


The gastrointestinal tract is host to a number of bacteria that reside within its lumen. The adult human harbours approximately 1014 microbial organisms belonging to 400–500 different species in the gastrointestinal tract( Reference O'Hara and Shanahan 1 – Reference Wallace, Guarner and Madsen 3 ). The gut is sterile while the fetus is in the uterus, and becomes colonised with microbes immediately after birth. The nature of the microbes that colonise the gut initially is thought to be important in conditioning the development of the host immune system( Reference Lee and Mazmanian 4 , Reference Bezirtzoglou and Stavropoulou 5 ). The establishment of the gut microbiota after birth was studied from the late 1950s through culture of the faeces in neonates( Reference Colgan 6 – Reference Rotimi and Duerden 9 ). Culture-based studies showed that facultative anaerobes (Enterobacteriaceae, streptococci and staphylococci) appear first. These establish an anaerobic luminal environment in the colon leading to the subsequent appearance of obligate anaerobes such as bifidobacteria( Reference Adlerberth and Wold 10 ). Differences in the faecal microbiota have been noted between vaginally delivered neonates and those delivered by Caesarean section( Reference Biasucci, Rubini and Riboni 11 ). By the second year of life, the gut microbiota composition stabilises with approximately 400 species of bacteria, although studies in southern India suggest there is further remodelling of the microbiota through childhood and adolescence( Reference Balamurugan, Janardhan and George 12 ). Traditional bacterial culture is not capable of detecting many of the fastidious obligate anaerobes present in the gut as they rapidly die on exposure to minimal amounts of oxygen. The use of molecular techniques to study the gut flora indicates that 50–70 % of the faecal microbiota is not fit for culture, and thus molecular techniques give us a more comprehensive representation of the microbiota composition. Several molecular studies have examined the development of the gut microbiota in high-income countries( Reference Eggesbø, Moen and Peddada 13 – Reference Hascoët, Hubert and Rochat 15 ).
The developing intestinal microbiome is important in intestinal immune conditioning and in nutrition( Reference Kau, Ahern and Griffin 2 , Reference Lee and Mazmanian 4 , Reference Bezirtzoglou and Stavropoulou 5 ). Specific microbial communities such as lactobacilli and bifidobacteria are used as probiotics to alter the gut microbiota profile in a manner that will favour health. Neonates in low-income countries are born in a contaminated environment. Infant feeding practices in these countries differ from those in high-income countries. The only information on development of the gut microbiota in neonates in low-income countries comes from limited culture-based studies done four decades ago( Reference Mata and Urrutia 7 , Reference Albert, Bhat and Rajan 16 ). The present studies were undertaken to examine the development of the gut microbiota in neonatal life, extending through to 6 months after birth, using molecular techniques directed at the 16S rRNA gene, specifically to elucidate the pattern of development of the gut microbiota in a developing country and to ascertain differences in microbiota composition associated with mode of delivery, socio-economic status, feeding practices and birth weight.
Methods
The study was carried out in the Departments of Gastrointestinal Sciences and Neonatology of the Christian Medical College, Vellore. Infants delivered in the inpatient services of the hospital were the primary participants. Mothers admitted to the obstetric wards for delivery were informed of the study on admission. Inclusion criteria were infants born in the hospital after 28 weeks of gestation, hailing from families residing within 30 km of the hospital. Exclusion criteria were preterm infants born at gestational age <27 weeks, birth weight <900 g, infants with lethal anomalies and critically-ill infants requiring cardio-respiratory support on the first day of life with oxygen requirement of more than 80 % at 24 h of age. Focus discussions were held with mothers prior to obtaining informed written consent. This study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects/patients were approved by the Institutional Review Board of the Christian Medical College, Vellore (approval no. 13-19-03-2008). Written informed consent was obtained from all participating mothers.
