


Introduction
Glucose homeostasis is mainly controlled by the islets of Langerhans, which constitute the endocrine portion of the pancreas. About one million islets are distributed throughout the exocrine pancreas in an adult human. The size of these multicellular structures varies from 50 to 500 μm. They are composed of 1000–2000 different types of cells, which secrete several cell-specific hormones and peptides in response to nutrients: glucagon is released from α-cells, insulin from β-cells, somatostatin from δ-cells, pancreatic polypeptide from PP-cells and ghrelin from ɛ-cells( Reference Brissova, Fowler and Nicholson 1 , Reference Cabrera, Berman and Kenyon 2 ). α- and β-Cells are the most abundant islet cell types. In rodents, glucagon- and insulin-releasing cells constitute about 15–20 % and 60–80 % of the total islet population, respectively, although the proportion of each cell type may vary depending on the anatomical portion of the pancreas( Reference Brissova, Fowler and Nicholson 1 , Reference Cabrera, Berman and Kenyon 2 ). While β-cells are mostly integrated in the central core of the islet, α-cells are around the periphery forming a cell mantle. However, the α-cell population in humans is up to 45 % of the islet and is distributed in patches throughout the whole islet, frequently juxtaposed to β-cells. While intercellular communication via gap junctions is frequent in β-cells, leading to synchronised responses like a syncitium, α-cells work individually without apparent intercellular coupling( Reference Nadal, Quesada and Soria 3 , Reference Quesada, Todorova and Soria 4 ). These cytoarchitecture features along with results of microcirculatory flow studies suggest that there is an important intercellular communication conduit from β- to α-cells( Reference Smith, Rosen and Villa-Komaroff 5 ), in which paracrine interactions play a prominent role( Reference Unger and Orci 6 ). An extensive capillary network throughout the islet( Reference Cabrera, Berman and Kenyon 2 ) allows for a rapid sensing of changes in plasma nutrients and signalling molecules, triggering the subsequent secretory responses. Additionally, islet cells, particularly α-cells, are subjected to a precise regulation through a highly innervated sympathetic and parasympathetic system that extends through the perivascular spaces. This neural network has a critical function in rapid islet secretory responses, particularly under hypoglycaemic conditions( Reference Ahren 7 , Reference Taborsky and Mundinger 8 ). Thus, among the numerous signalling levels that regulate the secretory capacity of islet cells, nutrients are a major player.
Changes in plasma glucose levels are one of the main control inputs for the release of glucagon and insulin. In pancreatic β-cells, high glucose levels induce the intracellular uptake of the carbohydrate, triggering the coupling between intracellular metabolic signals and the insulin secretory output. Insulin receptors are then activated by increased plasma insulin levels, leading to the uptake of glucose and its accumulation as glycogen or fat in muscle, liver and adipose tissue. This process leads to a decrease in plasma glucose concentrations until they reach basal levels. In contrast, pancreatic α-cells release glucagon more actively in hypoglycaemic conditions. This hormone binds to its receptors in the liver, the main target organ of glucagon, and activates hepatic gluconeogenesis and glycogenolysis( Reference Quesada, Tuduri and Ripoll 9 , Reference Unger, Aguilar-Parada and Muller 10 ). As a consequence, hepatic glucose is released to the bloodstream, allowing for a restoration of plasma glucose levels. Since insulin and glucagon share some target organs but have opposite roles, the final effect of both hormones depends on the insulin:glucagon ratio. In addition to glucose, plasma lipids and amino acids also regulate hormonal release from both cell types and, at the same time, insulin and glucagon act on fat and protein metabolism( Reference Arafat, Kaczmarek and Skrzypski 11 – Reference Xiao, Pavlic and Szeto 14 ), as will be discussed later. All these processes are impaired in diabetes mellitus. This metabolic disease is not only the result of insulin secretory abnormalities, the loss of β-cell mass and peripheral insulin resistance but it is also the outcome of altered glucagon release as well as α-cell mass changes( Reference Cryer 15 ). Despite the importance of glucagon secretion, understanding of the function of α-cells in health and diabetes has remained elusive for a long time. This limited information was mainly due to the scarcity of α-cells within the islets of common animal models, the low availability of glucagon-releasing cell lines, the lack of physiological patterns for the recognition of this cell type and several limitations of the available techniques and methodologies. However, in the last decade, the biology of the pancreatic α-cell has experienced a renaissance due to technical advances along with the development of several experimental strategies based on the modulation of glucagon secretion and/or action that may be helpful in the treatment of diabetes and other endocrine disorders. In the present review, all these aspects will be discussed.
Nutrient regulation of pancreatic α-cells
Among the multiple control levels that regulate pancreatic α-cell function, nutrients exert a primary role (Fig. 1). Nutrients modulate pancreatic α-cell secretion, glucagon gene expression, cell proliferation and cell death.
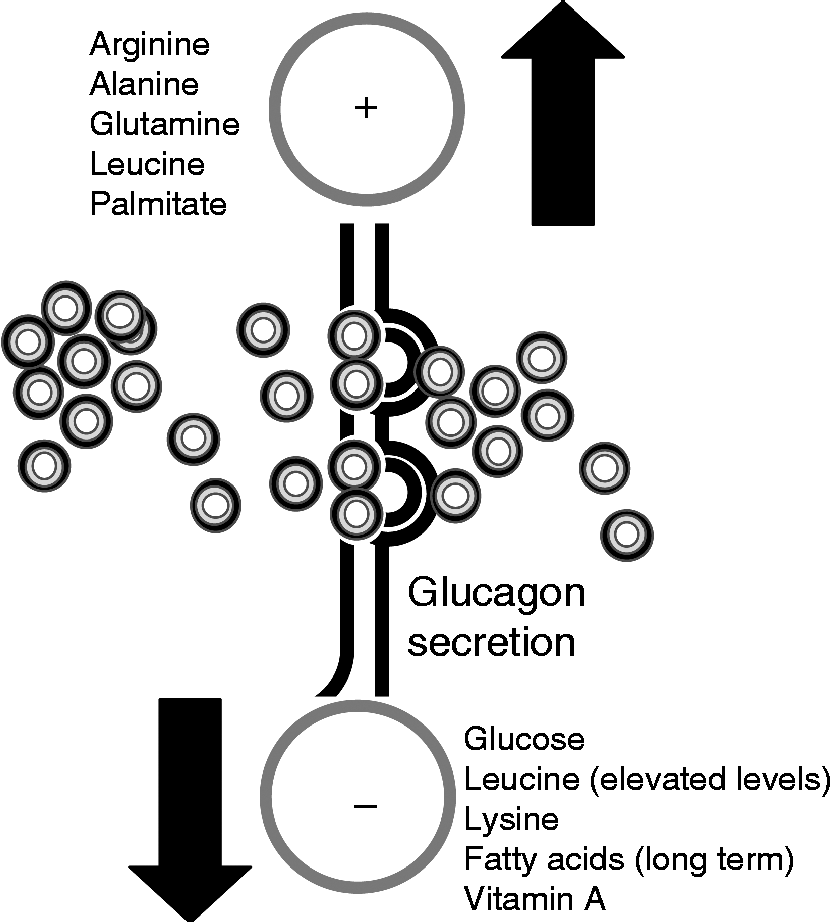
Fig. 1 Nutrient regulation of glucagon secretion. The stimulatory (+) and inhibitory (–) effects of different nutrients on glucagon release by pancreatic α-cells are shown. For further information, see the text.
