







1. Introduction
Consider a liquid drop floating in the air and evaporating. In its simplest formulation, this problem was examined more than 140 years ago by Maxwell (Reference Maxwell1877, Reference Maxwell1890), and its more elaborate versions are still explored now. Several of these are relevant to the present study: Sobac et al. (Reference Sobac, Talbot, Haut, Rednikov and Colinet2015), Cossali & Tonini (Reference Cossali and Tonini2019) and Finneran (Reference Finneran2021) argued that the dependence of the fluid's properties on the temperature can affect the evaporation rate strongly; Talbot et al. (Reference Talbot, Sobac, Rednikov, Colinet and Haut2016) examined transient deviations of drop evaporation from the quasi-steady regime; Finneran, Garner & Nadal (Reference Finneran, Garner and Nadal2021) showed that the transient effects can cause a noticeable departure from the so-called ‘ $d^{2}$ law’ (stating that the time of a drop's evaporation is proportional to its diameter squared); Long, Micci & Wong (Reference Long, Micci and Wong1996), Landry et al. (Reference Landry, Mikkilineni, Paharia and McGaughey2007), Jakubczyk et al. (Reference Jakubczyk, Kolwas, Derkachov, Kolwas and Zientara2012), Hołyst et al. (Reference Hołyst, Litniewski, Jakubczyk, Kolwas, Kolwas, Kowalski, Migacz, Palesa and Zientara2013), Hołyst, Litniewski & Jakubczyk (Reference Hołyst, Litniewski and Jakubczyk2017), Rana, Lockerby & Sprittles (Reference Rana, Lockerby and Sprittles2018, Reference Rana, Lockerby and Sprittles2019) and Zhao & Nadal (Reference Zhao and Nadal2023) showed that the ‘
$d^{2}$ law’ (stating that the time of a drop's evaporation is proportional to its diameter squared); Long, Micci & Wong (Reference Long, Micci and Wong1996), Landry et al. (Reference Landry, Mikkilineni, Paharia and McGaughey2007), Jakubczyk et al. (Reference Jakubczyk, Kolwas, Derkachov, Kolwas and Zientara2012), Hołyst et al. (Reference Hołyst, Litniewski, Jakubczyk, Kolwas, Kolwas, Kowalski, Migacz, Palesa and Zientara2013), Hołyst, Litniewski & Jakubczyk (Reference Hołyst, Litniewski and Jakubczyk2017), Rana, Lockerby & Sprittles (Reference Rana, Lockerby and Sprittles2018, Reference Rana, Lockerby and Sprittles2019) and Zhao & Nadal (Reference Zhao and Nadal2023) showed that the ‘ $d^{2}$ law’ can be violated by out-of-equilibrium gas kinetics. A more detailed review of the recent literature on evaporating drops can be found in Sazhin (Reference Sazhin2017).
$d^{2}$ law’ can be violated by out-of-equilibrium gas kinetics. A more detailed review of the recent literature on evaporating drops can be found in Sazhin (Reference Sazhin2017).
Most work on evaporation has been based on Maxwell's hypothesis that the air near the drop's surface – or, more generally, near any liquid/air interface – is close to saturation. This allows one to solve the problem without knowing anything about the interfacial dynamics. The only exceptions are models accounting for the so-called Knudsen layer – a layer where the out-of-equilibrium kinetics plays a part (e.g. Rana et al. Reference Rana, Lockerby and Sprittles2018, Reference Rana, Lockerby and Sprittles2019; Zhao & Nadal Reference Zhao and Nadal2023).
The physics behind Maxwell's hypothesis becomes clear if evaporation is viewed as a two-tier process: first, the interface emits molecules of the liquid into the air; second, the air passes them on to infinity. If the former emits more vapour than the latter can pass on, then the vapour accumulates near the drop – bringing the adjacent air layer close to saturation and the interface close to thermodynamic equilibrium. As a result, the interface reduces vapour emission in line with the air throughput. In terms of this mechanism, Maxwell's hypothesis amounts to an assumption that the vapour-emission capacity of the interface exceeds the throughput of air.
Maxwell could not have tested this assumption, as there were no quantitative models of interfaces at the time. Two such models exist now: the Enskog–Vlasov kinetic equation (de Sobrino Reference de Sobrino1967; Grmela Reference Grmela1971; Grmela & Garcia-Colin Reference Grmela and Garcia-Colin1980a,Reference Grmela and Garcia-Colinb; Frezzotti, Gibelli & Lorenzani Reference Frezzotti, Gibelli and Lorenzani2005; Barbante, Frezzotti & Gibelli Reference Barbante, Frezzotti and Gibelli2015; Frezzotti & Barbante Reference Frezzotti and Barbante2017; Frezzotti et al. Reference Frezzotti, Gibelli, Lockerby and Sprittles2018; Benilov & Benilov Reference Benilov and Benilov2018, Reference Benilov and Benilov2019a,Reference Benilov and Benilovb; Struchtrup & Frezzotti Reference Struchtrup and Frezzotti2022) and the diffuse-interface model (see a review by Anderson, McFadden & Wheeler Reference Anderson, McFadden and Wheeler1998). The latter is much simpler, thus it makes a better starting point.
The diffuse-interface model (DIM) is based on two assumptions regarding the van der Waals force.
(i) Van der Waals interactions are pairwise – hence the net force affecting a molecule is an algebraic sum of the forces exerted by the other molecules.
(ii) The thickness of the interface is much greater than the spatial scale of van der Waals interactions.
Admittedly, assumption (ii) does not hold well at room temperature, in which case the interfacial thickness rarely exceeds the molecular size by an order of magnitude (e.g. Liu, Harder & Berne Reference Liu, Harder and Berne2005; Verde, Bolhuis & Campen Reference Verde, Bolhuis and Campen2012; Pezzotti, Galimberti & Gaigeot Reference Pezzotti, Galimberti and Gaigeot2017; Pezzotti, Serva & Gaigeot Reference Pezzotti, Serva and Gaigeot2018; Dodia et al. Reference Dodia, Ohto, Imoto and Nagata2019; and references therein). Thus the DIM should be viewed as a means to estimate qualitatively the importance of interfacial dynamics for evaporation, whereas quantitative predictions should be left to the Enskog–Vlasov kinetic equation (which can handle thin interfaces). These two models are to each other what the first term of a Taylor series is to the exact value of the function.
The DIM has been used many times before – for phase transitions in ferroelectrics (Ginzburg Reference Ginzburg1960), spinodal decomposition (e.g. Cahn Reference Cahn1961; Lowengrub & Truskinovsky Reference Lowengrub and Truskinovsky1998), crystal growth (e.g. Collins & Levine Reference Collins and Levine1985; Tang, Carter & Cannon Reference Tang, Carter and Cannon2006; Heinonen et al. Reference Heinonen, Achim, Kosterlitz, Ying, Lowengrub and Ala-Nissila2016; Philippe, Henry & Plapp Reference Philippe, Henry and Plapp2020), solidification (e.g. Stinner, Nestler & Garcke Reference Stinner, Nestler and Garcke2004; Nestler, Garcke & Stinner Reference Nestler, Garcke and Stinner2005), polymers (e.g. Thiele, Madruga & Frastia Reference Thiele, Madruga and Frastia2007; Madruga & Thiele Reference Madruga and Thiele2009), electrowetting (e.g. Lu et al. Reference Lu, Glasner, Bertozzi and Kim2007), contact lines in liquids (e.g. Pismen & Pomeau Reference Pismen and Pomeau2000; Ding & Spelt Reference Ding and Spelt2007; Yue, Zhou & Feng Reference Yue, Zhou and Feng2010; Yue & Feng Reference Yue and Feng2011; Sibley et al. Reference Sibley, Nold, Savva and Kalliadasis2014; Ding et al. Reference Ding, Zhu, Gao and Lu2017; Borcia et al. Reference Borcia, Borcia, Bestehorn, Varlamova, Hoefner and Reif2019; Zhu et al. Reference Zhu, Kou, Yao, Wu, Yao and Sun2019, Reference Zhu, Kou, Yao, Li and Sun2020), Faraday instability (e.g. Borcia & Bestehorn Reference Borcia and Bestehorn2014; Bestehorn et al. Reference Bestehorn, Sharma, Borcia and Amiroudine2021), Rayleigh–Taylor instability (e.g. Zanella et al. Reference Zanella, Tegze, Tellier and Henry2020, Reference Zanella, Le Tellier, Plapp, Tegze and Henry2021), cavitation (e.g. Petitpas et al. Reference Petitpas, Massoni, Saurel, Lapebie and Munier2009), nucleation (e.g. Magaletti, Marino & Casciola Reference Magaletti, Marino and Casciola2015; Magaletti et al. Reference Magaletti, Gallo, Marino and Casciola2016; Gallo, Magaletti & Casciola Reference Gallo, Magaletti and Casciola2018; Gallo et al. Reference Gallo, Magaletti, Cocco and Casciola2020), liquid films (e.g. Benilov Reference Benilov2020, Reference Benilov2022a), tumour growth (e.g. Frigeri, Grasselli & Rocca Reference Frigeri, Grasselli and Rocca2015; Rocca & Scala Reference Rocca and Scala2016; Dai et al. Reference Dai, Feireisl, Rocca, Schimperna and Schonbek2017), classification of data (e.g. Bertozzi & Flenner Reference Bertozzi and Flenner2012, Reference Bertozzi and Flenner2016), and so on. The DIM has also been applied to evaporation and condensation (e.g. Pomeau Reference Pomeau1986; Barbante, Frezzotti & Gibelli Reference Barbante, Frezzotti and Gibelli2014; Benilov Reference Benilov2022a,Reference Benilovb, Reference Benilov2023b), but only in application to liquids evaporating into, or condensating from, their own vapour, not air.
In the present paper, the DIM is used to examine evaporation of liquids into air under ‘normal conditions’, i.e. when the temperature is between  $15\,^{\circ }\mathrm {C}$ and
$15\,^{\circ }\mathrm {C}$ and  $35\,^{\circ }\mathrm {C}$, and the air pressure is at
$35\,^{\circ }\mathrm {C}$, and the air pressure is at  $1$ atm. A generalised Maxwell boundary condition is derived for this case; to facilitate its use without going into the detail of the derivation, this condition is summarised here.
$1$ atm. A generalised Maxwell boundary condition is derived for this case; to facilitate its use without going into the detail of the derivation, this condition is summarised here.
The following notation will be used:  $\rho _{1}$ is the density of the liquid or its vapour,
$\rho _{1}$ is the density of the liquid or its vapour,  $\rho _{2}$ is that of air. Outside the drop, the air is almost homogeneous, i.e.