Demographic details and clinical details including mode of delivery and immediate postnatal interventions were entered into case report forms. Information regarding the infant's health and feeding was recorded. The infant's length and weight were recorded, at birth and at 3 and 6 months after birth. The socio-economic status of the family was scored using a standard scoring system based on education, possessions and income adjusted for 2007( Reference Kumar, Shekhar and Kumar 17 ). Infants were followed up to the age of 1 year. Samples of stool were obtained from each infant on postnatal days 1, 2, 4, 7, 14, 28, 90 and 180, transported immediately to the laboratory, divided into aliquots, identifier numbers allotted and stored at −80°C until processing. DNA was extracted from stool in batches using QiaAMP Stool DNA mini-kits (Qiagen), checked for quality, quantified using Qubit dsDNA BR assay kits (Invitrogen) in a Qubit fluorometer (Invitrogen) and stored at −20°C until analysis. The DNA was subjected to real-time PCR using primers (Table 1) which were targeted at 16S rRNA gene sequences specific to microbial communities at the species, genus or group level( Reference Suzuki, Taylor and DeLong 18 – Reference Ramakrishna 24 ). The microbial communities that were targeted included Bifidobacterium, Lactobacillus acidophilus group, Bacteroides–Prevotella group, Clostridium coccoides–Eubacterium rectale group, Clostridium leptum group, Enterococcus and Staphylococcus. These microbial communities were targeted because of their significance to human health. The DNA sequences of interest were amplified using SYBR green in a Chromo4 instrument (Biorad)( Reference Matsuda, Tsuji and Asahara 25 ). DNA of bacteria of interest was expressed as the relative difference relative to the amplification of ‘universal’ DNA sequences conserved throughout domain bacteria. Melting curve analysis was always done to check the specificity of the amplification. Quantification was based on the fluorescence intensity obtained from the intercalated SYBR Green dye. The cycle number at which the signal was first detected – the threshold cycle (Ct) – correlated with the original concentration of DNA template. 16S rRNA gene copy number was calculated for total bacteria in faeces using an external set of standard DNA copies for the universal bacteria sequences. For the other bacterial communities studied, relative DNA copy abundance was calculated from the Ct at which the target DNA was detected relative to the Ct at which ‘universal bacterial’ DNA was detected after amplification, using the formula 2−ΔCt and expressed as relative fold difference( Reference Balamurugan, Janardhan and George 26 ). In brief, the calculation of the relative fold difference was based on the difference between the PCR cycle number at which the ‘universal bacteria’ amplicon reached a set threshold (Ct) and the PCR cycle number at which the amplicon of interest reached the same threshold. Each PCR plate used in the quantitative PCR contained, besides the PCR for the amplicons of interest, PCR with primers for amplification of the ‘universal domain bacteria’ sequences that are strongly conserved in the 16S rRNA gene. The ratio calculated for each of the duplicate samples correlated well with the other, with an adjusted R 2 in the region of 0·80.
Table 1. Primers used for real-time PCR amplification of the bacterial communities of interest

Bacterial quantification was handled as a continuous variable. The data on bacterial quantification were expressed as the relative fold difference which is a logarithmic value, and significance of difference was evaluated using non-parametric tests. Repeated measures ANOVA (with post hoc Tukey's multiple comparison) was done to compare microbial abundance within the same infants at different time points. The Student's t test with Welch's correction was used to compare two groups at any time point.
Results
A total of eighty-three infants (forty-one male) participated in these studies. Their mean birth weight was 2·87 (sd 0·45) kg and birth length 48·4 (sd 2·5) cm. At 3 months age, their mean weight and length were 5·9 (sd 1·0) kg and 60·5 (sd 3·6) cm, respectively. Of the infants, ten were delivered by Caesarean section while the rest were vaginally delivered (eight had forceps or suction cup used during delivery); three infants were of very low birth weight, eight were of low birth weight and the remainder were of normal birth weight. The gestation period was 32–36 weeks for seven infants, while the rest were all born after 36 weeks of gestation; delivery was post-term in 18 mothers. A total of six infants received formula milk in the first week of life, while the others had exclusive breast-feeding for periods ranging from 1 to 15 months with the majority continuing exclusive breast feeds for 6 months. A total of eight infants were weaned to solid foods in the fourth month of life, and eighteen in the fifth month of life, while weaning foods were introduced in the remaining infants at the age of 6 months or later. The mean socio-economic score was 16 (sd 5) where a score of eleven–fifteen was classified as middle lower middle class and sixteen–twenty-five as upper middle class.