Glucose
One of the main stimuli that modulate glucagon secretion is plasma glucose. The ingestion, injection or infusion of glucose in animal models and human subjects has been proven to suppress glucagon secretion, while decreased plasma glucose levels are correlated with enhanced glucagon release( Reference Unger, Aguilar-Parada and Muller 10 , Reference Gerich, Lorenzi and Schneider 16 , Reference Ohneda, Aguilar-Parada and Eisentraut 17 ). Similarly, in vitro experiments have shown that glucagon release is stimulated in hypoglycaemic conditions, whereas high glucose levels down-regulate α-cell secretory activity( Reference Salehi and Vieira 18 , Reference Vieira and Salehi 19 ). Pancreatic α-cells are excitable cells able to respond to changes in extracellular glucose concentrations. At low glucose levels, these cells develop action potentials that trigger intracellular Ca signals and glucagon secretion in rodent and human islets( Reference Nadal, Quesada and Soria 3 , Reference Quesada, Todorova and Soria 4 , Reference Gopel, Zhang and Eliasson 20 – Reference Tuduri, Marroqui and Soriano 22 ). In contrast, hyperglycaemic conditions inhibit α-cell functional activity, lowering glucagon release( Reference Quesada, Tuduri and Ripoll 9 ). This glucose modulation may rely on different control levels: the neural sensing of glucose and the subsequent neural regulation of α-cells( Reference Taborsky, Ahren and Havel 23 , Reference Thorens 24 ), the sensing of glucose by neighbouring β- and δ-cells within the islet and the inhibition of pancreatic α-cells by paracrine regulation (see the following sections) and, lastly, the glucose sensing and intracellular metabolism directly by pancreatic α-cells( Reference Rorsman, Salehi and Abdulkader 25 ). The latter possibility has been the subject of intense debate. While some studies indicate that ATP-dependent K+ (KATP) channels play a central role in coupling glucose-modulated metabolic changes with pancreatic α-cell electrical activity and secretion( Reference Rorsman, Salehi and Abdulkader 25 , Reference MacDonald, De Marinis and Ramracheya 26 ), other reports support a KATP-independent glucose effect( Reference Gylfe 27 – Reference Quoix, Cheng-Xue and Mattart 29 ). Some of these controversies have emerged from studies of α-cell metabolism. The analysis of mitochondrial membrane potential, NADH autofluorescence or direct measurements of cytosolic ATP levels have shown both measurable and negligible glucose-induced changes in α-cell metabolism( Reference Quesada, Todorova and Soria 4 , Reference Quoix, Cheng-Xue and Mattart 29 – Reference Ravier and Rutter 31 ). In this regard, it has been described that glucose-induced ATP/ADP changes in α-cells are very low compared with the ones in β-cells, probably because of the biochemical differences existing between both islet cell types( Reference Detimary, Dejonghe and Ling 32 ). In experiments with either samples of non-β-cells or purified α-cells, it has been determined that GLUT-1 is the high-affinity, low-capacity GLUT present in α-cells, in contrast to GLUT-2, which is present in β-cells. Despite this difference, this metabolic step is not a limiting factor in α-cell glucose utilisation( Reference Gorus, Malaisse and Pipeleers 33 , Reference Heimberg, De Vos and Pipeleers 34 ). Other biochemical differences between both cell types also suggest that α-cells are less competent in the use they make of mitochondrial glucose oxidation( Reference Quesada, Todorova and Soria 4 , Reference Schuit, De Vos and Farfari 35 ). In fact, compared with pancreatic β-cells, non-β-cells exhibit a higher expression of the lactate/monocarboxylate transporter and a lower expression of pyruvate carboxylase( Reference Sekine, Ito and Hashimoto 36 , Reference Zhao, Wilson and Schuit 37 ). Additionally, non-β-cells present higher rates of lactate dehydrogenase activity, while mitochondrial glycerol phosphate dehydrogenase activity is low( Reference Sekine, Ito and Hashimoto 36 ). These characteristics may explain why the coupling between glycolysis and mitochondrial glucose oxidation is not high in α-cells. Recent reports, however, have shown that in pancreatic α-cells labelled with yellow fluorescent protein, glucose hyperpolarises the mitochondrial membrane potential, further indicating that it directly affects mitochondrial metabolism and pointing to a KATP-dependent modulation of glucagon secretion by glucose( Reference Allister, Robson-Doucette and Prentice 38 ). In contrast, another recent study demonstrates that glucose acts directly on α-cells to control glucagon release but through KATP-independent mechanisms( Reference Cheng-Xue, Gomez-Ruiz and Antoine 39 ). Additionally, the authors suggest that glucagon output may depend on the balance between the direct and indirect (paracrine) effects of glucose. In recent years, several molecules have been identified that may play a key role in the glucose-sensing capacity of α-cells and its transduction into glucagon secretion. These include the per-arnt-sim domain-containing protein kinase (PASK) and AMP-activated protein kinase (AMPK), which are well-known energy sensors, as well as Rev-erb α, a clock gene involved in metabolism( Reference da Silva Xavier, Farhan and Kim 40 – Reference Vieira, Marroquí and Figueroa 42 ). Thus, a larger consensus is required regarding the role of α-cell KATP channels and glucose metabolism in glucagon release. In addition to the role of glucose in glucagon secretion, it may also be involved in the regulation of α-cell survival, although few studies have been made regarding this. It has been documented that high levels of glucose (33.3 mm) combined with palmitate to simulate in vitro glucolipotoxicity conditions induce apoptosis in pancreatic α-cells( Reference Ellingsgaard, Ehses and Hammar 43 ).
Amino acids
Although amino acids are important modulators of α-cell secretion, little is known about this process at the molecular level and the mechanisms involved. Amino acids such as arginine, alanine and glutamine are potent stimulators of glucagon release, while leucine or lysine contribute to a lesser extent to α-cell secretion( Reference Gerich, Lorenzi and Schneider 16 , Reference Dumonteil, Magnan and Ritz-Laser 44 – Reference Pipeleers, Schuit and Van Schravendijk 46 ). It is postulated that the function of this increase in glucagon release is to physiologically prevent hypoglycaemia after protein intake, since amino acids also stimulate insulin secretion. An increased glucagon secretion is observed if α-cells are incubated in the presence of a mixture of different amino acids but not when exposed to each one of the nutrients separately( Reference Quoix, Cheng-Xue and Mattart 29 , Reference Cheng-Xue, Gomez-Ruiz and Antoine 39 , Reference Pipeleers, Schuit and Van Schravendijk 46 ). In fact, the effect of glucose on intracellular Ca signalling and glucagon release is further detected in the presence of mediums containing a mixture of amino acids( Reference Cheng-Xue, Gomez-Ruiz and Antoine 39 ), indicating that these nutrients are critical for α-cell function. Ostenson & Grebing( Reference Ostenson 47 ) showed that glutamine is a positive modulator of glucagon release. This effect involves the oxidation of this amino acid and is likely to be related to actions on glucose metabolism and ATP levels in the pancreatic α-cell( Reference Ostenson 47 ). Moreover, a recent study performed in human islets from healthy and type 2 diabetic individuals showed that glycine, acting via an α-cell-specific glycine receptor, was the predominant amino acid stimulating glucagon release via an intracellular Ca influx( Reference Li, Liao and Zhuo 48 ). Similarly, ingested glycine increases plasma glucagon in human subjects( Reference Gannon, Nuttall and Nuttall 49 ). Amino acids like arginine may be involved in the release of glucagon by direct plasma membrane depolarisation and Ca influx in the pancreatic α-cell. However, other amino acids act as negative modulators. For instance, isoleucine can inhibit α-cell secretion while leucine has a dual effect: it is a positive stimulus at physiological concentrations but becomes inhibitory at elevated levels( Reference Leclercq-Meyer, Marchand and Woussen-Colle 50 ). In In-R1-G9 glucagonoma cells, concentrations of alanine and glutamine in the micromolar range contribute to the triggering of intracellular Ca oscillations, whereas these signals are inhibited with millimolar levels of these amino acids( Reference Bode, Weber and Fehmann 51 ). This effect has been attributed to amino acid-induced hyperpolarisation of the plasma membrane by the modulation of the Na–K pump, and involves the intracellular metabolism of these nutrients. Therefore, divergent effects may take place depending on the type and concentration of amino acids.
