lipid storage and release( Reference Malagon, Cruz and Vazquez-Martinez 336 – Reference Pulido, Rabanal-Ruiz and Almabouada 338 ). In 3T3-L1 adipocytes, stimulation of lipolysis increases the association of Rab18 with lipid droplets, suggesting that Rab18 recruitment is regulated by the metabolic state of individual lipid droplets. Furthermore, Rab1a and its effector protein are reportedly involved in the CD36 trafficking signalling pathway( Reference Thompson, Lobo and Bernlohr 259 ).
Integral membrane proteins and transporters
While the main signalling cascades and regulators of lipolysis have been identified, the cellular interactions involved in lipid mobilisation and release still remain to be completely disentangled. Except in adipocytes, lipid droplets are normally small, mobile and interact with other cellular compartments in cells. On the contrary, fat cells are composed mainly of very large, immotile lipid droplets. The striking morphological differences between lipid droplets in adipocytes and non-adipocytes suggest that key differences must exist in the way in which lipid droplets in different cell types interact with other organelles to facilitate lipid transfer. A plethora of molecules involved in these interactions are now emerging, with integral membrane proteins and fatty acid transporters standing out as pivotal elements operating at the dynamic plasma membrane–lipid droplet interface.
Integral membrane proteins and transporters: aquaporin-7
Aquaporins (AQP) are integral membrane proteins that function mainly as water channels. AQP7 belongs to the subfamily of aquaglyceroporins, which are permeable to both glycerol and water, being expressed in adipocytes( Reference Frühbeck 339 – Reference Walker, Holness and Gibbons 341 ). Mouse and human AQP7 exhibit six prospective sites for PKA phosphorylation, suggesting a putative cAMP/PKA-dependent regulation. Aqp7-knockout mice show defective glycerol exit from fat cells, adipocyte hypertrophy due to TAG accumulation and moderate adult-onset obesity( Reference Hara-Chikuma, Sohara and Rai 342 , Reference Hibuse, Maeda and Nagasawa 343 ). Short-term regulation and translocation of AQP7 to the plasma membrane is stimulated by catecholamines, while insulin exerts a long-term negative control. More recently, in addition to AQP7, the presence and functionality of other members of the aquaglyceroporin subfamily, AQP3 and AQP9, have been identified in adipose tissue and shown to be regulated by insulin and leptin via the PI3K/Akt/mTOR signalling cascade( Reference Rodríguez, Catalán and Gómez-Ambrosi 344 ).
Integral membrane proteins and transporters: caveolin-1
Caveolae account for over 25 % of the adipocyte's membrane, being specialised plasma membrane microdomain invaginations involved in important cellular transport processes such as endo- and transcytosis as well as signal transduction( Reference Frühbeck, López and Diéguez 345 ). Three classes of caveolae formed by caveolin-1, the scaffolding hairpin-like protein facing the cytosol, have been identified, with high-density caveolae taking up exogenous fatty acids and converting them to TAG. These TAG-metabolising caveolae serve as a platform for FABP4, fatty acid transport protein (FATP) 1 and 4 (FATP1 and FATP4), long-chain acyl-CoA synthetase 1 (ACSL1) and CD36 (also known as fatty acid translocase). Noteworthy, these caveolae contain FATP1 and FATP4 together with the enzymes needed for TAG synthesis( Reference Trigatti, Anderson and Gerber 346 – Reference Meshulam, Breen and Liu 348 ). Furthermore, HSL and perilipin have been shown to be associated to these caveolae( Reference Cohen, Razani and Schubert 349 ), demostrating that TAG can be hydrolysed in them (Fig. 4). Caveolin-1 exerts an indirect structural role in caveolae formation, controlling surface availability or stability of CD36, a fatty acid transporter key to long-chain fatty acid uptake( Reference Covey, Brunet and Gandhi 350 ). In response to NEFA, caveolin-1 reportedly translocates from the plasma membrane to lipid droplets. Caveolin-1 knockout mice lack caveolae in adipocyte plasma membranes, exhibiting increased circulating NEFA and TAG, reduced adipocyte lipid droplet size and resistance to diet-induced obesity( Reference Cohen, Razani and Wang 351 ). Experiments with caveolin-1-null mouse embryonic fibroblasts indicate that caveolin-1 deficiency is followed by a total loss of caveolae, absence of CD36 plasma membrane expression and a reduction in fatty acid uptake, which is reverted by re-expression of caveolin-1( Reference Ring, Le Lay and Pohl 352 ). Interestingly, caveolin-1 has been shown to exert inhibitory interactions with various proteins such as PKA, endothelial NOS and insulin receptors, with knockout mice exhibiting an attenuated lipolytic activity and decreased perilipin phosphorylation( Reference Cohen, Razani and Schubert 349 ). Caveolin-1 potently inhibits cAMP-dependent signalling in vivo, with a direct interaction between caveolin-1 and the catalytic subunit of PKA having been demonstrated both in vitro and in vivo.
