



Introduction
The water dissociation process, also known as “water splitting,” provides a fascinating and promising technology to convert renewable electricity into storable chemical energy, including hydrogen and oxygen collected at the device anode and cathode, respectively. Development has exploded in this energy-harvesting technology. There are many materials innovations to explore breakthroughs in achieving cost-effective, highly efficient, and durable electrocatalysts to maximize the energy-conversion efficiency of water splitting.
In general, electrocatalysts can be rationally designed based on the feedback-loop approach as illustrated in Figure1, including (1) running a large number of design of experiments (DOEs) and establishing a materials library of catalytically active elements; (2) utilizing the library's information resources to fabricate different catalysts with desired properties via various synergistic strategies, followed by the systematic evaluation of their electrochemical parameters and corresponding electrocatalytic mechanisms; and (3) combining with complementary experimental and theoretical investigations to identify activity descriptors for the catalyst material system that can be employed to describe and/or predict the catalytic performance, thus further enhance catalytic performance and operational stability. These findings provide valuable feedback to the materials library to complete the design loop.
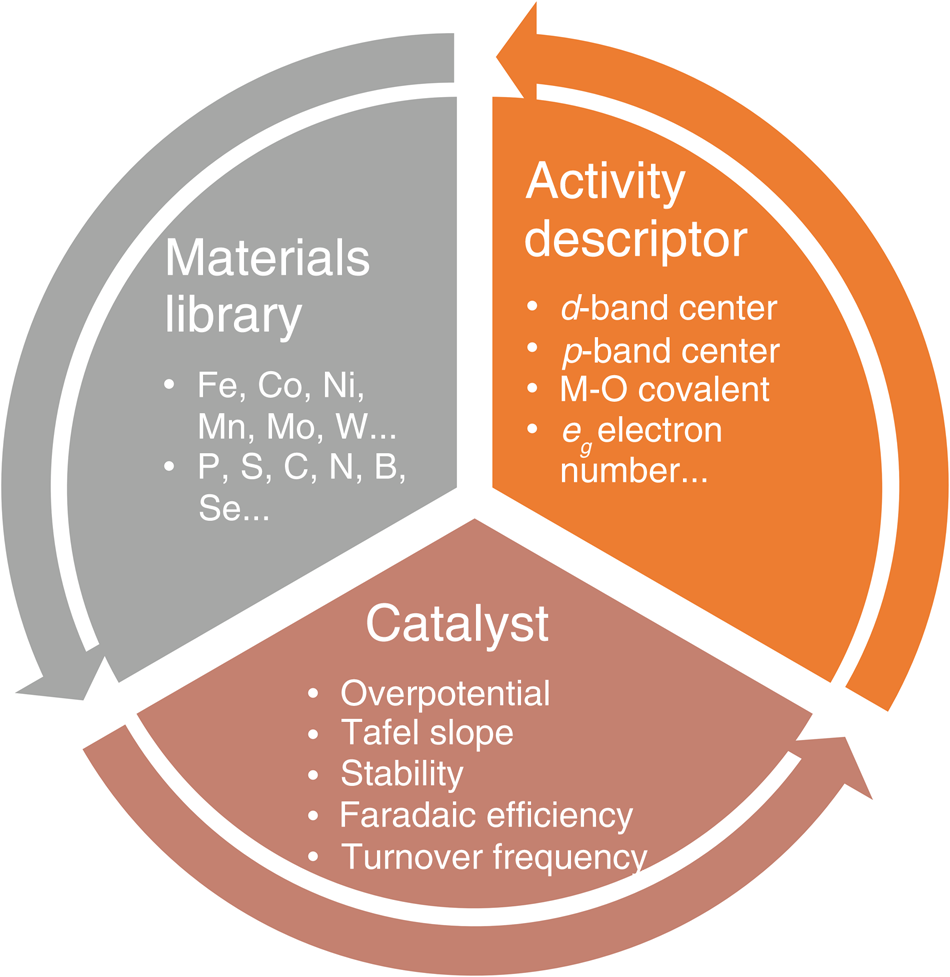
Figure 1. Schematic illustration of the electrocatalyst design approach. A design loop is established and follows the sequence: the preparation of a catalyst from the materials library, the evaluation of electrochemical performance, and confirmation of the activity descriptor. Note: M, metal; eg, orbital created when splitting occurs in the d orbital while making a complex.