Fatty acids
Even though lipotoxicity in pancreatic β-cells is one of the main factors related to obesity-induced diabetes, less is known about the role and potential toxic effect of fatty acids in the α-cell. It has been shown that the modulation of glucagon release by fatty acids depends on the chain length, spatial configuration and degree of saturation of the fatty acid as well as the incubation time, which may lead to different acute or chronic effects( Reference Collins, Luan and Thompson 52 – Reference Olofsson, Salehi and Gopel 54 ). The majority of studies show that short-term exposure to fatty acids stimulates glucagon release in isolated islets and clonal α-cell lines( Reference Olofsson, Salehi and Gopel 54 , Reference Bollheimer, Landauer and Troll 55 ). Palmitate increases glucagon secretion by enhancing α-cell intracellular Ca entry and also, by relieving the inhibitory paracrine action of somatostatin, which is secreted from δ-cells( Reference Olofsson, Salehi and Gopel 54 ). Linoleic acid increases glucagon secretion in isolated mouse and rat islets as well as in In-R1-G9 glucagonoma cells( Reference Flodgren, Olde and Meidute-Abaraviciene 56 , Reference Wang, Zhao and Gui 57 ). This effect depends on the activation of the NEFA receptor G-protein coupled receptor 40 (GPR40) and the intracellular release of InsP3. Similar findings have been reported in rat islets with oleic acid( Reference Fujiwara, Maekawa and Dezaki 58 ). The long-term effects of fatty acids have been examined alone or in the presence of high concentrations of glucose. When αTC1-6 clonal α-cells were cultured with palmitate and oleate for up to 3 d, glucagon secretion was enhanced by means of fatty acid oxidation and TAG accumulation in a time- and dose-dependent manner but cell proliferation was decreased( Reference Hong, Jeppesen and Nordentoft 59 ). Accordingly, the long-term culture of this clonal α-cell line with palmitate augmented glucagon release and enhanced glucagon expression and protein content, probably by activating the mitogenic mitogen-activated protein kinase (MAPK) pathway( Reference Piro, Maniscalchi and Monello 60 ). In contrast, it was observed that the inhibitory action of insulin on glucagon release was impaired in a long-term incubation with fatty acids. This was attributed to palmitate-induced insulin resistance due to defects in the insulin receptor substrate-1 (IRS-1)/phosphatidylinositol kinase (PI3K)/serine-threonine protein kinase (Akt) pathway( Reference Piro, Maniscalchi and Monello 60 ). In mouse isolated islets, long-term exposure to oleate and palmitate results in an over-secretion of glucagon at both low and high glucose concentrations but diminished glucagon protein content( Reference Collins, Luan and Thompson 52 ). Similar results were observed in rat islets: the chronic effects of fatty acids produced a marked increase in glucagon release, but decreased glucagon content and did not change glucagon gene expression( Reference Dumonteil, Magnan and Ritz-Laser 44 , Reference Gremlich, Bonny and Waeber 61 ). It has been recently reported that the survival of α-cells, like of β-cells, is also sensitive to glucolipotoxic conditions: palmitate (0.5 mm) combined with high glucose levels are able to induce apoptosis in rodent α-cells( Reference Ellingsgaard, Ehses and Hammar 43 ). In general, all these data support the hypothesis that chronic elevation of fatty acids might contribute to α-cell deregulation in type 2 diabetes.
Other nutrients
α-Cell function is also sensitive to vitamins. For instance, plasma glucagon levels and glucagon release have been found to be augmented in rats with a diet deficiency of vitamin D3 ( Reference Bourlon, Faure-Dussert and Billaudel 62 ) while no major effects have been reported in non-insulin-dependent diabetic patients supplemented with vitamin D( Reference Orwoll and Riddle 63 ). Glucagon secretion is markedly impaired in rats subjected to vitamin A dietary deficiencies( Reference Chertow, Driscoll and Blaner 64 ). Additionally, retinol- and retinoic acid-binding proteins have been detected in glucagon-secreting α-cell lines while retinol and retinoic acid have been demonstrated to inhibit glucagon secretion in cultured rat islets( Reference Chertow, Driscoll and Blaner 64 , Reference Chertow, Driscoll and Primerano 65 ). Important increases in plasma glucagon levels are also related to biotin deficiencies due to fasting( Reference Klandorf and Clarke 66 ).
Other levels of regulation
In addition to nutrients, pancreatic α-cells are subjected to other control levels. These include neural regulation as well as autocrine, paracrine and endocrine signalling pathways that interact with the different steps involved in glucagon release or synthesis( Reference Quesada, Tuduri and Ripoll 9 ). Although numerous neurotransmitters released by nerve endings situated within the islet are associated with a stimulatory effect on glucagon secretion, the molecular and cellular mechanisms are still not well understood( Reference Ahren 7 ). Parasympathetic neurotransmitters such as vasoactive intestinal polypeptide, gastrin-releasing peptide, pituitary adenylate cyclase-activating polypeptide and acetylcholine have a stimulatory effect on pancreatic α-cells( Reference Wilkes, Bailey and Thompson 67 , Reference Winzell 68 ). Similarly, sympathetic nerves within the islet have a glucagonotropic effect via neurotransmitters such as noradrenaline, galanin and neuropeptide Y( Reference Ahren 7 , Reference Miralles, Peiro and Degano 69 ). Sympathetic activation can also induce adrenaline release, which is a potent stimulator of pancreatic α-cell exocytosis( Reference De Marinis, Salehi and Ward 70 ). Sensory nerves containing calcitonin gene-related protein and substance P may also trigger glucagon release from the pancreatic islet( Reference Ahren 7 ). It has been also shown that α-cells can secrete neural factors such as acetylcholine or glutamate( Reference Cabrera, Jacques-Silva and Speier 71 , Reference Rodriguez-Diaz, Dando and Jacques-Silva 72 ). Neural regulation of islet function has been related to the control of the pulsatile release of islet hormones and the synchronisation of individual cell responses within the islet( Reference Quesada, Tuduri and Ripoll 9 ). Additionally, neural regulation allows for a further control of the glucagon secretory response to hypoglycaemia as well as the suppression of glucagon secretion in conditions of hyperglycaemia( Reference Quesada, Tuduri and Ripoll 9 , Reference Cryer 15 ). This neural control is mediated by glucose-sensing neurons located in the ventromedial hypothalamus( Reference Miki, Liss and Minami 73 ).