$\rho _{2}$ is that of air. Outside the drop, the air is almost homogeneous, i.e.  $\rho _{2}\approx \rho _{2}^{(a)}$. Let
$\rho _{2}\approx \rho _{2}^{(a)}$. Let  $\rho _{1}^{(v.sat)}(T)$ be the saturated vapour density at a temperature
$\rho _{1}^{(v.sat)}(T)$ be the saturated vapour density at a temperature  $T$, and let
$T$, and let  $\rho _{1}^{(l.sat)}(T)$ be the matching liquid density. Introduce also the vapour-in-the-air diffusivity
$\rho _{1}^{(l.sat)}(T)$ be the matching liquid density. Introduce also the vapour-in-the-air diffusivity  $\mathcal {D}$, the shear and bulk viscosities of air,
$\mathcal {D}$, the shear and bulk viscosities of air,  $\mu _{s}$ and
$\mu _{s}$ and  $\mu _{b}$, respectively, and the specific gas constant
$\mu _{b}$, respectively, and the specific gas constant  $R_{i}$ of the liquid or its vapour (
$R_{i}$ of the liquid or its vapour ( $i=1$), or the air (
$i=1$), or the air ( $i=2$). The interface as a whole is characterised by its curvature
$i=2$). The interface as a whole is characterised by its curvature  $C$ and surface tension
$C$ and surface tension  $\sigma$.
$\sigma$.
All these parameters are macroscopic, i.e. they characterise the fluid as a continuum, whereas the Korteweg matrix  $K_{ij}$ is a microscopic characteristic. It describes the intermolecular (van der Waals) force due to the liquid–liquid (
$K_{ij}$ is a microscopic characteristic. It describes the intermolecular (van der Waals) force due to the liquid–liquid ( $K_{11}$), air–air (
$K_{11}$), air–air ( $K_{22}$) and liquid–air (
$K_{22}$) and liquid–air ( $K_{12}=K_{21}$) interactions. Macroscopically,
$K_{12}=K_{21}$) interactions. Macroscopically,  $K_{ij}$ determine the capillary properties of the fluid and thus can be deduced from measurements of its surface tension (see Appendix B). The numerical values of
$K_{ij}$ determine the capillary properties of the fluid and thus can be deduced from measurements of its surface tension (see Appendix B). The numerical values of  $K_{ij}$ determined this way for water and air are presented later, in (3.12).
$K_{ij}$ determined this way for water and air are presented later, in (3.12).
Let  $\rho _{1}^{(+)}$ be the macroscopic vapour density immediately outside the interface, and let
$\rho _{1}^{(+)}$ be the macroscopic vapour density immediately outside the interface, and let  $( \boldsymbol {n}\boldsymbol {\cdot }\boldsymbol {\nabla }\rho _{1})^{(+)}$ be its normal derivative (where
$( \boldsymbol {n}\boldsymbol {\cdot }\boldsymbol {\nabla }\rho _{1})^{(+)}$ be its normal derivative (where  $\boldsymbol {n}$ is directed towards the air). Then the generalised Maxwell boundary condition has the form
$\boldsymbol {n}$ is directed towards the air). Then the generalised Maxwell boundary condition has the form
 \begin{equation} \rho_{1}^{(+)}=\rho_{1}^{(v.sat)}\exp\left[ -\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)+\frac{\sigma C}{\rho_{1}^{(l.sat)}R_{1}T}\right] ,\end{equation}
\begin{equation} \rho_{1}^{(+)}=\rho_{1}^{(v.sat)}\exp\left[ -\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)+\frac{\sigma C}{\rho_{1}^{(l.sat)}R_{1}T}\right] ,\end{equation}
where  $\xi$ is the relative vapour flux,
$\xi$ is the relative vapour flux,
 \begin{equation} \xi=\frac{-\mathcal{D}( \boldsymbol{n}\boldsymbol{\cdot}\boldsymbol{\nabla}\rho_{1})^{(+)}}{E_{0}},\end{equation}
\begin{equation} \xi=\frac{-\mathcal{D}( \boldsymbol{n}\boldsymbol{\cdot}\boldsymbol{\nabla}\rho_{1})^{(+)}}{E_{0}},\end{equation}and the parameter
 \begin{equation} E_{0}=\frac{K_{22}^{1/2}\rho_{2}^{(a)5/2}(R_{2}T)^{1/2}} {\frac{4}{3}\mu_{s}+\mu_{b}}\end{equation}
\begin{equation} E_{0}=\frac{K_{22}^{1/2}\rho_{2}^{(a)5/2}(R_{2}T)^{1/2}} {\frac{4}{3}\mu_{s}+\mu_{b}}\end{equation}
will be referred to as the ‘vapour-emission capacity’ of the interface. Physically,  $E_{0}$ is determined by a narrow zone at the outskirts of the interface, driven by the van der Waals force, pressure gradient and viscosity. The location of this zone suggests that it is a DIM analogue of the Knudsen layer, i.e. the Knudsen layer plus the van der Waals force – which is an order-one effect in the DIM, but is neglected by the standard Hertz–Knudsen kinetic model.
$E_{0}$ is determined by a narrow zone at the outskirts of the interface, driven by the van der Waals force, pressure gradient and viscosity. The location of this zone suggests that it is a DIM analogue of the Knudsen layer, i.e. the Knudsen layer plus the van der Waals force – which is an order-one effect in the DIM, but is neglected by the standard Hertz–Knudsen kinetic model.
The universal non-dimensional function  $A(\xi )$ that appears in (1.1) is also a characteristic of the above-mentioned zone at the outskirts of the interface. It can be approximated by the expression
$A(\xi )$ that appears in (1.1) is also a characteristic of the above-mentioned zone at the outskirts of the interface. It can be approximated by the expression
 \begin{equation} A(\xi)=\frac{\frac{2}{5}\ln(\xi+3.2867)-0.81571}{\xi+3.2867}+\frac {1.3074}{(\xi+3.2867)^{7/5}}, \end{equation}
\begin{equation} A(\xi)=\frac{\frac{2}{5}\ln(\xi+3.2867)-0.81571}{\xi+3.2867}+\frac {1.3074}{(\xi+3.2867)^{7/5}}, \end{equation}obtained through a combination of asymptotics, numerics and curve fitting (see Appendix C).
The term involving the curvature  $C$ in condition (1.1) describes the Kelvin effect, i.e. the shift of the saturated vapour pressure near a curved surface (e.g. Eggers & Pismen Reference Eggers and Pismen2010; Colinet & Rednikov Reference Colinet and Rednikov2011; Rednikov & Colinet Reference Rednikov and Colinet2013, Reference Rednikov and Colinet2017, Reference Rednikov and Colinet2019; Morris Reference Morris2014; Janeček et al. Reference Janeček, Doumenc, Guerrier and Nikolayev2015; Saxton et al. Reference Saxton, Vella, Whiteley and Oliver2017). If the interface is flat (
$C$ in condition (1.1) describes the Kelvin effect, i.e. the shift of the saturated vapour pressure near a curved surface (e.g. Eggers & Pismen Reference Eggers and Pismen2010; Colinet & Rednikov Reference Colinet and Rednikov2011; Rednikov & Colinet Reference Rednikov and Colinet2013, Reference Rednikov and Colinet2017, Reference Rednikov and Colinet2019; Morris Reference Morris2014; Janeček et al. Reference Janeček, Doumenc, Guerrier and Nikolayev2015; Saxton et al. Reference Saxton, Vella, Whiteley and Oliver2017). If the interface is flat ( $C=0$) and its vapour-emission capacity exceeds the diffusive flux,
$C=0$) and its vapour-emission capacity exceeds the diffusive flux,
 \begin{equation} E_{0}\gg\vert {-\mathcal{D}}( \boldsymbol{n}\boldsymbol{\cdot}\boldsymbol{\nabla}\rho_{1})^{(+)}\vert , \end{equation}
\begin{equation} E_{0}\gg\vert {-\mathcal{D}}( \boldsymbol{n}\boldsymbol{\cdot}\boldsymbol{\nabla}\rho_{1})^{(+)}\vert , \end{equation}then (1.1) reduces to the classical Maxwell boundary condition.
This paper has the following structure. In § 2, Maxwell's boundary condition is reviewed briefly in application to evaporation of floating drops. Its generalised version is derived in § 3, and applied to drops in § 4. In § 5, the proposed model is compared to the Hertz–Knudsen kinetic model, and further applications of the former are outlined.
2. The classical model
2.1. Formulation
Consider a mixture of two fluids with partial densities  $\rho _{i}$, where
$\rho _{i}$, where  $i=1,2$. The first fluid will be referred to as either ‘liquid’ or ‘vapour’ (depending on the phase it is in), and the second fluid will be referred to as ‘air’.
$i=1,2$. The first fluid will be referred to as either ‘liquid’ or ‘vapour’ (depending on the phase it is in), and the second fluid will be referred to as ‘air’.
Under the assumption of spherical symmetry,  $\rho _{i}$ generally vary with the radial coordinate
$\rho _{i}$ generally vary with the radial coordinate  $r$ and time
$r$ and time  $t$. In the classical model, however, one assumes that inside the drop
$t$. In the classical model, however, one assumes that inside the drop
 \begin{equation} \rho_{1}=\rho_{1}^{(l)},\quad\rho_{2}=0\quad\text{if}\ r\in(0,r_{d}) , \end{equation}
\begin{equation} \rho_{1}=\rho_{1}^{(l)},\quad\rho_{2}=0\quad\text{if}\ r\in(0,r_{d}) , \end{equation}
where  $\rho _{1}^{(l)}$ is a known constant, and the drop's radius
$\rho _{1}^{(l)}$ is a known constant, and the drop's radius  $r_{d}$ depends on
$r_{d}$ depends on  $t$.
$t$.
Outside the drop, both densities do vary. The air density is much larger than that of the vapour, but their variations are of the same order (as, physically, the pressure should be spatially uniform). As a result, the variations of  $\rho _{2}$ are relatively weak, so one assumes
$\rho _{2}$ are relatively weak, so one assumes
 \begin{equation} \rho_{2}=\rho_{2}^{(a)}\quad\text{if}\ r\in( r_{d},\infty), \end{equation}
\begin{equation} \rho_{2}=\rho_{2}^{(a)}\quad\text{if}\ r\in( r_{d},\infty), \end{equation}
where the dry-air density  $\rho _{2}^{(a)}$ is a known parameter. Note that, physically,
$\rho _{2}^{(a)}$ is a known parameter. Note that, physically,  $\rho _{1}^{(l)}$ and
$\rho _{1}^{(l)}$ and  $\rho _{2}^{(a)}$ are inter-related by the requirement that the pressure difference between the inside and outside of the drop equals the capillary correction due to the curvature of the interface.
$\rho _{2}^{(a)}$ are inter-related by the requirement that the pressure difference between the inside and outside of the drop equals the capillary correction due to the curvature of the interface.
The classical model of evaporation also involves the liquid's thermal conductivity  $\kappa ^{(l)}$, air's thermal conductivity
$\kappa ^{(l)}$, air's thermal conductivity  $\kappa ^{(a)}$, saturated vapour density
$\kappa ^{(a)}$, saturated vapour density  $\rho _{1}^{(v.sat)}$, vaporisation heat
$\rho _{1}^{(v.sat)}$, vaporisation heat  $\varDelta$, and vapour-in-the-air diffusivity
$\varDelta$, and vapour-in-the-air diffusivity  $\mathcal {D}$. The dependence of these parameters on the temperature
$\mathcal {D}$. The dependence of these parameters on the temperature  $T$ is supposed to be known.