Despite the progress achieved by the above mentioned strategies in recent years, several key issues remain for insufficient electrochemical performance of the obtained catalysts. For example, the use of strongly acidic and alkaline electrolytes as well as precious metal (i.e., Ir and Pt)-based active materials would restrict the large-scale practical utilization of electrocatalysts. When non-noble electrocatalysts are employed, the large overpotential along with high energy loss, poor stability, and narrow pH operating range would inevitably limit applications.
In order to overcome these obstacles, three methods are widely adopted to improve the electrochemical performance of catalysts. First, the intrinsic activity of catalyst active sites can be substantially increased with different electronic structure engineering strategies, including cation/anion doping, defect engineering, crystal phase and facet manipulation, and surface strain modification. Second, the exposure of these active sites can be greatly enlarged using various catalyst morphology designs. Finally, charge-transfer efficiency can be significantly improved by directly depositing catalysts onto hierarchical conducting support substrates.Reference Seh, Kibsgaard, Dickens, Chorkendorff, Nørskov and Jaramillo1 In this case, the advanced materials revolution would totally change the paradigm in electrocatalyst design to achieve efficient and robust water splitting.
The articles in this issue highlight recent advances in materials innovations of electrocatalysts. These include rational design spanning from transition-metal-based oxides and their derivatives, organic polymer nanomaterials, all the way to inorganic–organic nanocomposites. More importantly, the challenges and opportunities are compiled and discussed for possible future research and serve as important guidelines for electrocatalyst material design in water splitting.
Standard methodology
There has been substantial achievement and progress in excellent catalyst preparation methods and thorough mechanistic understanding. However, it is difficult to consistently compare the electrochemical performance of different catalysts for a given reaction. There is an urgent need to establish a standard methodology to benchmark catalyst performance parameters for sustainable development. At the same time, before any electrochemical testing is performed for a catalyst, there are several important areas that need special attention: (1) The potential contamination of active catalyst materials and electrolytes, especially when investigating iron-free oxygen evolution reaction (OER) catalysts, due to the residual iron ion impurities from multiple sources;Reference Liardet and Hu2 (2)the necessity to calibrate with Pt reference electrodes; (3) the comparison of electrochemical performance of catalysts to those of the bare substrate electrodes to ensure minimal activity from substrates; and (4) the inability to exploit Pt as counterelectrodes during hydrogen evolution reaction (HER), particularly in acidic conditions.Reference Dong, Fang, Wang, Yip, Cheung, Wang, Wong, Chu and Ho3 During electrochemical performance testing, the accurate measurement of an active surface area is extremely important for the evaluation of catalyst performance.
When comparing the surface area measured from the disk electrode surface area (i.e., geometric surface area), the Brunauer–Emmett–Teller area (specific surface area), and theelectrochemical surface area (ECSA), it is recommended to normalize the current density to ECSA, which can reflect the real intrinsic activity; however, it is sometimes not possible to determine the ECSA of new catalyst material systems.Reference Sun, Li and Xu4 The electrochemically active surface area represents the area of the electrode materials that is effectively accessible to the electrolyte and participates in the electrochemical reaction. The Tafel slope is another important catalytic performance parameter that reflects the rate of an electrochemical reaction to the overpotential and the reaction kinetics of catalysts. The overpotential is the potential difference (voltage) between a half-reaction's thermodynamically determined reduction potential and the potential at which the redox event is experimentally observed. It is especially meaningful when the overpotential values are larger than 118 mV, indicating the Tafel regime in the absence of mass-transfer effects.Reference Voiry, Chhowalla, Gogotsi, Kotov, Li, Penner, Schaak and Weiss5 Much care is taken to ensure the evaluation of Tafel slopes over a wide range of values up to 150 mV to obtain further insights into electrode kinetics.Reference Voiry, Chhowalla, Gogotsi, Kotov, Li, Penner, Schaak and Weiss5 Shinagawa et al. recently demonstrated that the applicability of the Tafel analysis in conjunction with the Butler–Volmer equation, which describes how the current on the electrode changes with the electrode potential, has been overlooked, leading to an inaccurate description of the interfacial reaction involving catalysts.Reference Shinagawa, Garcia-Esparza and Takanabe6 Finally, the evolution in turnover frequency (TOF), the number of reactions per unit time and unit active site at a given temperature, pressure, reactant ratio, and a certain degree of reaction, for different overpotential values should be carefully compiled when reporting the electrocatalytic activity of catalysts. All of these procedures would standardize the best practices for evaluating and reporting the catalytic performance of electrocatalysts.