Additionally, islet hormones such as insulin, glucagon and somatostatin or β-cell-secretory products such as ATP, γ-aminobutyric acid, amylin or Zn also act as paracrine and autocrine signals that affect α-cell physiology( Reference Quesada, Tuduri and Ripoll 9 , Reference Leibiger, Moede and Muhandiramlage 74 , Reference Tuduri, Filiputti and Carneiro 75 ). Extrapancreatic hormones such as leptin and gastrointestinal incretins can also modulate α-cell function( Reference Tuduri, Marroqui and Soriano 22 , Reference Ma, Zhang and Gromada 76 ). In this latter group of hormones, glucagon-like peptide-1 (GLP-1) is one of the most important bioactive peptides with numerous islet effects. GLP-1 is produced by intestinal ileum L-cells by post-translational processing of proglucagon and is secreted after a meal in response to nutrients( Reference Tuduri, Marroqui and Soriano 22 , Reference Ma, Zhang and Gromada 76 ). Given that GLP-1 has a potent glucose-induced insulinotropic action but reduces glucagon release, some pharmacological approaches in diabetes have led to the design of GLP-1 derivatives of improved duration and resistance to degradation compared with the natural hormone( Reference Tuduri, Marroqui and Soriano 22 , Reference Ma, Zhang and Gromada 76 ). In contrast to the suppressive effect on glucagon release observed in animal models and human subjects( Reference Dunning and Foley 77 ), initial experiments at the cellular level reported an enhanced protein kinase A (PKA)-dependent exocytosis in rat pancreatic α-cells, which was associated with increased glucagon secretion( Reference Ding, Renstrom and Rorsman 78 ). In light of these findings, it was proposed that the GLP-1 inhibitory effect on pancreatic α-cells may rely on paracrine mechanisms( Reference Ding, Renstrom and Rorsman 78 , Reference de Heer, Rasmussen and Coy 79 ). However, recent studies in mice have shown that GLP-1 can indeed reduce glucagon secretion in α-cells by PKA-dependent inhibition of a specific subset of Ca channels via a small increase in intracellular cyclic AMP (cAMP) concentrations( Reference De Marinis, Salehi and Ward 70 ). These last observations support a molecular basis for the studies showing an inhibitory GLP-1 action on glucagon release. Unlike GLP-1, a stimulatory effect has been reported for the gastrointestinal hormone glucose-dependent insulinotropic polypeptide (GIP), which is produced by intestinal K-cells. Studies with perifused mouse pancreatic islets showed that GIP was able to reverse the suppressive effect of high glucose levels on glucagon release( Reference Opara, Burch and Taylor 80 ). In primary rat α-cells and perfused rat pancreas, GIP stimulated glucagon release at low glucose concentrations( Reference de Heer, Rasmussen and Coy 79 ).
Actions of glucagon on nutrient metabolism
Glucagon synthesis
The preproglucagon gene is mainly expressed in pancreatic α-cells, in intestinal L-cells and in the central nervous system and is closely regulated by nutritional status. The preproglucagon promoter contains up to six regulatory sequences called G1–5 and CRE (cAMP response element), which confers responsiveness to cAMP. The promoter consists of a region comprising the regulatory sequences G1 and G4 while the remaining sequences are part of the enhancer regions. Numerous transcription factors are involved in the control of glucagon expression such as LIM-homeobox transcription factor islet-1 (Isl1), paired box gene 6 (Pax6), musculoaponeurotic fibrosarcoma oncogene homolog (Mafs), forkhead box protein A1 (Foxa1) and forkhead box protein A2 (Foxa2) among others( Reference Gosmain and Cheyssac 81 , Reference Jin 82 ). The transcription of the preproglucagon gene results in a peptide of 160 amino acids. The processing of this peptide into glucagon, GLP-1 and GLP-2 hormones depends on the post-translational cleavage mediated by different prohormone convertase (PC) subtypes. The differential cell-specific expression of PC generates different peptides in each tissue( Reference Mojsov, Heinrich and Wilson 83 ). PC2 is responsible in α-cells for the production of glucagon in addition to other products such as glicentin, glicentin-related pancreatic polypeptide, intervening peptide 1 and the major proglucagon fragment( Reference Dey, Lipkind and Rouille 84 – Reference Patzelt, Tager and Carroll 86 ). The importance of PC2 for the correct processing of glucagon has been proven in experiments using PC2 knockout (KO) mice( Reference Furuta, Zhou and Webb 87 ). In contrast to the results in some clonal α-cell lines( Reference Dumonteil, Ritz-Laser and Magnan 88 , Reference McGirr, Ejbick and Carter 89 ), studies in isolated rat islets indicate that the effect of glucose on glucagon gene expression is not direct but occurs via paracrine mechanisms that stimulate the inhibitory insulin signal( Reference Dumonteil, Magnan and Ritz-Laser 44 ). This hormone decreases glucagon gene expression in pancreatic α-cells by activating phosphatidylinositol kinase (PI3K) and PKB pathways( Reference Grzeskowiak, Amin and Oetjen 90 – Reference Schinner, Barthel and Dellas 92 ). In line with these findings, the enhanced glucagon expression found in rats with insulinopenic diabetes was corrected by administrating insulin( Reference Dumonteil, Ritz-Laser and Magnan 88 ). As mentioned earlier, it has been reported that lipids are able to modulate glucagon gene expression both in a paracrine way through their effects on β-cells( Reference Piro, Maniscalchi and Monello 60 ) and by direct mechanisms( Reference Bollheimer, Landauer and Troll 55 ). In the latter case, while short-term experiments indicate a down-regulation of the glucagon gene after being treated with palmitate( Reference Bollheimer, Landauer and Troll 55 ), no effect has been observed in long-term studies( Reference Dumonteil, Magnan and Ritz-Laser 44 , Reference Gremlich, Bonny and Waeber 61 ). Amino acids may also play a role in glucagon gene regulation, although much further investigation is still necessary: while arginine has been shown in some studies to increase glucagon expression and protein synthesis via protein kinase C activation( Reference Yamato, Noma and Tahara 93 ), other researchers have failed to observe these changes( Reference Dumonteil, Magnan and Ritz-Laser 44 ). In clonal αTC1-6 cells, the removal of histidine from the culture leads to a decrease in preproglucagon gene expression similar to what is observed in an amino acid-free medium( Reference Paul, Waegner and Gaskins 94 ).