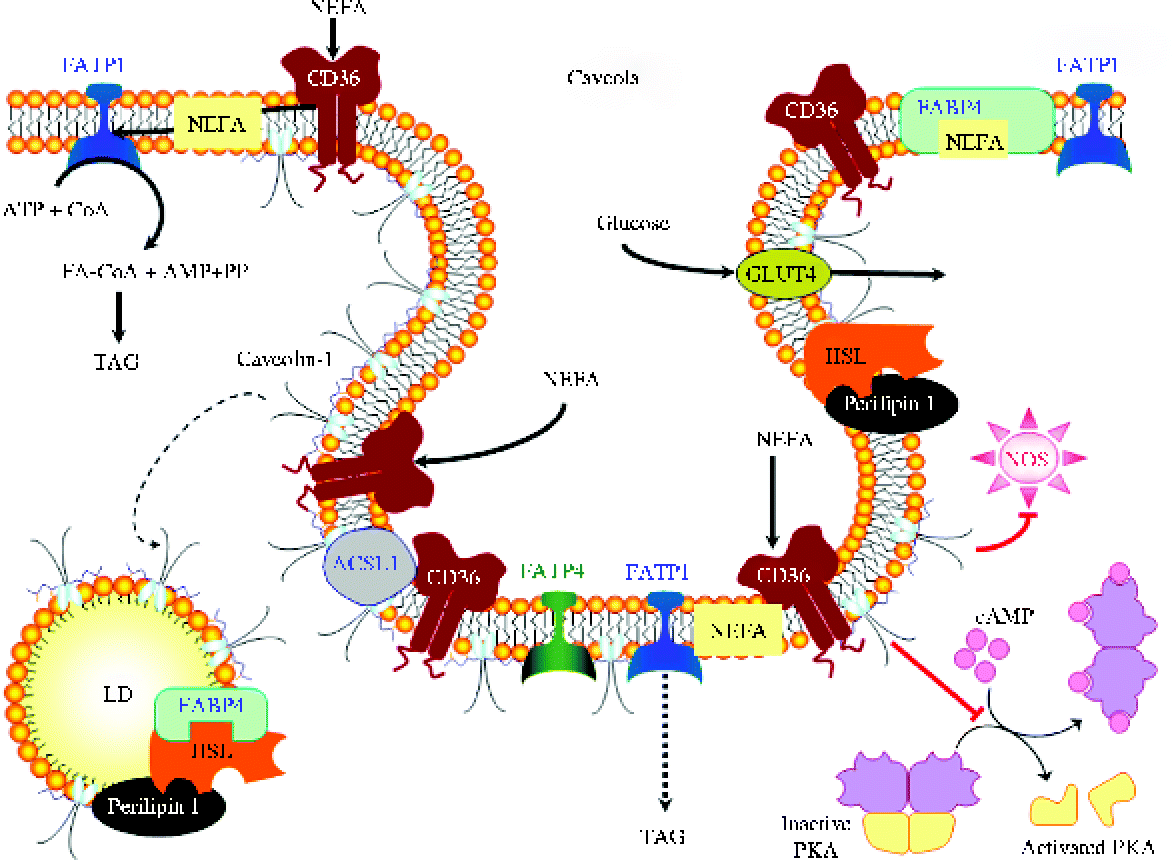
Fig. 4 Schematic diagram of a caveola present in the adipocyte's membrane and its participation in lipolysis. ACSL1, acyl coenzyme A synthetase 1; cAMP, cyclic AMP; CD36, fatty acid translocase; FA, fatty acid; FABP, fatty acid binding protein; FATP, fatty acid transport protein; HSL, hormone-sensitive lipase; LD, lipid droplet; NOS, NO synthase; PKA, protein kinase A; PP1, pyrophosphate. (A colour version of this figure can be found online at http://www.journals.cambridge.org/nrr)
Integral membrane proteins and transporters: fatty acid translocase (CD36)
As mentioned above, CD36 localises to caveolae as well as to intracellular vesicles. CD36 is a glycoprotein belonging to the family of class B scavenger receptors predicted to have two transmembrane domains at the N- and C-terminal, a large extracellular domain loop and two short intracellular cytoplasmic tails( Reference Thompson, Lobo and Bernlohr 259 ). CD36 is expressed in organs with high fatty acid metabolism rates, such as adipose tissue, operating as a NEFA scavenger. Insulin activation of the forkhead transcription factor and AMPK stimulation trigger CD36 translocation from intracellular stores to the plasma membrane, thereby enhancing NEFA uptake. CD36 deficiency is associated with increased basal lipolysis and responsiveness to the anti-lipolytic effect of insulin, with Cd36-null mice exhibiting an impaired fatty acid uptake in metabolic tissues (including adipocytes) and increased plasma NEFA and TAG concentrations( Reference Zhou, Samovski and Okunade 353 , Reference Vroegrijk, van Klinken and van Diepen 354 ). Knockdown of CD36 by RNAi in 3T3-L1 adipocytes resulted in a profound reduction of both basal and insulin-stimulated NEFA uptake. Conversely, overexpression of CD36 led to mice with decreased adiposity and low circulating levels of NEFA, TAG and cholesterol, suggesting that a strict control of these molecules for an effective lipolysis is required.