Surface electronic structure modification
In essence, all surface modification schemes would change the electronic structure of catalyst active sites, which in turn, affects the adsorption and desorption capabilities of active sites for intermediate products. Before modifying the catalyst, it is essential to first understand the reaction process of target catalyst systems, particularly determining the rate-limiting step. Several methods can be employed to alter the surface electronic structure of catalysts, including cation/anion doping, defect manipulation, crystal phase and facet engineering, and surface strain modification.
Cation/anion doping is one of the most commonly used techniques in the modification of nanomaterials. Typically, elements with excellent catalytic activity are selected as dopants (e.g., Fe, Co, Ni, P, and S). Even elements without any significant catalytic activity (e.g., Zn, Ga, and Cu) are implemented due to their ability to modify the electronic structure of the active sites, thus enabling good catalytic performance of host materials.Reference Zou and Zhang7 In most cases, this associated catalytic activity enhancement is attributed to optimized absorption/desorption behavior, increased electrical conductivity, and improved ECSA after surface modification. Using this doping scheme, metal-Nx-C-based single-atom catalysts have been widely demonstrated as efficient catalytic materials because they help improve the efficiency of atomic utilization and have a unique coordination environment.Reference Bayatsarmadi, Zheng, Vasileff and Qiao8 Indeed, active metal centers surrounded by macrocyclic ligands exist in various kinds of complexes. The Li and Bai article in this issue mainly introduces the preparation of metal-Nx-C based catalyst based on porphyrin and macrocycle derivatives.Reference Li, Bao and Bai9
In terms of catalytic mechanisms, recent studies have attempted to justify the performance enhancement by introducing new energy levels in the host. When the relative position of a catalyst conduction-band edge or Fermi level is manipulated below the redox level of O2/H2O, the water oxidation reaction becomes easier. For instance, in the case of Zn-MoS2, its energy level has been observed to align properly and match with the reduction potential of H2O as compared with that of pristine MoS2 (Figure 2a). The result is improved catalytic performance.Reference Shi, Zhou, Yang, Xu, Wang, Wang, Xu, Xia and Chen10 It is also noted that the conductivity of semiconductors has a significant influence on their electrocatalytic characteristics. n-type and p-type semiconductors are more inclined to facilitate efficient HER and OER processes, respectively, while their bipolar counterparts are more capable of effectively enabling both HER and OER procedures.Reference He, He, Wang, Zhu, Golani, Handoko, Yu, Gao, Ding, Wang, Liu, Zeng, Yu, Guo, Yakobson, Wang, Seh, Zhang, Wu, Wang, Zhang and Liu11 Regardless, due to the existence of a double layer, the contact between a catalyst and an electrolyte is more complicated than that of gas–solid catalyst systems. The effect of this double layer must be carefully considered when introducing the concept of energy-level matching.

Figure 2. Surface electronic structure engineering. (a) Conduction-band edge profiles of Zn-MoS2, Fe-MoS2, and pristine MoS2 as compared with the reaction potential.Reference Shi, Zhou, Yang, Xu, Wang, Wang, Xu, Xia and Chen10 Compared with the reduction potential of H2O, the energy level of Zn-MoS2 is properly positioned to permit electron transfer with much lower applied energy (lower overpotential), thus confirming Zn-MoS2 as a promising electrocatalyst for HER. (b) The preparation of single-crystalline Co3O4 nanosheets with oxygen vacancies.Reference Cai, Bi, Hu, Liu, Dwarica, Tian, Li, Kuang, Li, Yang, Wang and Sun13 (c) Atomic force microscope image of strained MoS2 with S vacancy (SV-MoS2) fabricated on Au nanocone arrays.Reference Li, Tsai, Koh, Cai, Contryman, Fragapane, Zhao, Han, Manoharan, Abild-Pedersen, Nørskov and Zheng23 (d)Tafel slopes of CoOx and MnOx on different supports.Reference Frydendal, Busch, Halck, Paoli, Krtil, Chorkendorff and Rossmeisl28 The dashed line corresponds to the original sample while the solid line represents the gold-containing compound. Dramatic improvements can be achieved for manganese and cobalt-based oxides if gold is added to the surface or used as substrate. Note: EG, ethylene glycol; NHE, normal hydrogen electrode; E, potential; GC, glassy carbon electrode; j geo, current densities normalized to the geometric area; η, overpotential.