Glucagon receptor and intracellular signalling
The glucagon receptor is a protein belonging to the secretin–glucagon receptor II class family of the G protein-coupled receptors and consists of 477 amino acids in humans and 485 amino acids in rodents with a primary sequence homology of more than 80 % between them( Reference Mayo, Miller and Bataille 95 ). This receptor is characterised by seven transmembrane-spanning domains connected by three intracellular and extracellular loops. Its activation is coupled to GTP-binding heterotrimeric G proteins of the Gαs type that stimulate adenylate cyclase and PKA( Reference Weinstein, Yu and Warner 96 ). Although glucagon-induced PKA signalling is the most important biochemical cascade, this hormone can also activate the phospholipase C/inositol phosphate pathway via Gq proteins, resulting in the release of Ca2+ from intracellular stores( Reference Mayo, Miller and Bataille 95 ). In addition, glucagon has also been implicated in signalling via 5′-AMPK( Reference Longuet, Sinclair and Maida 13 , Reference Kimball, Siegfried and Jefferson 97 ), p38 MAPK and c-Jun N-terminal kinase (JNK)( Reference Longuet, Sinclair and Maida 13 , Reference Chen, Ishac and Dent 98 ) (Fig. 2). Although the glucagon receptor is mainly expressed in the liver, its expression is also observed in multiple tissues including the pancreas, heart, kidney, brain, smooth muscle, adipocytes, lymphoblast, spleen, retina, adrenal gland and gastrointestinal tract( Reference Svoboda, Tastenoy and Vertongen 99 ). Although a preferential expression of the glucagon receptor is found in pancreatic β-cells, it is also expressed, to a lesser degree, in non-β-cells, including glucagon-secreting α-cells( Reference Kedees, Grigoryan and Guz 100 , Reference Kieffer, Heller and Unson 101 ). Mice lacking the glucagon receptor exhibit several phenotypic alterations such as pancreatic α-cell hyperplasia, hyperglucagonaemia, mild hypoglycaemia, increased number of islets per pancreas and increased GLP-1 plasma levels( Reference Gelling, Du and Dichmann 102 ). Additionally, these mice show resistance to high-fat diet-induced hyperinsulinaemia and streptozotocin-induced diabetes, while exhibit improved insulin sensitivity compared with wild-type controls( Reference Conarello, Jiang and Mu 103 – Reference Vuguin, Kedees and Cui 106 ). However, a specific over-expression of the glucagon receptor in pancreatic β-cells also improves glucose tolerance in mice exposed to high-fat diets, suggesting an improvement in β-cell function( Reference Gelling, Vuguin and Du 107 ).

Fig. 2 Glucagon intracellular signalling and actions in different tissues. Intracellular signalling and biochemical pathways activated by glucagon are illustrated in the top of the figure. The different target tissues of glucagon as well as its effects are summarised in the lower part of the figure. For further information, see the text. PLC, phospholipase c; PIP2, phosphatidylinositol 4,5-bisphosphate; AC, adenylate cyclase; Pi, inorganic phosphate; IP3, inositol 1,4,5-trisphosphate; AMPK, AMP-activated protein kinase; JNK, c-Jun N-terminal kinase; cAMP, cyclic AMP; PKA, protein kinase A; MAPK, mitogen-activated protein kinase.
Effects of glucagon on hepatic nutrient metabolism
The primary target organ of glucagon is the liver where the insulin:glucagon ratio controls multiple key steps of hepatic metabolism (Fig. 2). The action of glucagon on the liver plays a key role in glucose homeostasis, particularly in adaptive and counter-regulatory responses to hypoglycaemic conditions, fasting and starvation( Reference Longuet, Sinclair and Maida 13 ) as well as in situations of increased fuel demand such as vigorous exercise( Reference Berglund, Lustig and Baheza 108 ) or states of metabolic stress such as trauma, inflammation or sepsis( Reference Jones, Tan and Bloom 109 ). Given that the brain relies on a continuous glucose supply, several lines of defence against hypoglycaemia take place to counterbalance this situation. These include glucose-sensitive neural responses, an increased release of hyperglycaemic hormones such as adrenaline and an enhanced glucagon secretion. Additionally, glucagon opposes numerous insulin actions. Glucagon has a dual effect on the liver: on the one hand, it stimulates gluconeogenesis and glycogenolysis increasing hepatic glucose output and, on the other hand, it decreases glycogenesis and glycolysis( Reference Quesada, Tuduri and Ripoll 9 , Reference Yoon, Puigserver and Chen 110 ). These effects along with decreased plasma insulin levels allow for the restoration of normoglycaemia by favouring hepatic glucose release. Although the glucagon receptor is highly selective for glucagon, it can also bind to other glucagon-related peptides( Reference Hjorth, Adelhorst and Pedersen 111 ). One of the main effects of glucagon to modulate gluconeogenesis is the up-regulation of key enzymes involved in this process such as glucose-6-phosphatase and phosphoenolpyruvate carboxykinase through the activation of the cAMP response element-binding protein and PPARγ-coactivator-1( Reference Yoon, Puigserver and Chen 110 , Reference Herzig, Long and Jhala 112 ). The modulatory effect of glucagon on pyruvate kinase accounts for the down-regulation of glycolysis( Reference Quesada, Tuduri and Ripoll 9 ). Glucagon also regulates glycogen synthesis and lysis by modulating the activity of glycogen synthase and glycogen phosphorylase via the phosphorylation of these enzymes( Reference Band and Jones 113 ).
Glucagon is not only implicated in glucose homeostasis but in hepatic lipid metabolism as well. Several reports indicate that TAG production is decreased in perfused livers treated with glucagon( Reference De Oya, Prigge and Grande 114 , Reference Penhos, Wu and Daunas 115 ). The potential hypolipidaemic actions of glucagon after its administration also include a reduction in plasma and hepatic TAG, decreased VLDL and cholesterol levels, as well as increased fatty acid oxidation( Reference Longuet, Sinclair and Maida 13 , Reference Xiao, Pavlic and Szeto 14 , Reference Aubry, Marcel and Davignon 116 , Reference Eaton 117 ). Moreover, reduced glucagon signalling has been related to the development of fatty liver( Reference Charbonneau, Couturier and Gauthier 118 ). Experiments using KO and wild-type mice for the glucagon receptor showed that administering glucagon not only reduces the synthesis and secretion of TAG and their plasma levels but also stimulates fatty acid oxidation( Reference Longuet, Sinclair and Maida 13 ). These effects depend on the activation of PPARα and are necessary for the adaptive metabolic response to fasting. In line with these results, these KO mice rapidly develop hepatosteatosis compared with wild-type controls when exposed to a high-fat diet( Reference Longuet, Sinclair and Maida 13 ). This finding in glucagon receptor KO mice contrasts with the resistance to hepatosteatosis found in this model by others( Reference Conarello, Jiang and Mu 103 ). In diet-induced hepatic steatosis, glucagon signalling may be impaired as a consequence of a reduced number of hepatic plasma membrane glucagon receptors( Reference Charbonneau, Melancon and Lavoie 119 ). Recently, it has been demonstrated in db/db mice that the inhibition of hepatic glucagon signalling leads to an increase in LDL due to an over-expression of hepatic lipogenic genes and elevated de novo lipid synthesis( Reference Han, Akiyama and Previs 120 ). In addition to lipid metabolism, glucagon can also stimulate the hepatic uptake of amino acids. Accordingly, hyperglucagonaemia may be related to plasma hypoaminoacidaemia, particularly with those amino acids involved in gluconeogenesis, such as alanine, glycine and proline( Reference Cynober 121 ).