$T$ is supposed to be known.
The classical model is usually combined with the quasi-steady approximation, based on an assumption that the diffusivity and thermal conductivity are sufficiently large (which they are indeed for water under normal conditions). Then the term  $\partial \rho _{1}/\partial t$ in the diffusion equation and the term
$\partial \rho _{1}/\partial t$ in the diffusion equation and the term  $\partial T/\partial t$ in the heat-conduction equation can both be neglected. The simplest version of the classical model also neglects the Stefan flow (which carries the vapour away from the surface of the liquid), the Soret effects (the mass flux due to the temperature gradient), and the Dufour effect (the heat flux due to the density gradient).
$\partial T/\partial t$ in the heat-conduction equation can both be neglected. The simplest version of the classical model also neglects the Stefan flow (which carries the vapour away from the surface of the liquid), the Soret effects (the mass flux due to the temperature gradient), and the Dufour effect (the heat flux due to the density gradient).
Then, for the spherically symmetric case, one obtains
 \begin{gather} \frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial T}{\partial r}\right) =0\quad\text{if}\ r\in( 0,r_{d}) , \end{gather}
\begin{gather} \frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial T}{\partial r}\right) =0\quad\text{if}\ r\in( 0,r_{d}) , \end{gather} \begin{gather}\frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial\rho_{1}}{\partial r}\right) =0,\quad \frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial T}{\partial r}\right) =0\quad\text{if}\ r\in( r_{d} ,\infty) . \end{gather}
\begin{gather}\frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial\rho_{1}}{\partial r}\right) =0,\quad \frac{\partial}{\partial r}\left( r^{2}\,\frac{\partial T}{\partial r}\right) =0\quad\text{if}\ r\in( r_{d} ,\infty) . \end{gather}At the drop's centre, the usual boundary condition applies:
 \begin{equation} \frac{\partial T}{\partial r}=0\quad\text{at}\ r=0.\end{equation}
\begin{equation} \frac{\partial T}{\partial r}=0\quad\text{at}\ r=0.\end{equation}To formulate the boundary conditions at the drop's surface, introduce
 \begin{equation} T^{({\pm})}=\lim_{r\rightarrow r_{d}\pm0}T,\quad\left( \frac{\partial T}{\partial r}\right)^{({\pm})}=\lim_{r\rightarrow r_{d}\pm0} \frac{\partial T}{\partial r}, \end{equation}
\begin{equation} T^{({\pm})}=\lim_{r\rightarrow r_{d}\pm0}T,\quad\left( \frac{\partial T}{\partial r}\right)^{({\pm})}=\lim_{r\rightarrow r_{d}\pm0} \frac{\partial T}{\partial r}, \end{equation}
where  $^{(+)}$ and
$^{(+)}$ and  $^{(-)}$ mean ‘just outside’ and ‘just inside’ the drop, respectively. Then the continuity of the temperature and heat flux imply
$^{(-)}$ mean ‘just outside’ and ‘just inside’ the drop, respectively. Then the continuity of the temperature and heat flux imply
 \begin{gather} T^{(-)}=T^{(+)}\quad\text{at}\ r=r_{d}, \end{gather}
\begin{gather} T^{(-)}=T^{(+)}\quad\text{at}\ r=r_{d}, \end{gather} \begin{gather}\kappa^{(l)}\left( \frac{\partial T}{\partial r}\right)^{(-)}-\kappa ^{(a)}\left( \frac{\partial T}{\partial r}\right)^{(+)}=\dot{r}_{d}\rho _{1}^{(l)}\varDelta\quad\text{at}\ r=r_{d}, \end{gather}
\begin{gather}\kappa^{(l)}\left( \frac{\partial T}{\partial r}\right)^{(-)}-\kappa ^{(a)}\left( \frac{\partial T}{\partial r}\right)^{(+)}=\dot{r}_{d}\rho _{1}^{(l)}\varDelta\quad\text{at}\ r=r_{d}, \end{gather}where
 \begin{equation} \dot{r}_{d}=\frac{\mathrm{d}r_{d}}{\mathrm{d}t}. \end{equation}
\begin{equation} \dot{r}_{d}=\frac{\mathrm{d}r_{d}}{\mathrm{d}t}. \end{equation}As discussed in the Introduction, the classical model assumes the Maxwell boundary condition for the vapour density at the drop's surface, i.e.
 \begin{equation} \rho_{1}=\rho_{1}^{(v.sat)}\quad\text{at}\ r=r_{d},\end{equation}
\begin{equation} \rho_{1}=\rho_{1}^{(v.sat)}\quad\text{at}\ r=r_{d},\end{equation}and the following conditions should be imposed on the vapour–air mixture far away from the drop:
 \begin{equation} \rho_{1}\rightarrow\rho_{1}^{(a)},\quad T\rightarrow T^{(a)}\quad \text{as}\ r\rightarrow\infty.\end{equation}
\begin{equation} \rho_{1}\rightarrow\rho_{1}^{(a)},\quad T\rightarrow T^{(a)}\quad \text{as}\ r\rightarrow\infty.\end{equation}To ensure that the liquid is evaporating – not condensating or being in equilibrium – let the vapour at infinity be undersaturated:
 \begin{equation} \rho_{1}^{(a)}<\rho_{1}^{(a.sat)(a)}, \end{equation}
\begin{equation} \rho_{1}^{(a)}<\rho_{1}^{(a.sat)(a)}, \end{equation}
where  $\rho _{1}^{(a.sat)(a)}=\rho _{1}^{(a.sat)}(T^{(a)})$. Finally, the velocity of the drop's (receding) surface is related to the evaporative flux by
$\rho _{1}^{(a.sat)(a)}=\rho _{1}^{(a.sat)}(T^{(a)})$. Finally, the velocity of the drop's (receding) surface is related to the evaporative flux by
 \begin{equation} \rho_{1}^{(l)}\dot{r}_{d}=\mathcal{D}\,\frac{\partial\rho}{\partial r} \quad\text{at}\ r=r_{d}.\end{equation}
\begin{equation} \rho_{1}^{(l)}\dot{r}_{d}=\mathcal{D}\,\frac{\partial\rho}{\partial r} \quad\text{at}\ r=r_{d}.\end{equation}
Boundary-value problem (2.3)–(2.13) governs  $\rho _{1}(r,t)$,
$\rho _{1}(r,t)$,  $T(r,t)$ and
$T(r,t)$ and  $r_{d}(t)$.
$r_{d}(t)$.
2.2. The dependence of  $\rho _{1}^{(v.sat)}$ on $T$
$\rho _{1}^{(v.sat)}$ on $T$


One's experience with raindrops sitting on ones's skin suggests that evaporative cooling does not exceed several degrees. This seems to enable one to neglect the dependence of the external parameters on the temperature – say, ‘freeze’ them at  $T=T^{(a)}$. Such an approximation is indeed applicable to all parameters but one – namely, the saturated vapour density
$T=T^{(a)}$. Such an approximation is indeed applicable to all parameters but one – namely, the saturated vapour density  $\rho _{1}^{(v.sat)}$.
$\rho _{1}^{(v.sat)}$.
This claim is illustrated for water in figure 1. One can see that the relative change of  $\rho _{1}^{(v.sat)}$ across the room temperature range exceeds those of the remaining parameters.
$\rho _{1}^{(v.sat)}$ across the room temperature range exceeds those of the remaining parameters.

Figure 1. The temperature dependencies of the saturated vapour density  $\rho _{1}^{(v.sat)}$, vapour-in-the-air diffusivity
$\rho _{1}^{(v.sat)}$, vapour-in-the-air diffusivity  $\mathcal {D}$, thermal conductivity
$\mathcal {D}$, thermal conductivity  $\kappa ^{(a)}$ of the air, and vaporisation heat
$\kappa ^{(a)}$ of the air, and vaporisation heat  $\varDelta$ (all non-dimensionalised on their reference values): (1)
$\varDelta$ (all non-dimensionalised on their reference values): (1)  $\rho _{1}^{(v.sat)}(T)/\rho _{1}^{(v.sat)}(25\,^{\circ }\mathrm {C})$, (2)
$\rho _{1}^{(v.sat)}(T)/\rho _{1}^{(v.sat)}(25\,^{\circ }\mathrm {C})$, (2)  $\mathcal {D}(T)/\mathcal {D}(25\,^{\circ }\mathrm {C})$, (3)
$\mathcal {D}(T)/\mathcal {D}(25\,^{\circ }\mathrm {C})$, (3)  $\kappa ^{(a)}(T)/\kappa ^{(a)}(25\,^{\circ }\mathrm {C})$, and (4)
$\kappa ^{(a)}(T)/\kappa ^{(a)}(25\,^{\circ }\mathrm {C})$, and (4)  $\varDelta (T)/\varDelta (25\,^{\circ }\mathrm {C})$. Curves (1)–(4) correspond to the empirical formulae of Wagner & Pruss (Reference Wagner and Pruss1993), Engineering ToolBox (2018), White (Reference White2005) and Lindstrom & Mallard (Reference Lindstrom and Mallard1997), respectively.
$\varDelta (T)/\varDelta (25\,^{\circ }\mathrm {C})$. Curves (1)–(4) correspond to the empirical formulae of Wagner & Pruss (Reference Wagner and Pruss1993), Engineering ToolBox (2018), White (Reference White2005) and Lindstrom & Mallard (Reference Lindstrom and Mallard1997), respectively.
In this paper, the dependence  $\rho _{1}^{(v.sat)}(T)$ is approximated by Antoine's equation (Antoine Reference Antoine1888). If written in terms of the density – as opposed to the pressure, as in the original formulation – it takes the form
$\rho _{1}^{(v.sat)}(T)$ is approximated by Antoine's equation (Antoine Reference Antoine1888). If written in terms of the density – as opposed to the pressure, as in the original formulation – it takes the form
 \begin{equation} \frac{\rho_{1}^{(v.sat)}}{\rho_{A}}=\frac{T_{A}}{T}\exp\left( -\frac{T_{A} }{T}\right) ,\end{equation}
\begin{equation} \frac{\rho_{1}^{(v.sat)}}{\rho_{A}}=\frac{T_{A}}{T}\exp\left( -\frac{T_{A} }{T}\right) ,\end{equation}where, for water,
 \begin{equation} \rho_{A}=6.6326\times10^{4}\ \mathrm{kg\ m}^{{-}3},\quad T_{A}=5292\ \mathrm{K}.\end{equation}
\begin{equation} \rho_{A}=6.6326\times10^{4}\ \mathrm{kg\ m}^{{-}3},\quad T_{A}=5292\ \mathrm{K}.\end{equation}
These values have been obtained by fitting (2.14) to the high-accuracy IAPWS empirical formula (Wagner & Pruss Reference Wagner and Pruss1993) for the room temperature range. The relative deviation of the two formulae is less than 0.23 %, which is much better than the accuracy of Antoine's equation with  $\rho _{A}$ and
$\rho _{A}$ and  $T_{A}$ taken from a thermodynamics handbook.
$T_{A}$ taken from a thermodynamics handbook.