Besides doping, introducing defects into the host lattice not only modifies the electronic structure, but sometimes also changes the physical properties, such as in the case of porous nanomaterials. There are several methods to introduce defects, including thermal reduction in H2, NH3, or inert atmospheres, chemical reduction in NaBH4 or N2H4 ambient atmosphere, plasma treatment, and mechanical ball milling.Reference Yan, Li, Huo, Chen, Dai and Wang12 It is worth mentioning that it is challenging to introduce vacancies into the lattice with controlled concentration and minimized crystal damage. Recently, single-crystalline ultrathin Co3O4 nanosheets with numerous oxygen vacancies were prepared by soaking the nanosheets in a mixed solution of NaOH and ethylene glycol (Figure 2b).Reference Cai, Bi, Hu, Liu, Dwarica, Tian, Li, Kuang, Li, Yang, Wang and Sun13
Moreover, since cations often have a higher defect formation energy than anions, anion vacancies are easier to generate on the surface. Ni cation defects have been successfully incorporated into Ni(OH)2 catalyst via a propylene oxide-mediated alkalization precipitation method, which can precisely control the hydrolysis process of Ni2+ in the precursor solution.Reference He, Wan, Jiang, Pan, Wu, Wang, Wu, Ye, Ajayan and Song14 Based on density functional theory (DFT) calculations, the reduction of Bohr magnetons indicates a decrease in the electron spin filling of eg orbitals, which in turn enhances the strength of associated metal–oxygen bonds, leading to enhanced OER catalytic activity. Using a similar technique, Co cation defects can be reliably introduced into typical cobalt oxides via thermal calcination in order to enhance their corresponding electrochemical activity toward efficient oxygen evolution.Reference Zhang, Zhang, Pan, Shen, Mahmood, Ma, Shi, Jia, Wang, Zhang, Xu and Zou15
For phase and facet engineering in crystals, the use of non-noble metals is more difficult than precious metals because of the lower reduction potential, yielding faster and less controllable nucleation and crystallization. Also, amorphous materials have been shown to possess better electrocatalytic performance than their crystalline counterparts.Reference Indra, Tallarida, Schmeißer, Menezes, Strasser, Driess, Das, Bergmann and Sahraie16 Fortunately, amorphous materials can be easily prepared by controlling the crystallinity by varying solvent and reaction time. Increasing the solvent viscosity, reducing the processing temperature, and shortening the reaction time are more conducive to the formation of amorphous structures.Reference Yang, Guo, Huang, Xi, Gao, Su, Wang, Cao and Dong17 Transition-metal dichalcogenides with 1T-phase (octahedral with tetragonal symmetry) (e.g., MoS2 and WS2) have demonstrated superior electrocatalytic performance over their 2H-phase (trigonal prism with hexagonal symmetry) counterparts owing to the substantially reduced charge-transfer resistance in the metallic phase.Reference Yu, Nam, He, Wu, Zhang, Yang, Chen, Ma, Zhao, Liu, Ran, Wang, Li, Huang, Li, Xiong, Zhang, Liu, Gu, Du, Huang and Zhang18
There have been extensive investigations of different crystalline structures of MnO2 for their catalytic characteristics. Multiple results show that MnO2 materials exhibit decreasing electrochemical activity depending on their crystal phase, following an order of α → β → γ-MnO2.Reference Cheng, Su, Liang, Tao and Chen19 Due to the particularity of electrochemical processes, in situ phase transitions often occur during catalyst operations. The in situ phase transition of WO3 to HxWO3 has been identified in WO3/Pt nanocomposites through in situ characterization in acid electrolytes, where fast hydrogen and electron transfer ability promote high HER activity of Pt.Reference Xie, Chen, Du, Yan, Zhang, Chen, Liu and Wang20 Importantly, atomic-level engineering on the surface of nanomaterials is employed to precisely control the exposure of active sites, subsequently enhancing the corresponding electrocatalytic activity. Wei et al. prepared porous Co3O4 nanosheets with three different exposure facets of {110}, {111}, and {112}.Reference Wei, Fang, Dong, Lan, Shu, Zhang, Bu and Ho21 Their results indicate that the {112} crystal face exhibits the best OER performance, which can be attributed to the higher density of Co2+ active sites and oxygen vacancies on the surface.
Strain engineering is recognized as one of the powerful methods to modify the surface electronic structure by manipulating the tensile or compressive strain on the material surface. Stress is a measure of forces acting on materials commonly observed in alloys or core–shell structures because of lattice mismatch.Reference Strasser, Koh, Anniyev, Greeley, More, Yu, Liu, Kaya, Nordlund, Ogasawara, Toney and Nilsson22 Nonetheless, controlling these stress quantities of single-component catalysts is complicated and not cost effective. Currently, depositing target catalyst materials directly on structure-tunable substrates is the main preparation scheme utilized to accurately control the associated surface strain. An example is strained MoS2 monolayer films that can be effectively prepared on prefabricated Au nanocone arrays via the vapor deposition method (Figure 2c). Complementary DFT and experimental studies confirm that the hydrogen adsorption free energy can be manipulated by varying the surface strain on MoS2.Reference Li, Tsai, Koh, Cai, Contryman, Fragapane, Zhao, Han, Manoharan, Abild-Pedersen, Nørskov and Zheng23 These illustrations provide strong evidence of the effectiveness of modifying surface electronic structures of catalysts for enhanced electrocatalytic performance by the previously discussed approaches.