Effects of glucagon on food intake, body weight and body energy
Glucagon is able to exert several actions on food intake behaviour as well as on the regulation of weight and body energy (Fig. 2). However, the precise signalling mechanisms that allow this hormone to perform these actions are still unclear. It has been shown that glucagon receptors are present in the rat brain, including the hypothalamus( Reference Hoosein and Gurd 122 ), and that glucagon is able to bind to mouse astrocyte suspensions and induce cAMP production( Reference Cockram, Kum and Ho 123 ). Additionally, this hormone is able to suppress the electrical activity of glucose-sensitive hypothalamic neurons( Reference Inokuchi, Oomura and Nishimura 124 ). Intraventricular infusion of glucagon leads to hyperglycaemia( Reference Amir 125 ) but it also decreases food intake, an effect that is less potent when glucagon is administered peripherally( Reference Inokuchi, Oomura and Nishimura 124 ). In addition to a potential direct action on central regions, it has been reported that glucagon may be sensed by peripheral vagal nerves that communicate the signal to satiety control areas of the hypothalamus to exert an anorexigenic action. This is supported by experiments in which a glucagon-induced decrease in food intake was suppressed when animals were subjected to hepatic vagotomy( Reference Geary and Smith 126 ). This reduced food intake when glucagon is infused has been observed not only in rodents but in human subjects as well( Reference Geary and Smith 127 – Reference Penick and Hinkle 129 ). The anorectic action of glucagon has been related to its ability to reduce meal size( Reference Woods, Lutz and Geary 130 ), an effect that can be abolished with the infusion of neutralising glucagon antibodies( Reference Langhans, Zeiger and Scharrer 131 ). Severe anorexia is also found in rats when transplanted with glucagonomas, further supporting the satiating role of this hormone( Reference Jensen, Blume and Mikkelsen 132 ). In contrast with these findings, glucagon receptor KO mice fed with high-fat diets exhibit lower food intake and body-weight increase than wild-type controls( Reference Conarello, Jiang and Mu 103 ). In addition to the central actions mentioned above, it has been suggested that the satiety effect of glucagon may involve the suppression of the orexigenic hormone ghrelin via the hypothalamic–pituitary axis( Reference Arafat, Perschel and Otto 133 ). It has recently been reported that hypothalamic glucagon signalling inhibits hepatic glucose production and that the central resistance to this hormone might be involved in the hyperglycaemia that characterises diabetes( Reference Mighiu, Yue and Filippi 134 ).
Considerable evidence supports the role of glucagon as a thermogenic agent, thereby favouring the body's energy expenditure. In rats, an energy-increasing effect of glucagon was reported some time ago( Reference Davidson, Salter and Best 135 ). In healthy volunteers, RMR increases after glucagon infusion in conditions of insulin deficiency( Reference Nair 136 ). The presence of insulin seems to counterbalance this glucagon action( Reference Calles-Escandon 137 ). Recently, indirect calorimetry measurements in obese, non-diabetic individuals showed that glucagon infusion alone or in combination with GLP-1 is able to increase resting energy expenditure( Reference Tan, Field and McCullough 138 ). These thermogenic effects have been associated with direct actions on brown adipose tissue due to increased oxygen consumption, metabolism and heat production in both in vitro and in vivo experiments( Reference Billington, Briggs and Link 139 – Reference Yahata and Kuroshima 141 ). Additionally, chronic glucagon administration in rats increases the mass of brown adipose tissue as well as its thermogenic capacity and fatty acid uptake( Reference Billington, Bartness and Briggs 142 ). Several reports suggest that these effects are mediated by the sympathetic nervous system via catecholamines( Reference Filali-Zegzouti, Abdelmelek and Rouanet 143 ). This is supported by experiments in which the glucagon effect is prevented or reduced in conditions of brown adipose tissue denervation or pharmacological β-adrenergic inhibition with propranolol( Reference Billington, Bartness and Briggs 142 , Reference Dicker, Zhao and Cannon 144 ). It has also been shown that glucagon reduces body weight in both humans and normal rats( Reference Billington, Briggs and Link 139 , Reference Schulman, Carleton and Whitney 145 ) as well as in obese Zucker rats( Reference Chan, Mackey and Snover 146 ), a model of genetic obesity. These effects may probably be due to the above-mentioned glucagon action on food intake and energy expenditure. Recent results in mice confirm that glucagon not only induces a loss of body weight and fat mass but also lowers plasma cholesterol levels( Reference Habegger, Stemmer and Cheng 147 ). All these processes are attributed to the enhanced secretion of fibroblast growth factor 21 (FGF21) in plasma, whose levels are increased when glucagon is infused in mice and human subjects. Thus, glucagon may have potential therapeutic implications not only for diabetes but also for obesity.
Other effects of glucagon
Although the effect of glucagon on the liver is the best known, this hormone also affects other extra-hepatic organs and tissues (Fig. 2). Glucagon-receptor KO mice exhibit α-cell hyperplasia( Reference Gelling, Du and Dichmann 102 ), which is consistent with in vitro experiments showing that glucagon down-regulates α-cell proliferation in an autocrine manner( Reference Liu, Kim and Chen 148 ). The secretion of this hormone also increases in mouse and rat pancreatic α-cells via cAMP production( Reference Ma, Zhang and Gromada 76 ). The over-expression of the glucagon receptor in pancreatic β-cells improves glucagon- and glucose-stimulated insulin secretion, and enhances β-cell mass as well as glucose tolerance( Reference Gelling, Vuguin and Du 107 ). The direct effect of glucagon on β-cells is further supported by experiments showing that this hormone increases insulin release in perfused pancreas and isolated β-cells( Reference Kieffer, Heller and Unson 101 , Reference Moens, Heimberg and Flamez 149 ). Additionally, glucagon produces cardiac ionotropic and chronotropic actions, partly by stimulating Ca currents via cAMP production and the inhibition of phosphodiestarases( Reference Mery, Brechler and Pavoine 150 ), thus leading to increased contractility( Reference Gonzalez-Munoz, Nieto-Ceron and Cabezas-Herrera 151 ). It has also been indicated that this hormone regulates cardiac metabolism at the level of glucose utilisation( Reference Harney and Rodgers 152 ). The role of glucagon in adipose tissue has been controversial for a long time. Glucagon has been reported to augment lipolysis in isolated rodent and human adipocytes( Reference Heckemeyer, Barker and Duckworth 153 , Reference Perea, Clemente and Martinell 154 ). The physiological meaning of these results has been disputed based on findings that show no effect of glucagon on the abdominal fat tissue of healthy individuals submitted to microdialysis( Reference Gravholt, Moller and Jensen 155 ). However, new studies have reported glucagon-induced lipolysis in healthy volunteers, diabetic patients, diabetic animal models and isolated adipocytes from these animals( Reference Arafat, Kaczmarek and Skrzypski 11 ). All these effects were attributed to a glucagon-induced release of FGF21, which has lipolytic activity. Actually, recent reports demonstrate that FGF21 also mediates other different glucagon actions, as we previously commented( Reference Habegger, Stemmer and Cheng 147 ). Given that the lipolytic action of glucagon is blunted in denervated rats, its effect on adipose tissue has also been associated with sympathetic signals( Reference Lefebvre, Luyckx and Bacq 156 ). Kidneys are also a target site for glucagon, since its intravenous infusion is related to changes produced in urea synthesis and excretion as well as in water conservation( Reference Ahloulay, Bouby and Machet 157 ). In in vitro perfused inner medullary collecting ducts, glucagon induces cAMP production and changes in aquaporin 2 expression that affect water excretion( Reference Yano, Cesar and Araujo 158 ). Glucagon is also used as an anti-peristaltic agent because of its effects on small intestine motility( Reference Gutzeit, Binkert and Koh 159 , Reference Mochiki, Suzuki and Takenoshita 160 ). Moreover, it has recently been reported that glucagon also enhances sweet taste responses by acting directly on the mouse gustatory epithelium( Reference Amanda, Manuela and Antonia 161 ).