Integral membrane proteins and transporters: adipose fatty acid binding protein
FABP4, also known as ALBP and aP2, is a cytosolic lipid-binding protein highly expressed in adipocytes involved in fatty acid and retinoic acid intracellular trafficking( Reference Thompson, Lobo and Bernlohr 259 ). It acts as a molecular chaperone, facilitating NEFA uptake and lipolysis, interacting with HSL and shuttling fatty acids out of adipocytes (Fig. 4). Upon PKA activation the HSL–FABP4 complex translocates to lipid droplets. Consistently with this, in Fabp4-knockout mice basal and stimulated lipolysis are attenuated( Reference Kienesberger, Oberer and Lass 291 , Reference Scheja, Makowski and Uysal 355 – Reference Wei, Zan and Wang 357 ). Interestingly, Fabp4-null mice have been shown to compensate FABP4 deletion by increasing the expression of other FABP, thereby highlighting that lipolysis seems to be linked to total FABP content rather than to a specific FABP type( Reference Lafontan and Langin 4 ).
Integral membrane proteins and transporters: fatty acid transport protein 1
The underlying mechanism for fatty acid uptake by FATP1, an integral membrane protein of about 71 kDa with a hydrophobic domain at the N-terminal that may be membrane-anchored and other membrane-associated domains peripherally associated with the inner leaflet of the membrane, is still unknown. In response to insulin, FATP1 may translocate to structurally disordered non-lipid raft regions of the plasma membrane. Subsequently, FATP1 may extract fatty acid from the inner membrane leaflet and esterify it to CoA, thereby preventing its efflux and driving a NEFA concentration gradient across the membrane( Reference Wu, Ortegon and Tsang 358 , Reference Richards, Harp and Ory 359 ). Most of the incoming fatty acids are converted into acyl-CoA and preferentially shunted into TAG synthesis (Fig. 4). Noteworthy, the conversion of incoming long-chain fatty acids to TAG takes place on or around the plasma membrane in rat adipocytes, plausibly linking in a mechanistic way fatty acid influx to TAG synthesis( Reference Thompson, Lobo and Bernlohr 259 , Reference Liu, Gauthier and Sun 360 ). Knockdown and knockout experiments revealed an absolute requirement for FATP1 in insulin-stimulated fatty acid uptake, whereas FATP1 overexpression led to a fatty acid uptake increase.
Integral membrane proteins and transporters: fatty acid transport protein 4
FATP4 presents a 60 % identity to FATP1 and is expressed in adipose tissue, skin, heart, skeletal muscle, liver, as well as in the small intestine, where it was observed to work in intestinal lipid absorption( Reference Thompson, Lobo and Bernlohr 259 , Reference Lenz, Marx and Chamulitrat 361 ). FATP4 knockdown in 3T3-L1 adipocytes by RNAi did not affect basal and insulin-stimulated fatty acid uptake. FATP4 knockouts exhibit perinatal lethality due to restrictive dermopathy, suggesting a key role in the formation of the epidermal barrier rather than in fatty acid uptake and intestinal lipid absorption.
Integral membrane proteins and transporters: acyl-CoA synthetase long-chain 1
ACSL1, a 78-kDa membrane protein expressed in adipocytes and localised to various subcellular sites including the plasma membrane, lipid droplets, and GLUT4-containing vesicles, co-localises with FATP1( Reference Thompson, Lobo and Bernlohr 259 ). ACSL1 was found to be involved in the reacylation of fatty acids released from the lipid droplets during basal and hormone-induced lipolysis( Reference Richards, Harp and Ory 359 ). Overexpression of ACSL1 in fibroblasts is followed by an increase in NEFA uptake, thereby supporting a co-operative role in fatty acid transport across the adipocyte plasma membrane( Reference Phillips, Goumidi and Bertrais 362 ). However, knockdown of ACSL1 expression by RNAi in 3T3-L1 adipocytes points to a role in fatty acid efflux but not influx.