Not only does Antoine's equation provide an accurate approximation, but it also has physical meaning. As shown by Benilov (Reference Benilov2020), (2.14) describes the generic van der Waals fluid in the low-temperature limit – i.e. when  $T$ is much closer to the freezing point than to the critical point. Observe also that
$T$ is much closer to the freezing point than to the critical point. Observe also that  $T_{A}$ is large, which is what makes the dependence of
$T_{A}$ is large, which is what makes the dependence of  $\rho _{1}^{(v.sat)}$ on
$\rho _{1}^{(v.sat)}$ on  $T$ strong.
$T$ strong.
Equation (2.14) can be rearranged conveniently using a reference temperature (as done by Sobac et al. Reference Sobac, Talbot, Haut, Rednikov and Colinet2015). Choosing that to be  $T^{(a)}$, one obtains
$T^{(a)}$, one obtains
 \begin{equation} \frac{\rho_{1}^{(v.sat)}}{\rho_{1}^{(v.sat)(a)}}=\frac{T^{(a)}}{T}\exp\left( -\frac{T_{A}}{T}+\frac{T_{A}}{T^{(a)}}\right) ,\end{equation}
\begin{equation} \frac{\rho_{1}^{(v.sat)}}{\rho_{1}^{(v.sat)(a)}}=\frac{T^{(a)}}{T}\exp\left( -\frac{T_{A}}{T}+\frac{T_{A}}{T^{(a)}}\right) ,\end{equation}
where  $\rho _{1}^{(v.sat)(a)}$ is the saturated vapour density at infinity. One can further assume
$\rho _{1}^{(v.sat)(a)}$ is the saturated vapour density at infinity. One can further assume  $( T^{(a)}-T) /T^{(a)}\ll 1$ and reduce (2.16) to
$( T^{(a)}-T) /T^{(a)}\ll 1$ and reduce (2.16) to
 \begin{equation} \rho_{1}^{(v.sat)}=\rho_{1}^{(v.sat)(a)}\exp\left[ \frac{T_{A}}{T^{(a)2} }\,( T-T^{(a)}) \right] .\end{equation}
\begin{equation} \rho_{1}^{(v.sat)}=\rho_{1}^{(v.sat)(a)}\exp\left[ \frac{T_{A}}{T^{(a)2} }\,( T-T^{(a)}) \right] .\end{equation}
Observe that even if  $T$ is close to
$T$ is close to  $T^{(a)}$, the expression in the square brackets can still be order-one (because
$T^{(a)}$, the expression in the square brackets can still be order-one (because  $T_{A}\gg T^{(a)}$).
$T_{A}\gg T^{(a)}$).
2.3. Solution of boundary-value problem (2.3)–(2.13)
Equations (2.3)–(2.4) can be solved readily as
 \begin{gather} T=\textrm{const}_{1}+\frac{\textrm{const}_{2}}{r} \quad\text{if}\ r\in( 0,r_{d}) , \end{gather}
\begin{gather} T=\textrm{const}_{1}+\frac{\textrm{const}_{2}}{r} \quad\text{if}\ r\in( 0,r_{d}) , \end{gather} \begin{gather}\rho_{1}=\textrm{const}_{3}+ \frac{\textrm{const}_{4}}{r},\quad T=\textrm{const}_{5}+ \frac{\textrm{const}_{6}}{r} \quad\text{if}\ r\in( r_{d},\infty), \end{gather}
\begin{gather}\rho_{1}=\textrm{const}_{3}+ \frac{\textrm{const}_{4}}{r},\quad T=\textrm{const}_{5}+ \frac{\textrm{const}_{6}}{r} \quad\text{if}\ r\in( r_{d},\infty), \end{gather}where the constants of integration can be fixed via boundary conditions (2.5)–(2.11):
 \begin{gather} \left.\begin{gathered}
\textrm{const}_{1}=T^{(a)}+r_{d}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta
}{\kappa^{(a)}},\quad\textrm{const}_{2}=0,\quad\textrm{const}
_{3}=\rho_{1}^{(a)},\\ \textrm{const}_{4}=r_{d}\left[
\rho_{1}^{(v.sat)}\left( T^{(a)}
+r_{d}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta}{\kappa^{(a)}}\right)
-\rho _{1}^{(a)}\right], \quad\textrm{const}_{5}=T^{(a)},\\
\textrm{const}_{6}=r_{d}^{2}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta
}{\kappa^{(a)}}. \end{gathered}\right\} \end{gather}
\begin{gather} \left.\begin{gathered}
\textrm{const}_{1}=T^{(a)}+r_{d}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta
}{\kappa^{(a)}},\quad\textrm{const}_{2}=0,\quad\textrm{const}
_{3}=\rho_{1}^{(a)},\\ \textrm{const}_{4}=r_{d}\left[
\rho_{1}^{(v.sat)}\left( T^{(a)}
+r_{d}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta}{\kappa^{(a)}}\right)
-\rho _{1}^{(a)}\right], \quad\textrm{const}_{5}=T^{(a)},\\
\textrm{const}_{6}=r_{d}^{2}\dot{r}_{d}\,\frac{\rho_{1}^{(l)}\varDelta
}{\kappa^{(a)}}. \end{gathered}\right\} \end{gather}
Recalling (2.16) for  $\rho _{1}^{(v.sat)}$ and (2.13) for
$\rho _{1}^{(v.sat)}$ and (2.13) for  $\dot {r}_{d}$, one obtains
$\dot {r}_{d}$, one obtains
 \begin{equation} \underset{\text{diffusion}}{\underbrace{\frac{\rho_{1}^{(l)}}{\mathcal{D} \rho_{1}^{(v.sat)(a)}}\,\dot{r}_{d}r_{d}}}+\exp \underset{\text{non-isothermality}}{\underbrace{\dfrac{T_{A}\rho_{1}^{(l)}\varDelta}{T_{{}}^{(a)2}\kappa^{(a)}}\,\dot{r}_{d}r_{d}}}=H,\end{equation}
\begin{equation} \underset{\text{diffusion}}{\underbrace{\frac{\rho_{1}^{(l)}}{\mathcal{D} \rho_{1}^{(v.sat)(a)}}\,\dot{r}_{d}r_{d}}}+\exp \underset{\text{non-isothermality}}{\underbrace{\dfrac{T_{A}\rho_{1}^{(l)}\varDelta}{T_{{}}^{(a)2}\kappa^{(a)}}\,\dot{r}_{d}r_{d}}}=H,\end{equation}where
 \begin{equation} H=\frac{\rho_{1}^{(a)}}{\rho_{1}^{(v.sat)(a)}} \end{equation}
\begin{equation} H=\frac{\rho_{1}^{(a)}}{\rho_{1}^{(v.sat)(a)}} \end{equation}is the relative humidity of air far away from the drop.
Ordinary differential equation (2.21) determines  $r_{d}(t)$; the terms on its left-hand side are labelled according to their physical meanings.
$r_{d}(t)$; the terms on its left-hand side are labelled according to their physical meanings.
Equation (2.21) yields the following expression for a drop's time of evaporation:
 \begin{equation} t_{e}=r_{d}^{2}(0)\,\frac{x\rho_{1}^{(l)}}{2\mathcal{D}\rho_{1}^{(v.sat)(a)} },\end{equation}
\begin{equation} t_{e}=r_{d}^{2}(0)\,\frac{x\rho_{1}^{(l)}}{2\mathcal{D}\rho_{1}^{(v.sat)(a)} },\end{equation}
where  $r_{d}(0)$ is the drop's initial radius, and
$r_{d}(0)$ is the drop's initial radius, and  $x$ is the solution of the non-dimensional algebraic equation
$x$ is the solution of the non-dimensional algebraic equation
 \begin{equation} {-x}+\exp( -\alpha x) =H,\end{equation}
\begin{equation} {-x}+\exp( -\alpha x) =H,\end{equation}with
 \begin{equation} \alpha=\frac{T_{A}\mathcal{D}\rho_{1}^{(v.sat)(a)}\varDelta}{T^{(a)2}\kappa^{(a)}}.\end{equation}
\begin{equation} \alpha=\frac{T_{A}\mathcal{D}\rho_{1}^{(v.sat)(a)}\varDelta}{T^{(a)2}\kappa^{(a)}}.\end{equation}
Formula (2.23) is often referred to as the ‘ $d^{2}$ law’, where
$d^{2}$ law’, where  $d$ is the drop's diameter.
$d$ is the drop's diameter.
It is instructive to estimate  $\alpha$ for, say, the midpoint of the room temperature range,
$\alpha$ for, say, the midpoint of the room temperature range,  $T^{(a)}=25\,^{\circ }\mathrm {C}$, and pressure
$T^{(a)}=25\,^{\circ }\mathrm {C}$, and pressure  $1$ atm. Letting the liquid be water, one can use the NIST online calculator (Lindstrom & Mallard Reference Lindstrom and Mallard1997) to obtain
$1$ atm. Letting the liquid be water, one can use the NIST online calculator (Lindstrom & Mallard Reference Lindstrom and Mallard1997) to obtain
 \begin{equation} \rho_{1}^{(l)}=997.05\ \mathrm{kg\ m}^{{-}3},\quad\rho_{1}^{(v.sat)(a)} =0.023075\ \mathrm{kg\ m}^{{-}3},\quad \varDelta=2441.7\ \mathrm{kJ\ kg}^{{-}1}, \end{equation}
\begin{equation} \rho_{1}^{(l)}=997.05\ \mathrm{kg\ m}^{{-}3},\quad\rho_{1}^{(v.sat)(a)} =0.023075\ \mathrm{kg\ m}^{{-}3},\quad \varDelta=2441.7\ \mathrm{kJ\ kg}^{{-}1}, \end{equation}whereas for air, Engineering ToolBox (2003, 2009, 2018) yield
 \begin{equation} \rho_{2}^{(a)}=1.184\
\mathrm{kg\,m}^{{-}3},\quad\kappa^{(a)}
=0.02624\
\mathrm{W}\ \mathrm{m}^{{-}1}\
\mathrm{K}^{{-}1},\quad\mathcal{D}
=2.49\times10^{{-}5}\ \mathrm{m}^{2}\ \mathrm{s}^{{-}1}. \end{equation}
\begin{equation} \rho_{2}^{(a)}=1.184\
\mathrm{kg\,m}^{{-}3},\quad\kappa^{(a)}
=0.02624\
\mathrm{W}\ \mathrm{m}^{{-}1}\
\mathrm{K}^{{-}1},\quad\mathcal{D}
=2.49\times10^{{-}5}\ \mathrm{m}^{2}\ \mathrm{s}^{{-}1}. \end{equation}
Substituting values (2.26)–(2.27) into (2.25), and recalling the value in (2.15) for  $T_{A}$, one obtains
$T_{A}$, one obtains
 \begin{equation} \alpha\approx3.1828.\end{equation}
\begin{equation} \alpha\approx3.1828.\end{equation}2.4. Discussion
It is interesting to assess the importance of non-isothermal effects by comparing the above model with its isothermal reduction. It can be verified readily that the latter amounts to taking in (2.24) the limit  $\alpha \rightarrow 0$, so that
$\alpha \rightarrow 0$, so that  $x=1-H$. Denoting the isothermal time of evaporation by
$x=1-H$. Denoting the isothermal time of evaporation by  $t_{e}^{\prime }$, one obtains
$t_{e}^{\prime }$, one obtains
 \begin{equation} \frac{t_{e}}{t_{e}^{\prime}}=\frac{q}{1-H}. \end{equation}
\begin{equation} \frac{t_{e}}{t_{e}^{\prime}}=\frac{q}{1-H}. \end{equation}
It turns out that the isothermal approach noticeably underpredicts the evaporation time. The largest discrepancy is observed at high humidity, in which case (2.24) can be solved asymptotically. Assuming estimate (2.28) for  $\alpha$, one obtains
$\alpha$, one obtains
 \begin{equation} \frac{t_{e}}{t_{e}^{\prime}}\rightarrow\alpha+1\approx4.18\quad \text{as}\ H\rightarrow1. \end{equation}
\begin{equation} \frac{t_{e}}{t_{e}^{\prime}}\rightarrow\alpha+1\approx4.18\quad \text{as}\ H\rightarrow1. \end{equation}
The smallest discrepancy, in turn, is observed at low humidity. Solving (2.24) with  $H=0$ numerically, one obtains
$H=0$ numerically, one obtains
 \begin{equation} \frac{t_{e}}{t_{e}^{\prime}}\approx2.95\quad\text{for}\ H=0. \end{equation}
\begin{equation} \frac{t_{e}}{t_{e}^{\prime}}\approx2.95\quad\text{for}\ H=0. \end{equation}
Evidently, the isothermal model may yield a significant error in the whole range of  $H$, which agrees broadly with conclusions of Sobac et al. (Reference Sobac, Talbot, Haut, Rednikov and Colinet2015), Cossali & Tonini (Reference Cossali and Tonini2019) and Finneran (Reference Finneran2021).
