

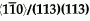 Grain Boundary In Al
Grain Boundary In Al
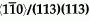 grain boundary (GB) in Al. The simulations were performed with Embedded Atom Method (EAM) potentials for Al and Al-Cu. The results predict that copper atoms tend to order along either side of the interface even at the pure symmetrical GB, forming alternating chains along the
grain boundary (GB) in Al. The simulations were performed with Embedded Atom Method (EAM) potentials for Al and Al-Cu. The results predict that copper atoms tend to order along either side of the interface even at the pure symmetrical GB, forming alternating chains along the  direction. The nucleation of the chains is driven by a change in the local atomic level stress induced by the pre-existing Cu atoms at the GB.
direction. The nucleation of the chains is driven by a change in the local atomic level stress induced by the pre-existing Cu atoms at the GB.Crossref Citations
This article has been cited by the following publications. This list is generated based on data provided by Crossref.
Liu, X.-Y.
Xu, Wei
Foiles, S. M.
and
Adams, J. B.
1998.
Atomistic studies of segregation and diffusion in Al-Cu grain boundaries.
Applied Physics Letters,
Vol. 72,
Issue. 13,
p.
1578.
Lezzar, B.
Khalfallah, O.
Larere, A.
Paidar, V.
and
Hardouin Duparc, O.
2004.
Detailed analysis of the segregation driving forces for Ni(Ag) and Ag(Ni) in the Σ=11 {113} and Σ=11 {332} grain boundaries.
Acta Materialia,
Vol. 52,
Issue. 9,
p.
2809.
Duparc, O. Hardouin
Larere, A.
Lezzar, B.
Khalfallah, O.
and
Paidar, V.
2005.
Comparison of the intergranular segregation for eight dilute binary metallic systems in the Σ 11′ {332} tilt grain boundary.
Journal of Materials Science,
Vol. 40,
Issue. 12,
p.
3169.