Morphology design
There are substantial studies that systematically investigate the preparation of nanomaterials with a high surface area, such as porous nanosheets, hollow nanotubes, nanocages, and 3D aerogels, which can increase the exposure area of active sites in electrolytes to further improve their catalytic performance.Reference Fang, Dong, Wei and Ho24 Among the various material synthesis schemes, the direct preparation of catalysts on 3D conductive and porous substrates, including nickel foam, copper foam, and carbon cloth, has been shown to be a good choice. Recent studies have shown that nickel foams not only act as a 3D substrate that increase the number of accessible active sites, they also act as a doping precursor to improve the conductivity and tune the electronic structure of active sites during hydrothermal synthesis.Reference Xu, Tian, Zhong, Kang and Yao25,Reference Zeng, Zhou, Yang, Du, Liu and Zhou26 However, the increase in the number of active catalyst sites (loading capacity) would be limited due to the need to ensure the required faster charge-transport and higher mass-transfer speeds. While the improvement of intrinsic activity is not affected by the above factors, the catalytic performance can be greatly and directly improved along with reduction of load and cost. For example, the cation exchange methodology has been explored to prepare strained and oxygen vacancy-enriched Co3O4 nanorod arrays with excellent OER characteristics.Reference Ling, Yan, Wang, Jiao, Hu, Zheng, Zheng, Mao, Liu, Du, Jaroniec and Qiao27 Morphology design and surface electronic structure engineering can thus work together to achieve the best catalyst performance.
Interface effects
Interface effects, which include the influence of the catalyst support and catalyst–catalyst interactions, have a great impact on the absorption and desorption characteristics of water-splitting intermediates for enhanced energy-conversion efficiency. Many promising materials are capable of functioning as supports, such as gold, carbon-based materials, and metal oxide compounds. Materials with good conductivity are not necessarily good supports. It has been observed that the measured TOF values of Co3O4 catalyst materials were highly dependent on the underlying metal supports following the trend of Au > Pt > Pd > Cu > Co (Figure 2d).Reference Frydendal, Busch, Halck, Paoli, Krtil, Chorkendorff and Rossmeisl28 Recent interest in nanocomposites has broadened the selection of catalyst materials to metal–organic framework (MOF)-based nanomaterials with large surface areas and tunable chemical compositions, in which they are usually constructed by coordination bonds connecting organic ligands and metal centers into 3D interconnected networks.Reference Yang, Xu and Jiang29 Through post-treatment, MOFs can then be converted in situ into a composite material consisting of metal-based active sites supported on carbon materials. Important studies on interface effects of metals and their compounds for water splitting are discussed in the Tang and Sunarticle in this issue.Reference Tang and Sun30 They illustrate the correlation between electronic interaction/strong bonding of composites and water-splitting performance, which is critical to the development of highly efficient and durable electrocatalysts.