Involvement of glucagon in diabetes pathophysiology
Role of glucagon in the control of glycaemia during diabetes mellitus
Type 1 diabetes is characterised by an autoimmune specific β-cell loss that results in insulin deficiency, leading to hyperglycaemia. These high plasma glucose levels are filtered by kidneys, which may result in glycosuria and, eventually, osmolar alterations that can produce a non-ketotic hyperosmolar coma. Since glucose utilisation as an energy substrate is limited as a consequence of insulin deficiency, increased levels of fatty acids are oxidised to acetyl CoA, increasing the production of ketonic bodies and ketonuria with the concomitant risk of ketoacidosis. Other symptoms that develop in poorly controlled diabetes are polyuria, polydipsia and polyphagia. Since the basic treatment for diabetes is insulin administration, another major complication may be hypoglycaemia (iatrogenic hypoglycaemia), which results from an excess of insulin in a condition of impaired homeostatic responses against the decline in plasma glucose levels( Reference Cryer 15 ). In the case of type 2 diabetes, hyperglycaemia is due to a combination of increased peripheral resistance to the action of insulin along with inappropriate insulin secretion. This situation can eventually progress to β-cell death( Reference Quesada, Tuduri and Ripoll 9 ), thus leading to a greater deterioration of glucose homeostasis and a worsening of hyperglycaemia. This type of diabetes is often associated with obesity. In both types of diabetes, poorly controlled glycaemia can lead to micro- and macrovascular, neural, retinal and renal complications as well as skin ulcers and, eventually, amputation.
In addition to the central role of insulin and pancreatic β-cells in the pathophysiology of diabetes, abundant evidence demonstrates that glucagon is also involved in this metabolic disorder. Absolute or relative (to insulin) hyperglucagonaemia is frequently found in diabetes during fasting and postprandial periods( Reference Larsson 162 – Reference Sherwin, Wahren and Felig 164 ). Hyperglucagonaemia in the context of insufficient insulin secretion and/or insulin resistance is associated with increased hepatic glucose output, which contributes to hyperglycaemia( Reference Li, Liao and Zhuo 48 , Reference Gastaldelli, Baldi and Pettiti 165 ). Additionally, α-cell function in response to changes in glucose is impaired in diabetes( Reference Siafarikas, Johnston and Bulsara 166 ). For instance, hyperglycaemia fails to suppress glucagon secretion in diabetic patients, which further exacerbates their high glucose levels during postprandial periods( Reference Dinneen, Alzaid and Turk 167 , Reference Shah, Vella and Basu 168 ). Although the mechanism for this failure is not clearly known, it has been suggested that the α-cell might be refractory to the insulin paracrine signal or insensitive to high glucose levels( Reference Quesada, Tuduri and Ripoll 9 , Reference Cryer 15 ). Patients with a glucagonoma develop hyperglucagonaemia and the characteristic hyperglycaemia of diabetes, which disappears when the tumour is removed, further suggesting that high levels of glucagon are involved in this disease( Reference Eldor, Glaser and Fraenkel 169 ). Another problem of diabetes is that pancreatic α-cells do not respond adequately to hypoglycaemia, which is particularly important in insulin-treated patients, and leads to increased morbidity and mortality rates in this disease( Reference Taborsky and Mundinger 8 , Reference Taborsky, Ahren and Havel 23 ). This life-threatening situation seems to be the result of several altered processes: impaired α-cell sensing of falling glucose levels, an inefficient effect of the withdrawal of the insulin inhibitory signal during hypoglycaemia as well as a defective function of the autonomous nervous system and of the different defence mechanisms against hypoglycaemia( Reference Taborsky and Mundinger 8 , Reference Bolli, De Feo and Perriello 170 – Reference Zhou, Zhang and Oseid 172 ). In addition to an altered α-cell function, diabetes has been also associated with increases in either absolute or relative (to β-cells) α-cell mass in animal models and humans( Reference Deng, Vatamaniuk and Huang 173 – Reference Yoon, Ko and Cho 175 ), which may explain the higher plasma glucagon levels in these patients.
Glucagon is also involved in protein and fat metabolic alterations in diabetes. This hormone plays a catabolic role during a protein load( Reference Charlton, Adey and Nair 176 ) and hyperglucagonaemia induces phenylalanine oxidation in healthy humans( Reference Tessari, Inchiostro and Barazzoni 177 ). Individuals with a glucagonoma exhibit higher levels of free amino acids in muscles and the liver( Reference Roth, Muhlbacher and Karner 178 ). In type 1 diabetic patients, hyperglucagonaemia has been related to different catabolic effects including an increased RMR and increased leucine oxidation, effects that were independent of insulin deficiency( Reference Charlton and Nair 179 ). Because of the effect of this hormone on amino acid hepatic uptake and the stimulation of gluconeogenesis, hyperglucagonaemia can lead to marked hypoaminoacidaemia, particularly from those amino acids that participate in gluconeogenesis( Reference Cynober 121 , Reference Boden, Rezvani and Owen 180 ). Glucagon has also been involved in lipid metabolism. Although the role of glucagon in adipose tissue lipolysis has remained controversial for a long time, new evidence in animal models and human subjects, including diabetic patients, demonstrates this effect( Reference Arafat, Kaczmarek and Skrzypski 11 ). These actions may be the result of the activation of hormone-sensitive lipase( Reference Slavin, Ong and Kern 181 ) and probably involve the participation of FGF21( Reference Arafat, Kaczmarek and Skrzypski 11 ). In animal models, chronic administration of glucagon induces hypolipidaemic effects( Reference Guettet, Rostaqui and Mathe 182 , Reference Guettet, Rostaqui and Navarro 183 ). Furthermore, it has been recently shown that hyperglucagonaemia modulates hepatic lipoprotein particle metabolism in humans both by decreasing hepatic lipoprotein particle production and by inhibiting particle clearance( Reference Xiao, Pavlic and Szeto 14 ). In the context of insulin deficiency and high levels of fatty acids, glucagon can accelerate the formation of ketonic bodies from the liver( Reference Beylot, Picard and Chambrier 184 – Reference Okuda, Kawai and Yamashita 186 ). Consistent with this, it has been observed that diabetic ketoacidosis is associated with hyperglucagonaemia combined with hypoinsulinaemia( Reference Taborsky 187 , Reference Wahid, Naveed and Hussain 188 ). Thus, in view of the role that glucagon plays in the pathophysiology of diabetes, it would be advisable to develop therapeutic strategies to limit either glucagon secretion or action.