$H$, which agrees broadly with conclusions of Sobac et al. (Reference Sobac, Talbot, Haut, Rednikov and Colinet2015), Cossali & Tonini (Reference Cossali and Tonini2019) and Finneran (Reference Finneran2021).
It is also interesting to calculate the time of evaporation of a typical ‘cough drop’ (through which COVID-19, flu, etc. are spread). The radii of such drops are typically between  $0.5$ and
$0.5$ and  $16.0\ \mathrm {\mu } \mathrm {m}$ (Yang et al. Reference Yang, Lee, Chen, Wu and Yu2007). The largest drops from this range survive the longest – hence are the most dangerous when spreading the disease. The dependence
$16.0\ \mathrm {\mu } \mathrm {m}$ (Yang et al. Reference Yang, Lee, Chen, Wu and Yu2007). The largest drops from this range survive the longest – hence are the most dangerous when spreading the disease. The dependence  $t_{e}$ on
$t_{e}$ on  $T$ for this case is illustrated in figure 2.
$T$ for this case is illustrated in figure 2.

Figure 2. The time of full evaporation of a water drop of radius  $16\ \mathrm {\mu }\mathrm {m}$ versus the temperature. The curves are marked with the corresponding relative humidity (in percentage).
$16\ \mathrm {\mu }\mathrm {m}$ versus the temperature. The curves are marked with the corresponding relative humidity (in percentage).
3. The generalised Maxwell boundary condition as described by the diffuse-interface model
Unlike the classical model where the drop's boundary has zero thickness, the DIM assumes the liquid and air to be separated by a finitely thin interface where the partial densities  $\rho _{i}$ vary smoothly. In this section, the DIM will be used to derive a model of evaporation that takes into account interfacial physics.
$\rho _{i}$ vary smoothly. In this section, the DIM will be used to derive a model of evaporation that takes into account interfacial physics.
Mathematically, the interface should be viewed as an inner asymptotic region separating the outer regions of liquid and air. The DIM describes them all, but it can be simplified depending on the region to which it is applied.
First, one can take advantage of the fact that the temperature field associated with evaporative cooling occurs at a macroscopic scale (e.g. the drop's radius). The interface, on the other hand, is microscopic – hence the temperature change in it is negligible, and it can be examined via the isothermal version of the DIM.
One should still account for the temperature dependence of the saturated vapour density (which was found to be important in the classical model) – but there is no need to use the full non-isothermal equations just for this. Instead, one can use the classical (macroscopic) model to calculate the temperature field and then use it to prescribe the correct value of  $\rho _{1}^{(v.sat)}(T)$ near the drop. This workaround allows one to avoid cumbersome calculations associated with non-isothermality, yet obtain the same result.
$\rho _{1}^{(v.sat)}(T)$ near the drop. This workaround allows one to avoid cumbersome calculations associated with non-isothermality, yet obtain the same result.
One can also consider first a flat interface. Once the generalised Maxwell boundary condition has been derived for this case, the pressure difference due to the capillary effect can be incorporated readily into it.
3.1. The governing equations
Let  $z$ be the vertical Cartesian coordinate, and let
$z$ be the vertical Cartesian coordinate, and let  $w$ be the vertical velocity of the mixture as a whole. Introduce also the pressure
$w$ be the vertical velocity of the mixture as a whole. Introduce also the pressure  $p$, and keep in mind that it is not an independent unknown: since the DIM assumes the fluid to be compressible,
$p$, and keep in mind that it is not an independent unknown: since the DIM assumes the fluid to be compressible,  $p$ is a known function of
$p$ is a known function of  $\rho _{1}$,
$\rho _{1}$,  $\rho _{2}$ and
$\rho _{2}$ and  $T$ (the equation of state). The shear and bulk viscosities –
$T$ (the equation of state). The shear and bulk viscosities –  $\mu _{s}$ and
$\mu _{s}$ and  $\mu _{b}$, respectively – are also known functions of the same arguments.
$\mu _{b}$, respectively – are also known functions of the same arguments.
Introduce the partial chemical potentials  $G_{1}$ and
$G_{1}$ and  $G_{2}$. Their thermodynamic definition is given in Appendix A.1 – but technically, one can simply treat them as given functions of
$G_{2}$. Their thermodynamic definition is given in Appendix A.1 – but technically, one can simply treat them as given functions of  $\rho _{1}$,
$\rho _{1}$,  $\rho _{2}$ and
$\rho _{2}$ and  $T$, such that
$T$, such that
 \begin{equation} \frac{\partial p}{\partial\rho_{j}}=\sum_{i}\rho_{i}\,\frac{\partial G_{i} }{\partial\rho_{j}}.\end{equation}
\begin{equation} \frac{\partial p}{\partial\rho_{j}}=\sum_{i}\rho_{i}\,\frac{\partial G_{i} }{\partial\rho_{j}}.\end{equation}
In the low-density limit,  $p$ and
$p$ and  $G_{i}$ tend to their ideal gas approximations:
$G_{i}$ tend to their ideal gas approximations:
 \begin{equation} p\sim\sum_{i}R_{i}\rho_{i},\quad G_{i}\sim R_{i}T\ln\rho_{i}\quad \text{as}\ \rho_{i}\rightarrow0,\end{equation}
\begin{equation} p\sim\sum_{i}R_{i}\rho_{i},\quad G_{i}\sim R_{i}T\ln\rho_{i}\quad \text{as}\ \rho_{i}\rightarrow0,\end{equation}
where  $R_{i}$ are the specific gas constants. For water and air, they are
$R_{i}$ are the specific gas constants. For water and air, they are
 \begin{equation} R_{1}=461.53\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2}\ \mathrm{K}^{{-}1},\quad R_{2}=289.41\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2}\ \mathrm{K}^{{-}1}, \end{equation}
\begin{equation} R_{1}=461.53\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2}\ \mathrm{K}^{{-}1},\quad R_{2}=289.41\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2}\ \mathrm{K}^{{-}1}, \end{equation}
with  $R_{2}$ calculated as the
$R_{2}$ calculated as the  $70/30$ weighted average of the gas constants of nitrogen and oxygen.
$70/30$ weighted average of the gas constants of nitrogen and oxygen.
The DIM comprises the standard hydrodynamic equations involving an undetermined external force  $F_{i}$, which will be identified later with the van der Waals force. Under the slow-flow (Stokes) approximation, a compressible isothermal binary fluid is governed by
$F_{i}$, which will be identified later with the van der Waals force. Under the slow-flow (Stokes) approximation, a compressible isothermal binary fluid is governed by
 \begin{gather} \frac{\partial( \rho_{1}+\rho_{2}) }{\partial t}+\frac {\partial[ ( \rho_{1}+\rho_{2}) w] }{\partial z}=0,\quad \frac{\partial\rho_{1}}{\partial t}+\frac{\partial( \rho_{1}w+J) }{\partial z}=0, \end{gather}
\begin{gather} \frac{\partial( \rho_{1}+\rho_{2}) }{\partial t}+\frac {\partial[ ( \rho_{1}+\rho_{2}) w] }{\partial z}=0,\quad \frac{\partial\rho_{1}}{\partial t}+\frac{\partial( \rho_{1}w+J) }{\partial z}=0, \end{gather} \begin{gather}\frac{\partial p}{\partial z}=\frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\rho_{1}F_{1}+\rho_{2}F_{2}, \end{gather}
\begin{gather}\frac{\partial p}{\partial z}=\frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\rho_{1}F_{1}+\rho_{2}F_{2}, \end{gather}where the effective viscosity is
 \begin{equation} \eta=\frac{4\mu_{s}}{3}+\mu_{b}, \end{equation}
\begin{equation} \eta=\frac{4\mu_{s}}{3}+\mu_{b}, \end{equation}the diffusive flux is (e.g. Giovangigli Reference Giovangigli2021)
 \begin{equation} J=D\left( F_{1}-\frac{\partial G_{1}}{\partial z}-F_{2}+\frac{\partial G_{2} }{\partial z}\right) , \end{equation}
\begin{equation} J=D\left( F_{1}-\frac{\partial G_{1}}{\partial z}-F_{2}+\frac{\partial G_{2} }{\partial z}\right) , \end{equation}
and  $D$ is the diffusion coefficient.
$D$ is the diffusion coefficient.