pH operation range
In principle, electrocatalysts usually facilitate an efficient OER process in alkaline electrolytes while effectively enabling the HER process under acidic conditions. Inevitably, there are many technological reactions that require the catalyst to function well across all pH conditions. For example, HER is one of the main reactions in hydrogen-based fuel cells and chlor-alkali and chlorate cells, whereas alkaline working electrolytes are required for cell operation. Fortunately, alkaline HER catalysts have been well developed, where their electrocatalytic performance is already comparable to or even better than those of precious metal catalysts operating in their optimal conditions.Reference Safizadeh, Ghali and Houlachi31 Due to the thermodynamic instability and low corrosion resistance of non-noble metals functioning in acid solutions, the development of efficient and durable electrocatalysts that can perform well in acid electrolytes has attracted much attention. Among the various catalyst material candidates, MnOx has been shown to be a stable OER catalyst in acid solution because of its self-healing ability, which is derived from MnOx redeposition that offsets catalyst dissolution during turnover.Reference Huynh, Bediako and Nocera32 Simultaneously, carbon and non-precious metal-based composites have shown encouraging prospects since carbon materials can serve as the buffer layer between electrolytes and catalysts.Reference Bennett33 One concern is that the use of composites cannot effectively prevent sharp fluctuations in local pH, where changes in pH near the electrode can be varied in a range of 5–9 pH units as compared with the electrolyte, even with current densities less than 10 mA cm−2.Reference Katsounaros, Meier, Klemm, Topalov, Biedermann, Auinger and Mayrhofer34 Recent development of carbon-based hybrids is stated in the Niu et al. article in this issue.Reference Niu, Sun, An, Qu, Wang and Sun35 More importantly, since employment of strong acid and alkaline electrolytes certainly consumes more energy and induces a higher operating cost as well as potential damage to water electrolysis devices, utilizing pure distilled water or even seawater as the electrolyte will become a major development in the near future.Reference Tong, Forster, Dionigi, Dresp, Sadeghi Erami, Strasser, Cowan and Farràs36 In this issue, the Janani et al. article summarizes the performance of various catalysts for water splitting and details the corresponding mechanisms in all pH ranges.Reference Janani, Choi, Surendran and Sim37
Activity descriptors
In order to explore high-performance electrocatalysts with appropriate material composition, morphology, and electrochemical characteristics, a large number of DOEs are necessary. Theoretical studies can provide complementary and valuable information to facilitate experimental investigations in a more effective manner for proper catalyst design. Specifically, using DFT calculation and other theoretical methods, the scaling relationships between the electronic structure of catalysts and the binding capacity of intermediates on the catalyst surface can be established.
The d-band theory is one of the earliest and most important electronic descriptors, where higher (lower) d-band center position of the metal ions would yield stronger (weaker) affinity toward adsorbates (Figure 3a).Reference Hammer and Nørskov38 In most cases, the performance enhancement mechanism of electrocatalysts can be understood by using d-band theory. The first example shows that when compared with the HER process in acid media, the “volcano” curve (plot of electrochemical activity as a function of free energy of hydrogen absorption that is in the shape of a volcano) of catalysts operating in alkaline conditions moved vertically rather than horizontally (Figure 3b–c), clearly indicating the effect of d-band center position of the metal ions.Reference Danilovic, Subbaraman, Strmcnik, Stamenkovic, Markovic and Serbian39 The second example shows that the occupation of the antibonding state of eg orbits is commonly considered a type of descriptor in perovskite oxides, suggesting again the influence of a different d-band center of metal ions.Reference Hwang, Rao, Giordano, Katayama, Yu and Shao-Horn40 Last, the effect of strain engineering on catalysts can be attributed to the movement of the d-band centers of their constituents, which subsequently affects their chemisorption potential energies or the energy barriers of intermediates.Reference Hammer and Nørskov38 However, transition-metal ions are the only type of materials that are considered as the active sites, as this descriptor of d-band theory is based on molecular orbital theory.

Figure 3. Activity descriptors. (a) Schematic illustration of the d-band theory.Reference Hammer and Nørskov38 x-axis is electron/eV. (b, c) Hydrogen evolution reaction (HER) activity volcano plots in acid and alkaline conditions, respectively.Reference Danilovic, Subbaraman, Strmcnik, Stamenkovic, Markovic and Serbian39 The metal-Had binding energy controls the activity treads both in acid and alkaline condition while extra energy is needed to dissociate water molecules. (d, e) Proposed absorbates evolution mechanism and lattice-oxygen participated mechanism (LOM) in the oxygen evolution reaction, respectively.Reference Rong, Parolin and Kolpak44 There are four steps in the normal oxygen evolution reaction mechanism: the reaction starts with the absorption of OH− on the active center (M-OH) followed by the direct combination of OH− anion to form M-O intermediate and H2O. The next step involves the production of M-OOH species, which subsequently decomposes to O2. For the LOM, the reaction can proceed on the surface oxygen vacancy sites with a similar process. A0 and A1 are two different structure configurations. A0: −O on 1/4 of surface Ni; A1: an unprotonated oxygen is removed from the NiO2 surface plane and placed on top of −O. (f) Formation energy of oxides as the descriptor, showing that the most active catalysts tend to be the least stable ones during reactions.Reference Calle-Vallejo, Díaz-Morales, Kolb and Koper50 Note: Had, hydrogen intermediates; M, metal; ε, energy; εf, Fermi level; ΔG form, formation energy of monoxides; η, overpotential.