As seen in Fig. 1, there were a mean of 4·1 × 109 (sem 1·9 × 109) bacterial rRNA gene copies/g stool on the first day of life that increased to 1·2 × 1010 (sem 3·1 × 109) copies/g stool by day 4 of life (NS) and remained stable at those levels for the first 6 months. All bacterial groups tested were detected in the stool from the first day of life and all could be detected until the end of 6 months of age. Enterobacteriaceae predominated in the first week of life (Fig. 2). Bifidobacteria were low in numbers on day 1 after birth but increased 10-fold or more from day 4 and remained constant thereafter (Fig. 2). Staphylococci also increased by day 4 and then returned to earlier levels by the end of the first month. The Bacteroides–Prevotella group, a prominent microbial community in the adult gut, was present in small numbers after birth and increased around 6 months of age to become one of the more abundant communities in the faecal microbiota. Enterococci were less abundant constituents of the faecal microbiota, remaining at somewhat similar levels from day 1 to 6 months of age. C. coccoides–E. rectale, a prominent carbohydrate-fermenting bacterial community, were found in very low relative numbers in the faeces of these infants until the age of 6 months (Fig. 2). The C. leptum group of bacteria was not detected in over a half of infants in the study.
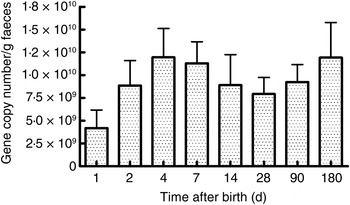
Fig. 1. Total bacterial count in faeces estimated using real-time PCR amplification of universal 16S rDNA sequences conserved for domain bacteria. Quantification was done with reference to an external standard of plasmid DNA copies. Values are means, with standard errors represented by vertical bars. None of the values was statistically significantly different from the others.
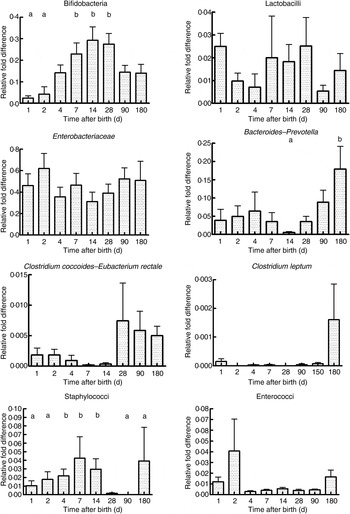
Fig. 2. Abundance of specific microbial communities in the faeces of neonates followed longitudinally at 6 months. Data were acquired by real-time PCR targeting specific sequences of the 16S rRNA gene and are expressed as relative difference in comparison with amplification of the ‘universal’ Bacteria domain sequence. Values are means, with standard errors represented by vertical bars. a,bMean values with unlike letters were significantly different (P < 0·05; repeated measures ANOVA with post hoc Tukey's test).