Therapeutic potential of modulating glucagon secretion and action in diabetes
Numerous studies have shown that restrained glucagon action is beneficial for the control of hyperglycaemia and/or diabetes management. For instance, glucagon-receptor KO mice exhibit reduced fasting hypoglycaemia and improved glucose tolerance( Reference Gelling, Du and Dichmann 102 ). Additionally, these mice are resistant to diet-induced obesity, as well as streptozotocin-induced β-cell loss and hyperglycaemia( Reference Conarello, Jiang and Mu 103 ). Similarly, lower hepatic glucose production and improved glucose tolerance are observed in mice that are deficient in the α-cell transcription factor ARX (aristaless-related homeobox), leading to the loss of glucagon-producing α-cells( Reference Hancock, Du and Liu 189 ). More recently, it has been shown that the metabolic and clinical alterations caused by type 1 diabetes are absent in glucagon receptor KO mice treated with streptozotocin and that the restoration of the hepatic glucagon receptor in this model leads to the reappearance of hyperglycaemia( Reference Lee, Wang and Du 104 , Reference Lee, Berglund and Wang 190 ). However, a limited glucagon action could interfere with the role of glucagon in hepatic lipid metabolism and lead to an increased susceptibility to hepatosteatosis after a high-fat diet( Reference Longuet, Sinclair and Maida 13 ), or it could also interfere with the hepatic survival function of glucagon( Reference Sinclair, Yusta and Streutker 191 ). Additionally, in this kind of KO mice the exocrine and endocrine cell masses are augmented, increasing the risk of developing tumours( Reference Gelling, Du and Dichmann 102 ). Thus, all these aspects require further examination in human subjects. Consistent with these findings in KO mice, other strategies such as glucagon antibodies, glucagon receptor antisense oligonucleotides and glucagon receptor antagonists revealed similar findings in hepatic glucose production and showed an antihyperglycaemic effect in diabetic animal models as well as in a few human studies( Reference Quesada, Tuduri and Ripoll 9 , Reference Parker, McPherson and Andrews 192 – Reference Winzell, Brand and Wierup 195 ). The antagonism of glucagon action on the liver also partly mediates the glucose-lowering effect of biguanides( Reference Miller, Chu and Xie 196 ). It has been recently demonstrated that the design of peptides with dual agonism for both the glucagon and the GLP-1 receptors is useful to fight against obesity in mice without any apparent adverse effects( Reference Day, Ottaway and Patterson 197 , Reference Pocai, Carrington and Adams 198 ). This is mainly the result of the antihyperglycaemic, lipolytic, satiating and energy expenditure actions of this combined treatment. In human subjects, the co-administration of GLP-1 during glucagon infusion leads to increased resting energy expenditure and lower hyperglycaemia( Reference Tan, Field and McCullough 138 ). In the present study, the augmented energy expenditure was attributed to glucagon alone. Similarly, chronic glucagon receptor agonism in diet-induced obese mice lowered food intake, body weight, fat mass and cholesterolaemia while increasing energy expenditure( Reference Habegger, Stemmer and Cheng 147 ). The actions of glucagon were mediated by FGF21. This growth factor has also been involved in the lipolytic action of glucagon in diabetic and non-diabetic animals and humans( Reference Arafat, Kaczmarek and Skrzypski 11 ). Additionally, peripherally administered glucagon inhibits food intake, presumably by the neural activation of appetite-regulating brain centres( Reference Parker, McCullough and Field 199 ). Thus, the combined activation of glucagon and GLP-1 receptors has been proved to be useful for the design of strategies against obesity and diabetes.
The modulation of glucagon secretion with a therapeutic potential has also been explored. Exogenous GLP-1 administration is known to stimulate insulin release while inhibiting glucagon secretion in isolated rodent islets, in perfused rat pancreas and in human subjects, which allows for the improvement of hyperglycaemia( Reference Quesada, Tuduri and Ripoll 9 , Reference Nauck, Heimesaat and Behle 200 ). The effect on glucagon is mediated by the direct action on pancreatic α-cells( Reference De Marinis, Salehi and Ward 70 ). Accordingly, GLP-1 agonists widely used in diabetes treatment such as exenatide or liraglutide also exert hypoglycaemic effects by lowering plasma glucagon levels( Reference Dupre, Behme and McDonald 201 ). Similar actions have been reported for inhibitors of the GLP-1-degrading enzyme dipeptidyl peptidase 4 (DPP-4)( Reference Edgerton, Johnson and Cherrington 202 ). DPP-4 inhibitors commonly used in diabetes such as vildagliptin or sitagliptin suppress hepatic glucose production by augmenting insulin release and lowering glucagon secretion( Reference Balas, Baig and Watson 203 ). Other drugs such as pramlintide, a synthetic analogue of islet amyloid polypeptide, also decreases glucagon secretion after a meal in diabetic individuals and may act on reducing hepatic glucose output to improve hyperglycaemia( Reference Edgerton, Johnson and Cherrington 202 , Reference Lebovitz 204 ).
Conclusions and future directions
The hyperglycaemic hormone glucagon has a key role in the control of glucose homeostasis by acting on the liver to stimulate gluconeogenesis and glycogenolysis and inducing hepatic glucose release. This hormone opposes the actions of insulin and is one of the main lines of defence against hypoglycaemic episodes, fasting and starvation, maintaining normoglycaemia. Less information is available about the function of glucagon during exercising or situations of metabolic stress. Although the main signalling pathway related to glucagon is mediated primarily by the activation of adenylate cyclase/PKA and, secondarily by phospholipase C/inositol 1,4,5-trisphosphate, recent studies have shown that AMPK, p38 MAPK and c-Jun N-terminal kinase (JNK) are also signalling conduits for glucagon. It would, therefore, be interesting to explore other potential signalling routes. In addition to the well-known effect of glucagon on hepatic glucose, this hormone also contributes to the regulation of lipid metabolism in the liver. Moreover, although glucagon receptors are present in numerous tissues including the heart, adipose tissue, kidneys and the brain among others, the effects and molecular pathways involved still require further examination, particularly those that glucagon produces at the central level. Despite the importance of pancreatic α-cells in the secretion of glucagon, the understanding of their regulation at molecular and cellular levels has been scarce for a long time. However, research on these cells and their role in nutrient metabolism and body energy has experienced a renewed impetus in recent years. It has been demonstrated that alterations in pancreatic α-cell function and glucagon actions are part of the pathophysiological events related to the development of diabetes. In this regard, several experimental approaches to improve diabetic symptoms include the modulation of glucagon release or its actions. Additionally, glucagon alone or in combination with other hormones may be relevant in the control of appetite, body weight and the treatment of obesity. Accordingly, this renewed interest in the pancreatic α-cell and glucagon may reveal new therapeutic strategies for metabolic disorders such as obesity and diabetes.
Acknowledgements
The authors acknowledge the work of current and former researchers of the authors' laboratory for their experimental support. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is an initiative of the Instituto de Salud Carlos III.
The present review was supported by grants from the Ministerio de Ciencia e Innovación (no. BFU2010-21773 and BFU2011-28358), Generalitat Valenciana (no. PROMETEO/2011/080 and ACOMP/2013/022) and the European Foundation for the Study of Diabetes (EFSD/BI Basic Programme). Funders had no role in the design, analysis or writing of the present review.
L. M. and I. Q. wrote the manuscript. B. M. designed the figures. L. M., P. A.-M., B. M., E. F., A. N. and I. Q. contributed to the discussion and revision of the text and approved the final manuscript.
There are no conflicts of interest to declare.