Depot-specific differences
The main anatomical fat depots in humans include intra-abdominal (greater and lesser omental and mesenteric depots, also known as visceral fat), lower-body (gluteal, subcutaneous leg and intramuscular fat) and upper-body subcutaneous fat( Reference Rodríguez, Catalán and Gómez-Ambrosi 363 , Reference Tchkonia, Thomou and Zhu 364 ). Subcutaneous adipose tissue constitutes the largest site for fat storage (about 80 % of total body fat), while under normal circumstances visceral adipose tissue accounts for a small fraction of body fat (about 20 % in men, and 5–8 % in women)( Reference Lafontan and Berlan 365 ). Regional differences, including preadipocyte replication and differentiation, adipocyte size, blood supply, gene expression, basal metabolic activities and hormonal responsiveness, contribute to regional fat distribution( Reference Rodríguez, Catalán and Gómez-Ambrosi 363 – Reference Caesar, Manieri and Kelder 366 ). Increased NEFA availability, resulting from increased effective adipose tissue lipolysis, plausibly undelies some of the visceral obesity-associated metabolic alterations( Reference Martin and Jensen 367 , Reference Nielsen, Guo and Johnson 368 ). Owing to its anatomical distribution, NEFA released from visceral fat are drained directly to the liver through the portal vein, whereas venous drainage of NEFA from subcutaneous adipose tissue is through systemic veins( Reference Ibrahim 369 ). The venous drainage of fat via the portal system directly provides NEFA as substrates for hepatic lipoprotein metabolism or glucose production. Excess NEFA favours the onset of dyslipidaemia, hyperinsulinaemia and insulin resistance by reducing hepatic degradation of apoB and insulin as well as by increasing VLDL production( Reference Lafontan and Langin 4 ).
Table 1 summarises regional variations in adipocyte lipolysis leading to increased NEFA release from visceral as compared with subcutaneous fat during hormone stimulation. Visceral adipocytes show the highest lipolytic responsiveness to catecholamines due to an increased function of the lipolytic β1-, β2- and β3-adrenoceptors( Reference Arner 370 , Reference Hoffstedt, Arner and Hellers 371 ). On the other hand, as mentioned above, several mechanisms have been linked to the weak lipolytic response to catecholamines in subcutaneous adipocytes, such as enhanced anti-lipolytic α2-adrenoceptor activity, decreased lipolytic β2-adrenoceptor responsiveness as well as reduced expression or function of HSL, FABP4 or perilipin( Reference Rodríguez, Catalán and Gómez-Ambrosi 363 , Reference Arner 370 ).
Table 1 Depot-specific differences of diverse factors regulating adipocyte lipolysis
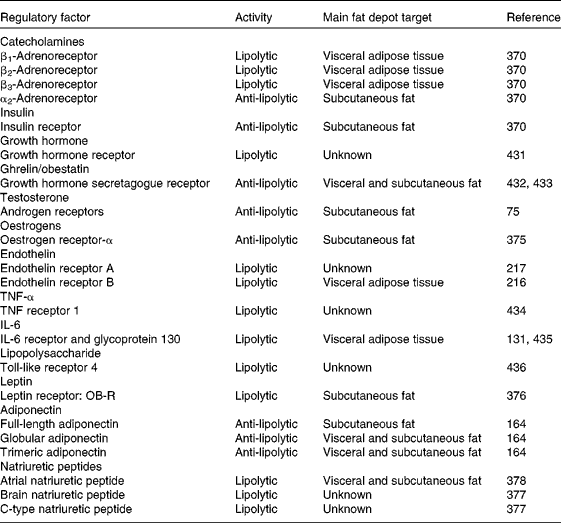
The anti-lipolytic effect of insulin is more prominent in subcutaneous adipocytes compared with visceral fat cells( Reference Arner 370 , Reference Zierath, Livingston and Thorne 372 ). Regional differences involve insulin receptor affinity, which is partly caused by variations in the insulin dissociation rate, but also by reduced insulin receptor phosphorylation and signal transduction via the IRS-1/PI3K pathway( Reference Arner 370 , Reference Zierath, Livingston and Thorne 372 , Reference Bolinder, Kager and Ostman 373 ). Testosterone has been reported to show both stimulatory( Reference Rebuffe-Scrive, Lonnroth and Marin 374 ) (i.e. up-regulation of β2-adrenoreceptors in visceral fat cells) and inhibitory( Reference Dicker, Ryden and Naslund 75 ) (i.e. down-regulation of β2-adrenoceptors and HSL in subcutaneous adipocytes) effects on cathecolamine-induced lipolytic activity. Oestrogen attenuates the lipolytic response through up-regulation of a number of anti-lipolytic α2-adrenergic receptors( Reference Pedersen, Kristensen and Hermann 375 ).
Leptin and adiponectin, the most abundant adipocyte-secreted factors, show opposite actions on lipolysis regulation( Reference Unger, Scherer and Holland 12 ). Leptin produces a significantly greater stimulation of lipolysis in subcutaneous fat cells compared with omental adipocytes( Reference Frühbeck and Gómez-Ambrosi 376 ). Adiponectin has recently emerged as an anti-lipolytic factor on binding adiponectin receptor type 1 and 2 (AdipoR1 and AdipoR2). Full-length adiponectin exerts an anti-lipolytic action in subcutaneous adipose tissue in non-obese subjects, while exhibiting no effect on visceral fat( Reference Kovacova, Tencerova and Roussel 163 , Reference Wedellova, Kovacova and Tencerova 164 ). Atrial (ANP), brain (BNP) and C-type (CNP) natiruretic peptides also induce lipolysis in human abdominal adipocytes, with the potency order of the lipolytic effect being ANP > BNP > CNP( Reference Dessi-Fulgheri, Sarzani and Rappelli 377 ). ANP-induced lipolysis is not subjected to primary regional regulation in differentiated human subcutaneous and visceral fat cells( Reference Dicker, Astrom and Wahlen 378 ). Fat-depot differences in the lipolytic effect of BNP and CNP remain to be established.