To understand the relationship of  $D$ with the diffusivity
$D$ with the diffusivity  $\mathcal {D}$ from the classical model, assume that the fluid is diluted, so that the van der Waals force is negligible (
$\mathcal {D}$ from the classical model, assume that the fluid is diluted, so that the van der Waals force is negligible ( $F_{i}=0$) and the chemical potentials are given by (3.2). As a result, (3.7) reduces to
$F_{i}=0$) and the chemical potentials are given by (3.2). As a result, (3.7) reduces to
 \begin{equation} J\sim D\left( -\frac{R_{1}T}{\rho_{1}}\,\frac{\partial\rho_{1}}{\partial z}+\frac{R_{2}T}{\rho_{2}}\,\frac{\partial\rho_{2}}{\partial z}\right) \quad\text{as}\ \rho_{i}\rightarrow0. \end{equation}
\begin{equation} J\sim D\left( -\frac{R_{1}T}{\rho_{1}}\,\frac{\partial\rho_{1}}{\partial z}+\frac{R_{2}T}{\rho_{2}}\,\frac{\partial\rho_{2}}{\partial z}\right) \quad\text{as}\ \rho_{i}\rightarrow0. \end{equation}
Let the vapour density be much smaller than that of the air ( $\rho _{1}\ll \rho _{2}$), but let their variations be comparable (
$\rho _{1}\ll \rho _{2}$), but let their variations be comparable ( $\partial \rho _{1}/\partial z\sim \partial \rho _{2}/\partial z$) – so that the above expression reduces to
$\partial \rho _{1}/\partial z\sim \partial \rho _{2}/\partial z$) – so that the above expression reduces to
 \begin{equation} J\sim{-}D\,\frac{R_{1}T}{\rho_{1}}\,\frac{\partial\rho_{1}}{\partial z} \quad\text{as } \rho_{i}\rightarrow0, \frac{\rho_{1}}{\rho_{2} }\rightarrow0. \end{equation}
\begin{equation} J\sim{-}D\,\frac{R_{1}T}{\rho_{1}}\,\frac{\partial\rho_{1}}{\partial z} \quad\text{as } \rho_{i}\rightarrow0, \frac{\rho_{1}}{\rho_{2} }\rightarrow0. \end{equation}A comparison of this expression with that for the classical diffusive flux yields
 \begin{equation} D\sim\frac{\rho_{1}}{R_{1}T}\,\mathcal{D}\quad\text{as}\ \rho _{i}\rightarrow0, \frac{\rho_{1}}{\rho_{2}}\rightarrow0.\end{equation}
\begin{equation} D\sim\frac{\rho_{1}}{R_{1}T}\,\mathcal{D}\quad\text{as}\ \rho _{i}\rightarrow0, \frac{\rho_{1}}{\rho_{2}}\rightarrow0.\end{equation}
For a dense mixture of fluids with comparable densities,  $D$ cannot be deduced from
$D$ cannot be deduced from  $\mathcal {D}$, but its dependence on
$\mathcal {D}$, but its dependence on  $( \rho _{1},\rho _{2},T)$ is supposed to be known from measurements.
$( \rho _{1},\rho _{2},T)$ is supposed to be known from measurements.
For a multicomponent flow, the (one-dimensional) DIM approximation of the van der Waals force is (Benilov Reference Benilov2023a)
 \begin{equation} F_{i}=\sum_{j}K_{ij}\,\frac{\partial^{3}\rho_{j}}{\partial z^{3}}, \end{equation}
\begin{equation} F_{i}=\sum_{j}K_{ij}\,\frac{\partial^{3}\rho_{j}}{\partial z^{3}}, \end{equation} where the Korteweg matrix  $K_{ij}$ characterises the interaction of molecules of components
$K_{ij}$ characterises the interaction of molecules of components  $i$ and
$i$ and  $j$ (Newton's third law implies
$j$ (Newton's third law implies  $K_{ij}=K_{ji}$). For the water–air combination, the Korteweg matrix is (see Appendix B)
$K_{ij}=K_{ji}$). For the water–air combination, the Korteweg matrix is (see Appendix B)
 \begin{equation} \begin{bmatrix} K_{11} & K_{12}\\ K_{21} & K_{22} \end{bmatrix}=\begin{bmatrix} 1.87810 & 0.84978\\ 0.84978 & 1.36880 \end{bmatrix} \times10^{{-}17}\ \mathrm{m}^{7}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}. \end{equation}
\begin{equation} \begin{bmatrix} K_{11} & K_{12}\\ K_{21} & K_{22} \end{bmatrix}=\begin{bmatrix} 1.87810 & 0.84978\\ 0.84978 & 1.36880 \end{bmatrix} \times10^{{-}17}\ \mathrm{m}^{7}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}. \end{equation}Next, the pressure term in the momentum equation (3.5) can be rewritten conveniently in terms of the chemical potentials. Using identity (3.1), one obtains
 \begin{equation} \rho_{1}\,\frac{\partial G_{1}}{\partial z}+\rho_{2}\,\frac{\partial G_{2} }{\partial z}=\frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\rho_{1}F_{1}+\rho_{2}F_{2}. \end{equation}
\begin{equation} \rho_{1}\,\frac{\partial G_{1}}{\partial z}+\rho_{2}\,\frac{\partial G_{2} }{\partial z}=\frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\rho_{1}F_{1}+\rho_{2}F_{2}. \end{equation}
This equation and (3.7) can be viewed as a set of algebraic equations for  $F_{1}-\partial G_{1}/\partial z$ and
$F_{1}-\partial G_{1}/\partial z$ and  $F_{2}-\partial G_{2}/\partial z$. Solving these equations and recalling (3.11) for
$F_{2}-\partial G_{2}/\partial z$. Solving these equations and recalling (3.11) for  $F_{i}$, one obtains
$F_{i}$, one obtains
 \begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) ={-}\frac{1}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) -\frac{\rho_{2}J}{D}\right] , \end{gather}
\begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) ={-}\frac{1}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) -\frac{\rho_{2}J}{D}\right] , \end{gather} \begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) ={-}\frac{1}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\frac{\rho_{1}J}{D}\right] . \end{gather}
\begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) ={-}\frac{1}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\frac{\rho_{1}J}{D}\right] . \end{gather}
Equations (3.4) and (3.14)–(3.15) determine the unknowns  $\rho _{1}$,
$\rho _{1}$,  $\rho _{2}$,
$\rho _{2}$,  $w$ and
$w$ and  $J$.
$J$.
3.2. Non-dimensionalisation
In what follows, the vertical coordinate  $z$ will be non-dimensionalised on the interfacial thickness
$z$ will be non-dimensionalised on the interfacial thickness  $\bar {z}$, which implies that distances should be measured from the current height
$\bar {z}$, which implies that distances should be measured from the current height  $Z(t)$ of the interface. Mathematically, this corresponds to the change of variables
$Z(t)$ of the interface. Mathematically, this corresponds to the change of variables  $( z,t) \rightarrow ( z_{new},t_{new})$, where
$( z,t) \rightarrow ( z_{new},t_{new})$, where
 \begin{equation} z_{new}=z-Z(t),\quad t_{new}=t. \end{equation}
\begin{equation} z_{new}=z-Z(t),\quad t_{new}=t. \end{equation} Rewriting (3.4) in terms of the new variables, introducing  $\dot {Z}=\mathrm {d}Z/\mathrm {d}t$, and omitting the subscript
$\dot {Z}=\mathrm {d}Z/\mathrm {d}t$, and omitting the subscript  $_{new}$, one obtains
$_{new}$, one obtains
 \begin{gather} \frac{\partial( \rho_{1}+\rho_{2}) }{\partial t}-\dot{Z}\,\frac{\partial\rho_{2}}{\partial z}+\frac{\partial[ ( \rho_{1}+\rho_{2}) w] }{\partial z}=0, \end{gather}
\begin{gather} \frac{\partial( \rho_{1}+\rho_{2}) }{\partial t}-\dot{Z}\,\frac{\partial\rho_{2}}{\partial z}+\frac{\partial[ ( \rho_{1}+\rho_{2}) w] }{\partial z}=0, \end{gather} \begin{gather}\frac{\partial\rho_{1} }{\partial t}-\dot{Z}\,\frac{\partial\rho_{1}}{\partial z}+\frac{\partial( \rho_{1}w+J) }{\partial z}=0, \end{gather}
\begin{gather}\frac{\partial\rho_{1} }{\partial t}-\dot{Z}\,\frac{\partial\rho_{1}}{\partial z}+\frac{\partial( \rho_{1}w+J) }{\partial z}=0, \end{gather}whereas (3.14)–(3.15) remain the same as before.
There are three density scales in the problem – the liquid density, the air density and the saturated vapour density. Since they differ from each other by orders of magnitude, no single scaling of  $\rho _{i}$ would fit all the asymptotic regions. Still, a ‘generic’ non-dimensionalisation is needed, so let the density scale be that of air,
$\rho _{i}$ would fit all the asymptotic regions. Still, a ‘generic’ non-dimensionalisation is needed, so let the density scale be that of air,  $\bar {\rho }=\rho _{2}^{(a)}$.
$\bar {\rho }=\rho _{2}^{(a)}$.
Introducing the air viscosity scale  $\bar {\eta }$, define the characteristic pressure and velocity by
$\bar {\eta }$, define the characteristic pressure and velocity by
 \begin{equation} \bar{p}=\frac{\bar{K}\bar{\rho}^{2}}{\bar{z}^{2}},\quad \bar{w}=\frac{ \bar{p}\bar{z}}{\bar{\eta}},\end{equation}
\begin{equation} \bar{p}=\frac{\bar{K}\bar{\rho}^{2}}{\bar{z}^{2}},\quad \bar{w}=\frac{ \bar{p}\bar{z}}{\bar{\eta}},\end{equation}
respectively. These choices for  $\bar {p}$ and
$\bar {p}$ and  $\bar {w}$ correspond to an asymptotic regime where the pressure gradient, viscous stress and van der Waals force in the momentum equation (3.5) are of the same order.
$\bar {w}$ correspond to an asymptotic regime where the pressure gradient, viscous stress and van der Waals force in the momentum equation (3.5) are of the same order.
Let  $\bar {t}$ be the evaporation time scale, and introduce
$\bar {t}$ be the evaporation time scale, and introduce
 \begin{equation} \varepsilon=\sqrt{\frac{\bar{z}}{\bar{t}\bar{w}}}. \end{equation}
\begin{equation} \varepsilon=\sqrt{\frac{\bar{z}}{\bar{t}\bar{w}}}. \end{equation}This parameter is responsible for the quasi-steady approximation – hence it is expected to be small. Another important parameter is the advection/diffusion ratio, defined by
 \begin{equation} \delta=\frac{\bar{w}\bar{\rho}^{2}\bar{z}}{\bar{D}\bar{p}}, \end{equation}
\begin{equation} \delta=\frac{\bar{w}\bar{\rho}^{2}\bar{z}}{\bar{D}\bar{p}}, \end{equation}
where  $\bar {D}$ is the characteristic value of the diffusion coefficient
$\bar {D}$ is the characteristic value of the diffusion coefficient  $D$.
$D$.
The following non-dimensional variables will be used:
 $$\begin{gather} \left.\begin{gathered}
t_{nd}=\frac{t}{\bar{t}},\quad z_{nd}=\frac{z}{\bar{z}},\\
( \rho_{i})_{nd}=\frac{\rho_{i}}{\bar{\rho}},\quad w_{nd}
=\frac{w}{\varepsilon\bar{w}},\quad
J_{nd}=\frac{J}{\varepsilon\bar{\rho} \bar{w}},\quad
Z_{nd}=\varepsilon Z,\\
\eta_{nd}=\frac{\eta}{\bar{\eta}},\quad
D_{nd}=\frac{D}{\bar{D}} ,\quad (
K_{ij})_{nd}=\frac{K_{ij}}{\bar{K}},\quad
(G_{i})_{nd}=\frac{\bar{\rho}}{\bar{p}}G_{i}.