The mode of delivery influenced bacterial colonisation. On the first day of life, lactobacilli and Enterobacteriaceae were more abundant in the faeces of vaginally delivered infants compared with those delivered by Caesarean section (Fig. 3), but their abundance in subsequent days was not different between the two groups. Bacteroides–Prevotella were more abundant in the faeces of vaginally delivered infants compared with those delivered by Caesarean section from birth up to 90 d (Fig. 3). Bifidobacteria were more abundant in vaginally delivered infants at 90 d (Fig. 3). The socio-economic status of the family was associated with differences in the faecal microbiota at 90 and 180 d when lactobacilli and Bacteroides–Prevotella were noted to be more abundant in infants from families with higher socio-economic score than in those coming from a lower socio-economic stratum (Fig. 4). Birth weight did not have a significant association with microbiota composition at birth, although lactobacilli were less abundant at 90 d in low-birth-weight infants (<2·5 kg) compared with large (>3·5 kg) infants. The overwhelming majority of infants were breast-fed from birth, making it difficult to compare the effect of breast-feeding and bottle-feeding on the microbiota. Cessation of breast-feeding by day 90 was not associated with any significant alteration in abundance of these microbial communities (data not shown). Infants who received supplemental cows' milk (in addition to breast feeds) by day 90 had lower numbers of Enterobacteriaceae than infants who were exclusively breast fed. We did not find any association between faecal microbiota abundance at birth and length at birth or at the age of 1 year.
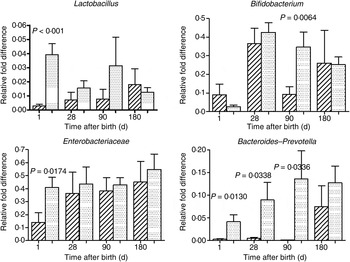
Fig. 3. Association of mode of delivery (normal vaginal delivery (![]() ); lower segment Caesarean section (
); lower segment Caesarean section (![]() )) and faecal microbial colonisation. Values are means, with standard errors represented by vertical bars. P values shown were calculated using the Student's t test with Welch's correction.
)) and faecal microbial colonisation. Values are means, with standard errors represented by vertical bars. P values shown were calculated using the Student's t test with Welch's correction.
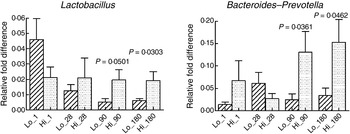
Fig. 4. Association of socio-economic status and microbial colonisation. Based on the socio-economic score, mothers were classified into three equal groups, with the lowest tertile being represented as Lo_ and the highest tertile as Hi_. Values are means, with standard errors represented by vertical bars. P values shown were calculated using Student's t test with Welch's correction. The other microbial communities tested did not significantly differ between low and high socio-economic groups (data not shown).
Discussion
This study provides a longitudinal documentation of the faecal microbial community composition of Indian infants from birth to 6 months of age. Faecal bacterial counts reached peak numbers by the fourth day of life and remained steady thereafter in the first 6 months of life. Enterobacteriaceae and lactobacilli were the most abundant bacteria in the first 2 d of life, while bifidobacteria and staphylococci increased in faeces by the fourth day, possibly accounting for the increase in faecal bacterial counts. Bacteroides–Prevotella group, another prominent component of the faecal microbiota, increased at the time of weaning. C. coccoides–E. rectale were low in abundance in the first 6 months of life, while the C. leptum group was not detected in significant numbers of infants. The study also confirms differences in the faecal microbiota between vaginally delivered infants and those delivered by Caesarean section, and demonstrates associations between gut colonisation patterns and socio-economic status of the family, exclusive breast-feeding and weaning.
The faecal microbiota is composed of obligate anaerobes with a smaller contribution from facultative anaerobes. The strict anaerobes are often difficult to culture. In the last two decades, the study of the faecal microbiota has been greatly enhanced by the advent of molecular methods( Reference Schmittgen and Livak 27 ). Most of these techniques depend upon targeting of the 16S rRNA gene, which aids in taxonomic identification of the microbes due to a large database of information available in public databases( Reference Ramakrishna 24 ). The present study used real-time PCR targeting these sequences in order to obtain quantitative abundance information of specific groups or genera of bacteria that are of biological significance to human health. This technique is very sensitive, being able to detect 2–1000 copies/ml of the target organism or group of organisms( Reference Ramakrishna 24 ). Recently deep sequencing and next-generation sequencing techniques have been used to examine the faecal microbiota. The latter techniques are particularly useful to identify the presence of novel or previously unknown microbes and are complementary to real-time PCR. For our purposes, the use of real-time PCR was more appropriate, providing quantitative information about bacterial groups or genera of interest.