In addition to the physiological depot-specific differences in the neuroendocrine control of adipose tissue, it is important to consider the role of body fat distribution in the development of cardiometabolic alterations( Reference Rodríguez, Catalán and Gómez-Ambrosi 363 – Reference Caesar, Manieri and Kelder 366 ). Adipose tissue distribution varies with sex, age, genetic background, nervous and endocrine factors, nutritional and pharmacological influences as well as disease state, which impinge on preadipocyte replication and differentiation, developmental gene expression, vascularity, inflammation, adipokine secretion and apoptosis. The excess visceral fat observed in obesity is closely linked with metabolic and cardiovascular co-morbidities, whereas increased subcutaneous fat may even exert protective effects. However, how interdepot differences in the molecular, cellular, histological and pathophysiological properties translate into co-morbidity development needs to be fully unravelled( Reference Karelis 379 – Reference Ahima and Lazar 381 ).
Lipophagy: role of autophagy in lipid metabolism
Autophagy is a self-digestive process that entails the formation of double-membrane vesicles, termed autophagosomes, that sequester and target cytoplasmic cargo for lysosomal degradation( Reference Czaja 382 – Reference Singh 384 ). In addition to quality control, autophagy also regulates lipid metabolism by degrading lipid droplets via lipophagy (Fig. 5). Small lipid droplets can be completely taken up by an autophagosome, or alternatively portions of large lipid droplets can be degraded( Reference Czaja 382 ). Depletion of nutrients during starvation activates a second important cellular energy sensor, AMPK, that further activates unc51-like kinase 1 (ULK1) phosphorylation. Active ULK1 induces autophagy via the phosphorylation of beclin-1, a protein that recruits regulatory proteins to the VPS34 complex (class III PI3K), which is essential for the activity of the phagophore( Reference Russell, Tian and Yuan 385 ). During the vesicle elongation process, ATG7 induces the conjugation of ATG12 to ATG5 as well as the conjugation of cytosolic light chain 3 (LC3)-I to phosphatidylethanolamine to generate LC3-II, one of the best-characterised components of autophagosomes. Once formed, autophagosomes engulf lipid droplets and eventually fuse with a hydrolase-containing lysosome, the lipases of which degrade lipids( Reference Czaja 382 ). This process generates fatty acids that are released into the cytoplasm and can be oxidised in the mitochondria to generate ATP to maintain energy homeostasis. Under basal fed conditions, nutrients (particularly amino acids) or insulin and growth factors trigger the activity of class I PI3K that, in turn, activates mTOR, the best-characterised negative regulator of autophagy, and blocks autophagosome formation( Reference Kanazawa, Taneike and Akaishi 386 , Reference Pattingre, Espert and Biard-Piechaczyk 387 ) (Fig. 5). As a result, lipid breakdown by autophagy is minimal in the fed state.

Fig. 5 Regulation of lipophagy. AMPK, AMP-activated protein kinase; Akt, protein kinase B; ATG, autophagy-related gene; IGF-1, insulin growth factor-1; IRS 1/2, insulin receptor substrate 1/2; LC3, light chain 3; mTOR, mammalian target of rapamycin; P, phosphate; PI3K, phospatidylinositol-3 kinase; ULK1, unc51-like kinase 1; VPS15, phosphoinositide-3-kinase, regulatory subunit 4; VPS34, class III phosphatidylinositol 3-kinase. (A colour version of this figure can be found online at http://www.journals.cambridge.org/nrr)
Autophagy also participates in adipocyte differentiation regulation( Reference Moscat and Diaz-Meco 388 ). Transgenic animals lacking the autophagy-related proteins ATG5 and ATG7 show a reduction in adipose mass, supporting that autophagy is essential for normal adipogenesis( Reference Singh, Xiang and Wang 389 , Reference Zhang, Goldman and Baerga 390 ). Analogously, Atg5 and Atg7 knockdown in 3T3-L1 adipocytes decrease intracellular lipid content and gene expression levels of the key adipogenic transcription factors, CCAAT/enhancer-binding protein α and β (C/EBPα and β) and PPAR-γ( Reference Singh, Xiang and Wang 389 ). White adipocytes of Atg7-deficient mice acquire some characteristics of brown adipocytes, such as higher mitochondrial content, multilocular lipid droplets and increased levels of the brown adipogenic factors PPAR-γ-coactivator 1α (PGC-1α) and uncoupling protein-1 (UCP-1), triggering adipose tissue fatty acid β-oxidation( Reference Zhang, Goldman and Baerga 390 ). Interestingly, loss of Atg7 disrupts brown fat differentiation and promotes the ‘beige’ (brown adipocyte-like) cell development in inguinal adipose tissue, thereby contributing to increased energy expenditure( Reference Wu, Bostrom and Sparks 391 , Reference Martinez-Lopez, Athonvarangkul and Sahu 392 ).