When DFT studies are combined with x-ray absorption spectroscopy (XAS) analysis, the P atoms of CoP catalysts are also identified as the effective active sites for the HER process.Reference Ha, Han, Risch, Giordano, Yao, Karayaylali and Shao-Horn41 As with CoP, both the metal and chalcogenide atoms can function as active sites on the pyrite-type transition-metal dichalcogenides (as detailed in the Gou et al. article in this issueReference Gou, Zhang, Wu, Dong and Qu42). Moreover, during the OER process, isotope labeling is employed to confirm that the lattice oxygen species participate in the reaction (Figure 3d–e).Reference Macounova, Makarova and Krtil43,Reference Rong, Parolin and Kolpak44 More reliable and useful activity descriptors based on the covalent bond nature between the metal and nonmetal sites of catalysts have been proposed, including the p-band theory, where the p band in the oxygen atom is taken as the activity descriptor, and metal-oxygen covalent mixing.Reference Zhao, Liu, Zha, Cheng, Studt, Henkelman and Gong45 Although the correlation between reaction energy and activation energy is established as an important parameter to predict the catalytic properties and binding strength of the active centers, it may not help facilitate further catalytic performance enhancement because it is difficult to achieve weak intermediate adsorption and low activation energy barriers at the same time. Tackling this dilemma is thus the ultimate challenge. Overall, in addition to the illustrated success of single-atom catalysts with a special coordination environment to break the scaling relationship, the development of more effective methods is needed.Reference Ye, Luo, Zhang, Zhang, Xu, Wang, He, Guo, Zhang, He, Ouyang, Gu, Liu and Sun46
Stability
Apart from catalytic activity, operation stability is another factor for the evaluation of electrocatalysts for practical applications. Typically, stability can be assessed in several hours of measurement using potentiodynamic or chronoamperometry techniques for testing the change in voltage with time, or chronopotentiometry to measure the change in current over time. The activity curve is then used as an indicator of the stability. To further enhance the stability, it is necessary to first understand the chemical and physical processes on the catalyst surface during reactions. This information can be obtained by utilizing in situ mass change and conversion analysis of active sites on the surface. The electrochemical quartz crystal microbalance (eQCM) with a detection limit of 1 ng as well as inductively coupled plasma (ICP) spectrometry can be employed to detect the change in mass.Reference Hillman47,Reference Frydendal, Paoli, Knudsen, Wickman, Malacrida, Stephens and Chorkendorff48 For example, eQCM proved that Ni- and Co-based oxides are more stable than FeOOH in alkaline media when the overpotential is 350 mV.Reference Hillman47 It should be noted that the mass change can also be caused by other reasons (such as ion intercalation). Therefore, as compared with eQCM, ICP is more reliable, although it cannot be used for real-time detection. Nevertheless, both eQCM and ICP can only provide limited information on catalyst stability due to the complex reaction process occurring in the electrolyte.
Considering that mass change normally results from the dissolution/deposition process of the catalyst and is accompanied by changes in composition and electronic structure,Reference Feng, Hong, Fong, Lee, Yacoby, Morgan and Shao-Horn49 it is essential to assess the direct link between active sites and stability. Even though there are many characterization methods that can provide clear evidence for changes in catalyst electronic structure before and after the reaction, such as high-resolution transmission electron microscopy and XAS, it is still difficult to correlate any direct changes between electronic structure and stability. Many researchers have tried to explain the relationship between stability and electronic structure from a theoretical perspective. The Koper group has demonstrated the intimate correlation between bulk thermochemistry and surface adsorption on the basis of transition-metal perovskites and monoxides for the OER process (Figure 3f).Reference Calle-Vallejo, Díaz-Morales, Kolb and Koper50 These results indicate that the most active compounds tend to behave as the least stable species during reactions, providing a valuable guideline for the strategy of further improving the activity and stability of a specific catalyst at the same time.
Opportunities and challenges
The water-splitting electrolyzer offers a green pathway to the cost-effective and high-purity production of hydrogen. Notably, several types of nanomaterials have demonstrated excellent electrocatalytic performance toward HER (e.g., NiMo nanowires,Reference Fang, Gao, Dong, Xia, Yip, Qin, Qu and Ho51 V8C7,Reference Xu, Wan, Zhang, Fang, Liu, Huang, Li, Gu and Wang52 and Co4N@nitrogen-doped carbonReference Yuan, Wang, Ma, Qiu, An and Cheng53), OER (e.g., NiFe LDH,Reference Lu and Zhao54 Fe-LiNiO2,Reference Zhu, Wu, Zhu, Li, Li, Lu, Wang, Zhu and Yang55 and NiFeB;Reference Han, Hong, Woo, Mhin, Kim, Kwon, Choi, Chung and Song56 LDH = layered double hydroxide), and overall water splitting (e.g., NiFe LDH@Cu nanowires@Cu foam,Reference Yu, Zhou, Sun, Qin, Yu, Bao, Yu, Chen and Ren57 NiFe MOFs@nickel foams,Reference Senthil Raja, Lin and Lu58 and NiFe LDH@Ni chains@nickel foamReference Cai, Bu, Wang, Su, Wei, Ho, Yang and Wang59) that is comparable to or better than that of noble metals-based materials. Nevertheless, several issues still need special attention for further enabling water splitting.