The development of the neonatal faecal microbiota has been studied by culture-dependent techniques from the 1950s onwards( Reference Colgan 6 – Reference Adlerberth and Wold 10 ). Rotimi & Duerden compared bacterial abundance in the first week of life by the percentage of children from whose faeces the bacteria could be isolated( Reference Rotimi and Duerden 9 ). These studies showed that Escherichia coli, enterococci, staphylococci and streptococci were the most prominent colonisers in the first week of life( Reference Adlerberth and Wold 10 ). It was postulated that these facultative anaerobes were able to survive in the neonatal colon where the abundance of oxygen does not permit obligate anaerobes to flourish, and that their growth led to a drop in oxygen tension which then allowed strict anaerobes, such as bifidobacteria, Bacteroides and Clostridium to colonise the gut( Reference Adlerberth and Wold 10 ). Colonisation by lactobacilli was reported to be uncommon in the first year of life( Reference Adlerberth and Wold 10 ). DNA-based studies of microbial colonisation suggest that Proteobacteria, Bacteroides, Firmicutes (dominated by Clostridium), Actinobacteria (and its constituent bifidobacteria) and Verrucomicrobia are the dominant microbiota in the first few months of life( Reference Palmer, Bik and Digiulio 28 ). The dominance of Enterobacteriaceae among the faecal microbiota in the first week of life has been described in both Western and Japanese neonates( Reference Adlerberth and Wold 10 , Reference Tsuji, Oozeer and Matsuda 29 ). Our study shows that they continued to be relatively abundant in the first 6 months of life. In the present study, faecal bacterial numbers did not significantly increase after day 1 and remained constant in the first 6 months of life, indicating very rapid colonisation to peak levels immediately after birth. This may be a feature of gut microbial colonisation in low-income countries. A culture-based study compared a low socio-economic status cohort of infants in Pakistan with a cohort in Sweden and found that colonisation of the intestine was significantly more rapid in the former( Reference Adlerberth, Carlsson and de Man 30 ).
In the present study lactobacilli, Bacteroides–Prevotella and Enterobacteriaceae were all significantly less abundant in faeces on the first day of life in neonates delivered by Caesarean section compared with those delivered vaginally. Our findings are corroborated by studies in which infants delivered vaginally had higher numbers of Bacteroides and bifidobacteria in their faecal microbiota compared with those delivered by Caesarean section( Reference Rotimi, Olowe and Ahmed 31 , Reference Penders, Thijs and Vink 32 ). Another study of 1-d-old infants using pyrosequencing of 16S rRNA genes suggested that vaginally delivered infants had greater numbers of Lactobacillus, Prevotella or Sneathia species, while faeces of neonates delivered by Caesarean section had greater abundance of Staphylococcus, Corynebacterium and Propionibacterium. Neonates in the latter study were not followed up after 1 d of life( Reference Dominguez-Bello, Costello and Contreras 33 ). In the case of delivery by Caesarean section, our findings suggest that colonisation of the neonatal gut by these specific microbial communities was delayed only by a few days at most.