Human adipose tissue contains autophagosomes and obesity is associated with an altered expression of the autophagy-related molecules LC3-I, LC3-II, beclin-1, ATG5 and ATG7( Reference Rodríguez, Gómez-Ambrosi and Catalán 200 , Reference Kovsan, Blüher and Tarnovscki 393 , Reference Nuñez, Rodrigues and Gomes 394 ). Markers of autophagy are correlated with whole-body adiposity, visceral fat distribution and adipocyte hypertrophy. However, the altered expression of autophagy in human obesity appears to be related to the degree of insulin resistance, rather than to excess adiposity( Reference Rodríguez, Gómez-Ambrosi and Catalán 200 ). In this sense, insulin constitutes a major inhibitor of autophagy, with insulin resistance being a potential activator of this process, since patients with type 2 diabetes show elevated formation of autophagosomes in subcutaneous adipose tissue( Reference Ost, Svensson and Ruishalme 395 ). Adipocyte autophagy is also regulated by TNF-α and ghrelin, showing opposite effects on the regulation of fat storage in human adipocytes( Reference Rodríguez, Gómez-Ambrosi and Catalán 200 ). TNF-α plays an important role in the pathophysiology of deranged lipid metabolism through both the suppression of LPL activity and enhancement of lipolysis in human fat cells( Reference Kawakami, Murase and Ogawa 396 ). In addition, TNF-α also triggers autophagy by increasing the transcript levels of BECN1 (beclin 1), required for the formation of the autophagosome initiation complex, as well as those of ATG5, and ATG7, the autophagy proteins involved in the conjugation cascades for autophagosome elongation in human adipocytes( Reference Rodríguez, Gómez-Ambrosi and Catalán 200 ). On the other hand, ghrelin is a gut-derived hormone that promotes adiposity through orexigenic and adipogenic actions( Reference Rodríguez, Gómez-Ambrosi and Catalán 199 , Reference López, Lage and Saha 397 ). Ghrelin isoforms (acylated and desacyl ghrelin) stimulate the expression of several fat storage-related proteins such as acetyl-CoA carboxylase, fatty acid synthase, LPL or perilipin through central mechanims( Reference López, Lage and Saha 397 ) and directly acting on human adipocytes( Reference Rodríguez, Gómez-Ambrosi and Catalán 199 ), thereby stimulating intracellular lipid accumulation. Besides its lipogenic action, acylated ghrelin reduces basal ATG5 and ATG7, while desacyl ghrelin inhibits TNF-α-induced expression of ATG5, ATG7 and BECN1. Taken together, ghrelin constitutes a negative regulator of basal and TNF-α-induced autophagy in human visceral adipocytes( Reference Rodríguez, Gómez-Ambrosi and Catalán 200 ).
Novel fascinating findings in the field of adipocyte apoptosis have been recently reported( Reference Murano, Rutkowski and Wang 398 , Reference Giordano, Murano and Mondini 399 ). White adipose tissue inflammation, a characteristic feature of obesity, results from the death of hypertrophic adipocytes that are subsequently cleared by macrophages, giving rise to crown-like structures (CLS). It has been recently shown that infiltrating macrophages actively take up remnant lipids of dead adipocytes( Reference Murano, Rutkowski and Wang 398 ). Upon induction of adipocyte apoptosis, inflammatory cells infiltrate adipose tissue initially consisting of neutrophils followed by macrophages that are involved in CLS formation. Moreover, subcutaneous and visceral hypertrophic adipocytes obtained from obese mice exhibit ultrastructural abnormalities (cholesterol crystals and Ca accumulation), being more common in the hyperglycaemic db/db v. normoglycaemic ob/ob mice and in the visceral v. subcutaneous depots. Data indicate that white adipocyte overexpansion induces a stress state that ultimately leads to death with NOD-like receptor family, pyrin domain containing 3 (NLRP3)-dependent caspase-1 activation in hypertrophic adipocytes probably inducing obese adipocyte death by pyroptosis, a proinflammatory programmed cell death( Reference Giordano, Murano and Mondini 399 ).
Lipolysis in human obesity
Obesity is characterised by a marked secretion of proinflammatory adipokines, including TNF-α, and a profound decrease in adiponectin synthesis( Reference Catalán, Gómez-Ambrosi and Ramírez 234 ). The increased TNF-α production in adipose tissue triggers MAP kinase activity in adipocytes, thus altering the action of perilipin and leading to an enhanced basal lipolytic rate( Reference Arner 2 , Reference Hotamisligil, Shargill and Spiegelman 400 ). Otherwise, adiponectin inhibits basal and cathecolamine-induced lipolysis in non-obese subjects, but this effect is lost in obesity( Reference Wedellova, Dietrich and Siklova-Vitkova 161 ). The isoform-specific ability to prevent lipolysis is modified in obesity. While full-length adiponectin exerts an anti-lipolytic action in subcutaneous fat, without effect on visceral fat, in non-obese individuals, the lower adiponectin isoforms (globular and trimeric) become important actors in obesity, showing anti-lipolytic activity in obese subcutaneous and visceral adipose tissue, respectively( Reference Wedellova, Kovacova and Tencerova 164 ).