Looking ahead, the following key challenges need to be addressed by the community. (1) The design of a catalyst with much higher intrinsic activity must be explored. Because good contact between catalyst and conductor is required for efficient charge-carrier collection, many powder catalysts with superior catalytic performance may not be applicable for industrial applications. Therefore, a functionally graded nanocomposite material, known as a traditional coating layer, may provide a promising direction for real-time applications, as discussed by the Coy article in this issue.Reference Coy60 (2) During catalyst material design, exploring the relationship between stability and electronic structure mainly under different electrolyte conditions is essential and provides important insights into further optimizing catalyst performance for reactions. (3) Simultaneously, it is also important to identify active sites and dynamic catalytic processes of the catalysts. Thus, in situ observations utilizing advanced characterization technologies as well as fundamental studies based on theoretical calculations are highly desirable in future research. (4) Since strong acidic and alkaline electrolytes may potentially degrade the electrolyzer cell performance, it is necessary to develop water-splitting technologies using the direct electrolysis of saline water or even seawater.
All of these indicate the importance of materials innovations for the realization of highly efficient and durable electrocatalysts for water splitting.
Acknowledgments
This work is financially supported by the General Research Fund (CityU 11211317) and the Theme-Based Research Scheme (T42–103/16-N) of the Research Grants Council of Hong Kong SAR, China, the National Natural Science Foundation of China (Grant 51672229), and the Science Technology and Innovation Committee of Shenzhen Municipality (Grant JCYJ20170818095520778).
 Xiuming Bu is a doctoral candidate in the Department of Materials Science and Engineering at the City University of Hong Kong. He received his MS degree in materials science and engineering from the University of Shanghai for Science and Technology, China, in 2017. His research interests include the fabrication of nanomaterials and their applications in electronics and energy conversion. Bu can be reached by email at [email protected].
Xiuming Bu is a doctoral candidate in the Department of Materials Science and Engineering at the City University of Hong Kong. He received his MS degree in materials science and engineering from the University of Shanghai for Science and Technology, China, in 2017. His research interests include the fabrication of nanomaterials and their applications in electronics and energy conversion. Bu can be reached by email at [email protected].
 Yanguang Li has been a professor at the Institute of Functional Nano and Soft Materials at Soochow University, China, since 2013. He obtained his BS degree in chemistry from Fudan University, China, in 2005, and his PhD degree in chemistry from The Ohio State University in 2010. He completed postdoctoral research at Stanford University. His research focuses on nanostructured functional materials for energy applications, particularly in the realm of electrocatalysis. Li can be reached by email at [email protected].
Yanguang Li has been a professor at the Institute of Functional Nano and Soft Materials at Soochow University, China, since 2013. He obtained his BS degree in chemistry from Fudan University, China, in 2005, and his PhD degree in chemistry from The Ohio State University in 2010. He completed postdoctoral research at Stanford University. His research focuses on nanostructured functional materials for energy applications, particularly in the realm of electrocatalysis. Li can be reached by email at [email protected].
 Johnny C. Ho is a professor of materials science and engineering at City University of Hong Kong. He received his BS degree in chemical engineering, and his MS and PhD degrees in materials science and engineering from the University of California, Berkeley, in 2002, 2005, and 2009, respectively. From 2009 to 2010, he was a postdoctoral research fellow in the Nanoscale Synthesis and Characterization Group at Lawrence Livermore National Laboratory. His research interests focus on synthesis, characterization, integration, and device applications of nanoscale materials for various technological applications, including nanoelectronics, sensors, and energy harvesting. Ho can be reached by email at [email protected].
Johnny C. Ho is a professor of materials science and engineering at City University of Hong Kong. He received his BS degree in chemical engineering, and his MS and PhD degrees in materials science and engineering from the University of California, Berkeley, in 2002, 2005, and 2009, respectively. From 2009 to 2010, he was a postdoctoral research fellow in the Nanoscale Synthesis and Characterization Group at Lawrence Livermore National Laboratory. His research interests focus on synthesis, characterization, integration, and device applications of nanoscale materials for various technological applications, including nanoelectronics, sensors, and energy harvesting. Ho can be reached by email at [email protected].