\end{gathered}\right\}
\end{gather}$$
$$\begin{gather} \left.\begin{gathered}
t_{nd}=\frac{t}{\bar{t}},\quad z_{nd}=\frac{z}{\bar{z}},\\
( \rho_{i})_{nd}=\frac{\rho_{i}}{\bar{\rho}},\quad w_{nd}
=\frac{w}{\varepsilon\bar{w}},\quad
J_{nd}=\frac{J}{\varepsilon\bar{\rho} \bar{w}},\quad
Z_{nd}=\varepsilon Z,\\
\eta_{nd}=\frac{\eta}{\bar{\eta}},\quad
D_{nd}=\frac{D}{\bar{D}} ,\quad (
K_{ij})_{nd}=\frac{K_{ij}}{\bar{K}},\quad
(G_{i})_{nd}=\frac{\bar{\rho}}{\bar{p}}G_{i}.
\end{gathered}\right\}
\end{gather}$$
Rewriting (3.14)–(3.17) in terms of the non-dimensional variables, and omitting the subscript  $_{nd}$, one obtains
$_{nd}$, one obtains
 \begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) ={-}\frac{\varepsilon}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) -\delta\,\frac{\rho_{2}J}{D}\right] , \end{gather}
\begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) ={-}\frac{\varepsilon}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) -\delta\,\frac{\rho_{2}J}{D}\right] , \end{gather} \begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) ={-}\frac{\varepsilon}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\delta\,\frac{\rho_{1}J}{D}\right] , \end{gather}
\begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) ={-}\frac{\varepsilon}{\rho_{1}+\rho_{2}}\left[ \frac{\partial}{\partial z}\left( \eta\,\frac{\partial w}{\partial z}\right) +\delta\,\frac{\rho_{1}J}{D}\right] , \end{gather} \begin{gather}\varepsilon\,\frac{\partial( \rho_{1}+\rho_{2}) }{\partial t} +\frac{\partial[ ( \rho_{1}+\rho_{2}) ( w-\dot {Z}) ] }{\partial z}=0,\quad\varepsilon\,\frac{\partial\rho_{1} }{\partial t}+\frac{\partial[ \rho_{1}( w-\dot{Z}) +J] }{\partial z}=0. \end{gather}
\begin{gather}\varepsilon\,\frac{\partial( \rho_{1}+\rho_{2}) }{\partial t} +\frac{\partial[ ( \rho_{1}+\rho_{2}) ( w-\dot {Z}) ] }{\partial z}=0,\quad\varepsilon\,\frac{\partial\rho_{1} }{\partial t}+\frac{\partial[ \rho_{1}( w-\dot{Z}) +J] }{\partial z}=0. \end{gather}
Evidently, the non-dimensional equations (3.22)–(3.24) can be transformed back to their dimensional versions, (3.14)–(3.17), by setting  $\varepsilon =\delta =1$. This shortcut will be used for re-dimensionalising the results obtained.
$\varepsilon =\delta =1$. This shortcut will be used for re-dimensionalising the results obtained.
In what follows, one will need the non-dimensional versions of the low-density expressions (3.2) for  $p$ and
$p$ and  $G_{i}$, and (3.10) for
$G_{i}$, and (3.10) for  $D$. To obtain these, introduce
$D$. To obtain these, introduce
 \begin{equation} T_{nd}=\frac{\bar{R}\bar{\rho}}{\bar{p}}\,T,\quad\mathcal{D}_{nd}=\frac {\bar{\rho}^{2}}{\bar{D}\bar{p}}\,\mathcal{D},\end{equation}
\begin{equation} T_{nd}=\frac{\bar{R}\bar{\rho}}{\bar{p}}\,T,\quad\mathcal{D}_{nd}=\frac {\bar{\rho}^{2}}{\bar{D}\bar{p}}\,\mathcal{D},\end{equation}
where  $\bar {R}$ is the characteristic gas constant. Rewriting (3.2) and (3.10) in terms of
$\bar {R}$ is the characteristic gas constant. Rewriting (3.2) and (3.10) in terms of  $T_{nd}$ and
$T_{nd}$ and  $\mathcal {D}_{nd}$, and omitting the subscript
$\mathcal {D}_{nd}$, and omitting the subscript  $_{nd}$, one can verify that the dimensional and non-dimensional versions of these expressions look exactly the same.
$_{nd}$, one can verify that the dimensional and non-dimensional versions of these expressions look exactly the same.
3.3. Non-dimensional parameters
Let the characteristic value of the Korteweg parameter be that of dry air,  $\bar {K}=K_{22}$, given by (3.12). The characteristic values of the rest of the parameters are given by (2.26)–(2.27) and (3.3).
$\bar {K}=K_{22}$, given by (3.12). The characteristic values of the rest of the parameters are given by (2.26)–(2.27) and (3.3).
For common fluids, the room temperature range is much closer to the freezing point than the critical point (e.g. for water, the former is  $0\,^{\circ }\mathrm {C}$ and the latter is
$0\,^{\circ }\mathrm {C}$ and the latter is  $374\,^{\circ }\mathrm {C}$). In such cases, the interfacial thickness
$374\,^{\circ }\mathrm {C}$). In such cases, the interfacial thickness  $\bar {z}$ is comparable to the liquid's molecular size – i.e. several ångströms – as confirmed by both experiments (e.g. Verde et al. Reference Verde, Bolhuis and Campen2012; Pezzotti et al. Reference Pezzotti, Galimberti and Gaigeot2017) and molecular simulations (e.g. Liu et al. Reference Liu, Harder and Berne2005; Pezzotti et al. Reference Pezzotti, Galimberti and Gaigeot2017; Dodia et al. Reference Dodia, Ohto, Imoto and Nagata2019; and references therein).
$\bar {z}$ is comparable to the liquid's molecular size – i.e. several ångströms – as confirmed by both experiments (e.g. Verde et al. Reference Verde, Bolhuis and Campen2012; Pezzotti et al. Reference Pezzotti, Galimberti and Gaigeot2017) and molecular simulations (e.g. Liu et al. Reference Liu, Harder and Berne2005; Pezzotti et al. Reference Pezzotti, Galimberti and Gaigeot2017; Dodia et al. Reference Dodia, Ohto, Imoto and Nagata2019; and references therein).
To obtain an estimate of  $\bar {z}$ from within the DIM, one can use the particular case of an equilibrium interface. It is examined briefly in Appendix A.2 for the parameters specific to the water–air combination listed in Appendix B. The resulting profiles of the water and air densities are shown in figure 3, suggesting that the interfacial thickness is
$\bar {z}$ from within the DIM, one can use the particular case of an equilibrium interface. It is examined briefly in Appendix A.2 for the parameters specific to the water–air combination listed in Appendix B. The resulting profiles of the water and air densities are shown in figure 3, suggesting that the interfacial thickness is
 \begin{equation} \bar{z}\sim5 \unicode{x00C5}.\end{equation}
\begin{equation} \bar{z}\sim5 \unicode{x00C5}.\end{equation}This value exceeds the water's molecular size by a factor of 2–3 (depending on how it is defined).

Figure 3. A schematic illustrating the asymptotic structure of the solution. The curves  $\rho _{1}(z)$ and
$\rho _{1}(z)$ and  $\rho _{2}(z)$ describe a flat equilibrium water/air interface at
$\rho _{2}(z)$ describe a flat equilibrium water/air interface at  $T=25\,^{\circ }\mathrm {C}$, computed using the DIM (see Appendix A.2). The scale of the horizontal axes in both panels is logarithmic. The height of the non-shaded region is used as an estimate for the interfacial thickness
$T=25\,^{\circ }\mathrm {C}$, computed using the DIM (see Appendix A.2). The scale of the horizontal axes in both panels is logarithmic. The height of the non-shaded region is used as an estimate for the interfacial thickness  $\bar {z}$.
$\bar {z}$.
According to (3.19),  $\varepsilon$ depends on the time scale
$\varepsilon$ depends on the time scale  $\bar {t}$ – which, in turn, depends on the evaporation rate – hence
$\bar {t}$ – which, in turn, depends on the evaporation rate – hence  $\varepsilon$ can be estimated only in application to a specific setting. Assuming that to be a drop of radius
$\varepsilon$ can be estimated only in application to a specific setting. Assuming that to be a drop of radius  $r_{d}$, one can use the continuity of the advective mass flux to relate
$r_{d}$, one can use the continuity of the advective mass flux to relate  $\dot {r}_{d}$ to
$\dot {r}_{d}$ to  $\bar {w}$, we have
$\bar {w}$, we have
 \begin{equation} \dot{r}_{d}\rho_{1}^{(l)}\sim\bar{w}\rho_{2}^{(a)}. \end{equation}
\begin{equation} \dot{r}_{d}\rho_{1}^{(l)}\sim\bar{w}\rho_{2}^{(a)}. \end{equation}
Expressing  $\bar {w}$ from this equality, substituting it into definition (3.19) of
$\bar {w}$ from this equality, substituting it into definition (3.19) of  $\varepsilon$, and approximating
$\varepsilon$, and approximating  $\dot {r}_{d}$ by
$\dot {r}_{d}$ by  $r_{d} /\bar {t}$, one obtains
$r_{d} /\bar {t}$, one obtains
 \begin{equation} \varepsilon=\sqrt{\frac{\rho_{2}^{(a)}\bar{z}}{\rho_{1}^{(l)}r_{d}}}. \end{equation}
\begin{equation} \varepsilon=\sqrt{\frac{\rho_{2}^{(a)}\bar{z}}{\rho_{1}^{(l)}r_{d}}}. \end{equation}
Luckily, this expression no longer involves  $\bar {t}$ (which is unknown without further calculations). Then for
$\bar {t}$ (which is unknown without further calculations). Then for  $r_{d}$ between 1
$r_{d}$ between 1  $\mathrm {\mu }$m and 1 nm, the above expression shows that
$\mathrm {\mu }$m and 1 nm, the above expression shows that  $\varepsilon$ is between
$\varepsilon$ is between  $0.771\times 10^{-3}$ and
$0.771\times 10^{-3}$ and  $2.44\times 10^{-2}$ – i.e. it is small.
$2.44\times 10^{-2}$ – i.e. it is small.