Circulating NEFA and glycerol concentrations are elevated in obesity, suggesting an increase in overall lipolysis during fasting( Reference Rodríguez, Catalán and Gómez-Ambrosi 344 ). Several impairments in the control of lipolysis have been reported in obese individuals, including an altered responsiveness to catecholamines( Reference Arner 2 , Reference Lafontan and Langin 4 , Reference Jocken and Blaak 53 ). Obese subjects show a lower lipolytic effect of catecholamines in subcutaneous adipose tissue through decreased action of lipolytic β2-adrenergic receptors and increased activity of the anti-lipolytic α2-adrenergic adrenoceptors( Reference Arner 370 , Reference Mauriege, Despres and Prud'homme 401 ). In this regard, a blunted lipolytic response has been shown in abdominal subcutaneous adipose tissue of obese individuals during intravenous infusion of the non-selective β-agonist isoprenaline( Reference Jocken, Goossens and van Hees 402 ). On the other hand, catecholamine-induced lipolysis is markedly increased in visceral fat due to increased activity of β3-adrenergic receptors and decreased activity of α2-adrenoceptors( Reference Arner 370 , Reference Mauriege, Despres and Prud'homme 401 ). In subjects with upper-body obesity these regional variations in the action of catecholamines on lipolysis are further enhanced( Reference Nielsen, Guo and Johnson 368 , Reference Arner 370 ). These abnormalities in catecholamine function promote the release of NEFA from the visceral adipocytes through the portal system and might cause several of the metabolic complications of upper-body obesity. In addition, several polymorphisms in genes encoding β1- (ADRB1), β2- (ADRB2) and β3- (ADRB3) adrenergic receptors have been associated with altered cathecolamine-induced adipocyte lipolysis and with obesity( Reference Dahlman and Arner 403 , Reference Terra, McGorray and Wu 404 ). The polymorphisms in the ADRB2 gene are highly frequent in obesity and associated with altered β2-adrenergic function (Arg16Gly and Gln27Glu) and catecholamine-induced lipolysis in subcutaneous fat cells (Arg16Gly and Thr164Ile)( Reference Large, Hellstrom and Reynisdottir 42 , Reference Hoffstedt, Iliadou and Pedersen 405 , Reference Eriksson, Dahlman and Ryden 406 ). However, the ADRB1 (Ser49Gly and Arg389Gly)( Reference Terra, McGorray and Wu 404 , Reference Ryden, Hoffstedt and Eriksson 407 , Reference Tafel, Branscheid and Skwarna 408 ) and ADRB3 (Trp64Arg)( Reference Li, Lönnqvist and Luthman 409 – Reference Snitker, Odeleye and Hellmer 411 ) polymorphisms do not appear to be major determinants of β1- and β3-adrenergic function for lipolysis or the pathophysiology of obesity.
It is not clear whether the anti-lipolytic effect of insulin is affected in obesity, since the altered catecholamine concentrations found in the obese state counteract the effect of insulin( Reference Arner 2 ). Consequently, normal, decreased and increased anti-lipolytic effects of insulin have been reported in obese patients( Reference Lafontan and Langin 4 ). Insulin sensitivity of adipose tissue lipolysis is normal or slightly impaired in the adipose tissue of obese individuals( Reference Lafontan and Langin 4 , Reference Jocken, Goossens and Boon 412 ). Modifications of other anti-lipolytic factors may also be altered in obesity.
The pathological enlargement of fat cells in obesity compromises angiogenesis and increases the formation of hypoxic areas that promote the apoptosis of adipocytes and induce the fibrotic and inflammatory programme( Reference Sun, Kusminski and Scherer 87 ). Apoptotic adipocytes are surrounded by M1-stage macrophages that form CLS in the adipose tissue. This process is accompanied by a chronic inflammation due to the secretion by adipose tissue-embedded immune cells and the dysfunctional adipocytes of proinflammatory cytokines and acute-phase reactants, such as TNF-α, C-reactive protein, IL-6, IL-8, leptin, serum amyloid A (SAA) and monocyte chemotactic protein (MCP)-1( Reference Gómez-Ambrosi, Salvador and Rotellar 232 , Reference Catalán, Gómez-Ambrosi and Ramírez 234 ). As detailed in the Cytokines and other ‘newcomers’ section, the increase in proinflammatory adipokines, such as TNF-α or leptin, might be responsible for the high basal rate of lipolysis in obese patients.