Breast-fed neonates have been reported to have higher faecal bacterial counts during the first week of life than bottle-fed neonates. In breast-fed infants, bifidobacteria accounted for 90 % of faecal bacteria, with lesser numbers of Enterobacteriaceae and enterococci( Reference Stark and Lee 34 , Reference Yoshioka, Iseki and Fujita 35 ). In bottle-fed infants, on the other hand, streptococci, Enterobacteriaceae and Bacteroides tended to predominate( Reference Benno, Sawada and Mitsuoka 36 ). Breast milk contains factors that promote the growth of Bifidobacterium bifidum, while formula feeds are considered to promote implantation and persistence of Clostridium perfringens ( Reference Solís, de Los Reyes-Gavilan and Fernández 37 ). Exclusive breast-feeding up to the age of 6 months is a message actively promoted to all young mothers in India. In our cohort, only six neonates received infant formula in the first week of life, in conditions where there was partial failure of breast-feeding. There was no significant difference noted in microbial colonisation of the gut in this group compared with breast-fed infants, but this must be interpreted with caution because of the very low numbers in the non-breast-fed group. The complete cessation of breast-feeding by 3 months did not have significant influence on abundance of faecal microbial communities. However, exclusive breast-feeding (no complementary feeds at all) was associated with a higher abundance of Enterobacteriaceae compared with infants who received complementary milk feeds in addition to breast milk. The significance of this observation is not clear and may need further investigation.
During weaning, the diet changes from a high-fat-milk-based diet to a diet rich in carbohydrate. Bacterial oxidative metabolism leads to changes in the redox potential of the colon and results in increased abundance of strict anaerobes such as Bacteroides and Clostridium. Bifidobacteria, C. coccoides and Bacteroides were reported to be more prominent after weaning in a multi-country European study( Reference Fallani, Amarri and Uusijarvi 38 ). In the present study, complementary feeding was introduced in a small number of infants before the age of 6 months and did not significantly alter microbiota composition.
This study had several shortcomings that need to be considered when interpreting the data in conjunction with other data available in the literature. Molecular techniques for examination of a complex microbial ecosystem as in the human gut do not usually correct for the multiplicity of 16S rRNA genes in an individual microbe. However, the gene copy number for total bacteria in our quantitative study was in the region of 109–1010 copies/g wet weight faeces, which is in excellent agreement with the results obtained by Palmer et al. using a similar method of quantitative real-time PCR( Reference Palmer, Bik and Digiulio 28 ). While the gene copy number is more or less constant in species, it may vary significantly across species and genera. The relative fold difference in this study thus does not give an absolute number, but since measurements were done in the same infants at different time points, the overall direction of change is an important indicator of changes in abundance of a particular microbial community. Secondly, the differences that we had anticipated due to influences such as Caesarean section, type of milk provided and socio-economic status, were relatively minor and not to the level that we had anticipated. This could partly be secondary to the small numbers of infants we had in some of the groups, but it is quite possible that they could truly represent the gut microbiota development pattern in developing countries.
These molecular studies show that the gastrointestinal tract was colonised by microbes even on the first day of life with representation of all major microbial communities. Colonisation reached peak levels almost immediately although there was a minor, non-significant increase in the next 2 d. There were differences in the proportions of the microbial communities in neonates delivered by Caesarean section and neonates born to mothers hailing from a lower socio-economic class but these differences did not persist beyond the second day of life. Introduction of supplemental feeding was associated with reduced abundance of Enterobacteriaceae. These studies provide an understanding of gastrointestinal colonisation between birth and weaning in low-income countries that will serve as background information for designing nutritional interventions targeted at the microbiota.
Acknowledgements
This study was supported by ad hoc research grant no. 5/8-1(223)/D/2008-ECD-II from the Indian Council of Medical Research, New Delhi. J. K. was supported by a Senior Research Fellowship from the Indian Council of Medical Research. The authors' contributions are as follows: J. K. was responsible for the laboratory analyses and statistical analyses; S. F. was responsible for participant recruitment, sample acquisition and sample preparation; R. B. was responsible for study design and initiation of the study; J. M. and R. V. were responsible for participant recruitment, sample acquisition and participant follow up; S. S. and A. K. J. were responsible for participant recruitment, study design and data acquisition; B. S. R. was responsible for study design, data analysis and presentation, and acquisition of funding. All authors read and approved the final manuscript. None of the authors has any conflict of interest to declare.






 Open access
Open access