Obesity is associated with a decreased expression and activity of HSL, but not ATGL, in visceral and subcutaneous adipocytes of obese individuals independently of age and sex, which may play an important role in the defective lipid mobilisation observed in obesity( Reference Mairal, Langin and Arner 413 – Reference Tinahones, Garrido-Sanchez and Miranda 415 ). Furthermore, a decreased access of lipases to TAG due to alterations in lipid droplet-associated proteins cannot be ruled out( Reference Gandotra, Le Dour and Bottomley 416 – Reference Magné, Aminoff and Sundelin 419 ). In humans CGI-58 mutations have been identified in patients with Chanarin–Dorfman syndrome, a disorder characterised by the accumulation of abnormally large amounts of lipid droplets in several organs( Reference Lass, Zimmermann and Haemmerle 420 , Reference Yamaguchi and Osumi 421 ). In these cases CGI-58 cannot be recruited to lipid droplets and fails to interact with perilipin, which may affect basal and PKA-stimulated lipolysis. Interestingly, CGI-58 gene silencing importantly reduces basal lipolysis by approximately 50 % but also completely abrogates PKA-stimulated lipolysis in a human white adipocyte model( Reference Bezaire and Langin 255 , Reference Bezaire, Mairal and Ribet 422 ). The exact and complex dynamics involving CGI-58, the diverse perilipins and ATGL in basal as well as PKA-stimulated lipolysis has yet to be completely unravelled.
Finally, changes in the molecules involved in lipolysis-derived metabolites, fatty acids and glycerol also contribute to lipolytic derangements in obesity. Several proteins like FABP, CD36 or FATP facilitate fatty acid transport across the membrane in adipocytes( Reference Rodríguez, Catalán and Gómez-Ambrosi 423 ). The transport of the other lipolysis-derived metabolite, glycerol, from adipocytes in response to the lipolytic stimuli is facilitated by AQP3 and AQP7 via their translocation from the cytosolic fraction (AQP3) or lipid droplets (AQP7) to the plasma membrane( Reference Walker, Holness and Gibbons 341 , Reference Rodríguez, Catalán and Gómez-Ambrosi 344 , Reference Kishida, Kuriyama and Funahashi 424 , Reference Yasui, Kubota and Iguchi 425 ). AQP7 expression is decreased in subcutaneous adipose tissue of obese subjects, resulting in an increase in intracellular glycerol accumulation, which is converted to glycerol-3-phosphate by the glycerol kinase enzyme and re-esterified into TAG, thereby promoting adipocyte hypertrophy( Reference Rodríguez, Catalán and Gómez-Ambrosi 344 , Reference Rodríguez, Catalán and Gómez-Ambrosi 426 ). On the other hand, the increased AQP3 and AQP7 expression in visceral fat in obese subjects suggests an overall increase in the lipolytic activity in this fat depot in obesity( Reference Rodríguez, Catalán and Gómez-Ambrosi 344 , Reference Rodríguez, Catalán and Gómez-Ambrosi 426 , Reference Catalán, Gómez-Ambrosi and Pastor 427 ).
Concluding remarks and future perspectives
While adipose tissue elicited scarce interest for many decades( Reference Lafontan 428 ), the identification in 1994 of leptin as an adipose-derived hormone( Reference Zhang, Proenca and Maffei 429 ) started a new era in adipobiology that recognises adipocytes as important dynamic endocrine cells. Essential lipolytic enzymes and a plethora of regulatory proteins and mechanisms have fundamentally changed our view of lipolysis and its impact, not only on adipose tissue but also more broadly on cellular metabolism( Reference Zierler, Jaeger and Pollak 430 ). Although the importance of lipolysis has been recognised for decades, many of the key proteins involved have been uncovered only recently. In this line, to further decipher the participation of lipolytic products and intermediates in many non-adipose tissues will be especially relevant to unravel previously underappreciated aspects of lipolysis and their relation to disease development. The regulation of lipolysis by numerous, and to some extent still incompletely identified, factors embodies the ‘lipolysome’, a complex metabolic network involved in ultimately controlling lipid mobilisation and fat storage. Information derived from the reactome linking the genome and metabolome via genome-sequence independent functional analysis of metabolic phenotypes and networks will be particularly fascinating. With the advent of systems biology a better integration of knowledge can be further expected to provide a more profound view of the true contribution of adipose tissue to health and disease.
Acknowledgements
The authors gratefully acknowledge the funding of the Spanish Instituto de Salud Carlos III, Fondo de Investigación Sanitaria – FEDER (project numbers CIBERobn CB06/03/1014, FIS PI10/01677 and PI12/00515) from the Ministerio de Economía y Competitividad, as well as the Plan de Investigación de la Universidad de Navarra (project PIUNA 2011-13). None of the funders had a role in the design, analysis or writing of this article.
All authors contributed fundamentally to the present paper. G. F. conducted the main review of the literature and drafted the manuscript. A. R. and L. M.-G. contributed significantly to the further drafting of the manuscript. All authors (G. F., L. M.-G., J. A. F.-F., S. F. and A. R.) made a critical review of the draft, provided input on data interpretation as well as commented on and approved the final manuscript.
The authors declare no conflicts of interest.