To estimate  $\delta$, estimate the diffusion coefficient by
$\delta$, estimate the diffusion coefficient by  $\bar {D}=\mathcal {D}\bar {\rho }^{2}/\bar {p}$, where
$\bar {D}=\mathcal {D}\bar {\rho }^{2}/\bar {p}$, where  $\mathcal {D}$ is the standard vapour-in-the-air diffusivity. Then (3.18) and (3.20) yield
$\mathcal {D}$ is the standard vapour-in-the-air diffusivity. Then (3.18) and (3.20) yield
 \begin{equation} \delta=\frac{\bar{K}\bar{\rho}^{2}}{\mathcal{D}\bar{\eta}}.\end{equation}
\begin{equation} \delta=\frac{\bar{K}\bar{\rho}^{2}}{\mathcal{D}\bar{\eta}}.\end{equation}
Estimating the viscosity of air at  $25\,^{\circ }\mathrm {C}$ using the results of Shang et al. (Reference Shang, Wu, Wang, Yang, Ye, Hu, Tao and He2019), one obtains
$25\,^{\circ }\mathrm {C}$ using the results of Shang et al. (Reference Shang, Wu, Wang, Yang, Ye, Hu, Tao and He2019), one obtains
 \begin{equation} \bar{\eta}=4.20\times10^{{-}5}\ \mathrm{kg}\ \mathrm{m}^{{-}1}\ \mathrm{s}^{{-}1},\end{equation}
\begin{equation} \bar{\eta}=4.20\times10^{{-}5}\ \mathrm{kg}\ \mathrm{m}^{{-}1}\ \mathrm{s}^{{-}1},\end{equation}
for which (3.29) yields  $\delta \approx 1.83\times 10^{-8}$. Thus one can assume that
$\delta \approx 1.83\times 10^{-8}$. Thus one can assume that
 \begin{equation} \delta\ll\varepsilon\ll1. \end{equation}
\begin{equation} \delta\ll\varepsilon\ll1. \end{equation}
In addition to  $\varepsilon$ and
$\varepsilon$ and  $\delta$, the problem involves two ‘hidden’ small parameters:
$\delta$, the problem involves two ‘hidden’ small parameters:
 \begin{equation} \epsilon^{(a/l)}=\frac{\rho_{2}^{(a)}}{\rho_{1}^{(l)}}\approx1.19\times 10^{{-}3},\quad\epsilon^{(v/a)}=\frac{\rho_{1}^{(v.sat)}}{\rho_{2}^{(a)} }\approx1.95\times10^{{-}2}. \end{equation}
\begin{equation} \epsilon^{(a/l)}=\frac{\rho_{2}^{(a)}}{\rho_{1}^{(l)}}\approx1.19\times 10^{{-}3},\quad\epsilon^{(v/a)}=\frac{\rho_{1}^{(v.sat)}}{\rho_{2}^{(a)} }\approx1.95\times10^{{-}2}. \end{equation}The presence of four independent small parameters makes the analysis awkward: they arise in various combinations, and each time, one has to check whether these are large, small or order-one. In particular, the following estimates will be needed:
 \begin{gather} \frac{\varepsilon^{2}\epsilon^{(a/l)}\epsilon^{(v/a)}}{\delta}\ll \frac{\varepsilon^{2}\epsilon^{(a/l)}}{\delta}\approx3.84\times10^{{-}2} \ll1, \end{gather}
\begin{gather} \frac{\varepsilon^{2}\epsilon^{(a/l)}\epsilon^{(v/a)}}{\delta}\ll \frac{\varepsilon^{2}\epsilon^{(a/l)}}{\delta}\approx3.84\times10^{{-}2} \ll1, \end{gather} \begin{gather}\frac{\delta}{\epsilon^{(v/a)}}\approx0.938\times10^{{-}6}\ll1. \end{gather}
\begin{gather}\frac{\delta}{\epsilon^{(v/a)}}\approx0.938\times10^{{-}6}\ll1. \end{gather}Thus all of the above parameter combinations will be assumed small.
3.4. The boundary/matching conditions
When applied to the interface (inner region), (3.22)–(3.24) need boundary conditions at large  $z$. These are to be obtained via matching the inner solution to those in the liquid and air (outer regions). The former is trivial: since the liquid is homogeneous and at rest, the interfacial solution should satisfy
$z$. These are to be obtained via matching the inner solution to those in the liquid and air (outer regions). The former is trivial: since the liquid is homogeneous and at rest, the interfacial solution should satisfy
 \begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l)}\quad\text{as}\ z\rightarrow -\infty, \end{gather}
\begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l)}\quad\text{as}\ z\rightarrow -\infty, \end{gather} \begin{gather}w\rightarrow0,\quad J\rightarrow0\quad\text{as}\ z\rightarrow -\infty. \end{gather}
\begin{gather}w\rightarrow0,\quad J\rightarrow0\quad\text{as}\ z\rightarrow -\infty. \end{gather}
Unlike the classical model where  $\rho _{i}^{(l)}$ are external parameters, the DIM determines them as functions of the pressure and humidity of air. The DIM also describes dissolution of air in liquid – hence
$\rho _{i}^{(l)}$ are external parameters, the DIM determines them as functions of the pressure and humidity of air. The DIM also describes dissolution of air in liquid – hence  $\rho _{2}^{(l)}\neq 0$. This quantity is very small and thus unimportant both dynamically and thermodynamically.
$\rho _{2}^{(l)}\neq 0$. This quantity is very small and thus unimportant both dynamically and thermodynamically.
The matching between the interface and air is less trivial: it can be carried out only after examining the large-distance behaviours of the inner solution and choosing the one that matches the outer solution. The properties of the latter are easy to guess: it should be such that  $\rho _{1}\ll \rho _{2}$ (small vapour concentration in the air), and the variations of
$\rho _{1}\ll \rho _{2}$ (small vapour concentration in the air), and the variations of 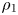 $\rho _{1}$ and
$\rho _{1}$ and  $\rho _{2}$ should be such that the total pressure is spatially uniform (e.g. Ricci & Rocca Reference Ricci and Rocca1984; Mills & Coimbra Reference Mills and Coimbra2016).
$\rho _{2}$ should be such that the total pressure is spatially uniform (e.g. Ricci & Rocca Reference Ricci and Rocca1984; Mills & Coimbra Reference Mills and Coimbra2016).
To verify that (3.22)–(3.24) admit such an asymptotic solution, let
 \begin{align} \rho_{i}=\rho_{i}^{(+)}+\varepsilon\delta\rho_{i}^{\prime(+)}z+{O} ( \varepsilon^{2}\delta,\varepsilon\delta^{2}) ,\quad w=w^{(+)}+{O}(\varepsilon),\quad J=J^{(+)}+{O}(\varepsilon ),\end{align}
\begin{align} \rho_{i}=\rho_{i}^{(+)}+\varepsilon\delta\rho_{i}^{\prime(+)}z+{O} ( \varepsilon^{2}\delta,\varepsilon\delta^{2}) ,\quad w=w^{(+)}+{O}(\varepsilon),\quad J=J^{(+)}+{O}(\varepsilon ),\end{align}
where  $\rho _{i}^{(+)}$,
$\rho _{i}^{(+)}$,  $\rho _{i}^{\prime (+)}$, etc. depend on
$\rho _{i}^{\prime (+)}$, etc. depend on  $t$ (but not on
$t$ (but not on  $z$) and may involve the hidden small parameters
$z$) and may involve the hidden small parameters  $\epsilon ^{(a/l)}$ and
$\epsilon ^{(a/l)}$ and  $\epsilon ^{(v/a)}$. It turns out that only
$\epsilon ^{(v/a)}$. It turns out that only  $\rho _{i}^{(+)}$ is independent in these expansions, with the other coefficients being expressible through
$\rho _{i}^{(+)}$ is independent in these expansions, with the other coefficients being expressible through  $\rho _{i}^{(+)}$.
$\rho _{i}^{(+)}$.
To find how  $w^{(+)}$ and
$w^{(+)}$ and  $J^{(+)}$ depend on
$J^{(+)}$ depend on  $\rho _{i}^{(+)}$, observe that to leading order, (3.24) can be integrated. Using boundary conditions (3.35)–(3.36) to fix the constants of integration, one obtains
$\rho _{i}^{(+)}$, observe that to leading order, (3.24) can be integrated. Using boundary conditions (3.35)–(3.36) to fix the constants of integration, one obtains
 \begin{equation} w={-}\dot{Z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2} }-1\right) +{O}(\varepsilon),\quad J={-}\dot{Z}\,\frac{\rho_{1}^{(l)}\rho_{2}-\rho_{1}\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}+{O} (\varepsilon),\end{equation}
\begin{equation} w={-}\dot{Z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2} }-1\right) +{O}(\varepsilon),\quad J={-}\dot{Z}\,\frac{\rho_{1}^{(l)}\rho_{2}-\rho_{1}\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}+{O} (\varepsilon),\end{equation}which entails
 \begin{equation} w^{(+)}={-}\dot{Z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}^{(+)}+\rho_{2}^{(+)}}-1\right) ,\quad J^{(+)}={-}\dot{Z}\,\frac{\rho_{1}^{(l)}\rho_{2}^{(+)}-\rho_{1}^{(+)}\rho_{2}^{(l)}}{\rho_{1}^{(+)}+\rho_{2}^{(+)}}. \end{equation}
\begin{equation} w^{(+)}={-}\dot{Z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}^{(+)}+\rho_{2}^{(+)}}-1\right) ,\quad J^{(+)}={-}\dot{Z}\,\frac{\rho_{1}^{(l)}\rho_{2}^{(+)}-\rho_{1}^{(+)}\rho_{2}^{(l)}}{\rho_{1}^{(+)}+\rho_{2}^{(+)}}. \end{equation}
To calculate  $\rho _{i}^{\prime (+)}$, one should substitute the expansions of
$\rho _{i}^{\prime (+)}$, one should substitute the expansions of  $w$ and
$w$ and  $J$ into (3.22)–(3.23). Since air is a diluted fluid, the effective viscosity does not depend on the density (e.g. Ferziger & Kaper Reference Ferziger and Kaper1972), i.e.
$J$ into (3.22)–(3.23). Since air is a diluted fluid, the effective viscosity does not depend on the density (e.g. Ferziger & Kaper Reference Ferziger and Kaper1972), i.e.
 \begin{equation} \eta\sim\eta_{0}\quad\text{as}\ \rho_{i}\rightarrow0,\end{equation}
\begin{equation} \eta\sim\eta_{0}\quad\text{as}\ \rho_{i}\rightarrow0,\end{equation}
where  $\eta _{0}$ depends only on
$\eta _{0}$ depends only on  $T$. This property implies that when expressions (3.38) are substituted into (3.22)–(3.23), the linear term in the expansion for
$T$. This property implies that when expressions (3.38) are substituted into (3.22)–(3.23), the linear term in the expansion for  $w$ cancels out. The quadratic term does not, but its contribution is much smaller than that of
$w$ cancels out. The quadratic term does not, but its contribution is much smaller than that of  $J^{(0)}$ (due to estimates (3.33)), and to leading order, one obtains the following algebraic equations for
$J^{(0)}$ (due to estimates (3.33)), and to leading order, one obtains the following algebraic equations for  $\rho _{i}^{\prime (+)}$:
$\rho _{i}^{\prime (+)}$:
 \begin{gather} \sum_{i}\left( \frac{\partial G_{1}}{\partial\rho_{i}}\right)^{(+)}\rho _{i}^{\prime(+)} ={-}\frac{\rho_{2}^{(+)}J^{(+)}}{D^{(+)}( \rho_{1}^{(+)}+\rho_{2}^{(+)}) }, \end{gather}
\begin{gather} \sum_{i}\left( \frac{\partial G_{1}}{\partial\rho_{i}}\right)^{(+)}\rho _{i}^{\prime(+)} ={-}\frac{\rho_{2}^{(+)}J^{(+)}}{D^{(+)}( \rho_{1}^{(+)}+\rho_{2}^{(+)}) }, \end{gather} \begin{gather}\sum_{i}\left( \frac{\partial G_{2}}{\partial\rho_{i}}\right)^{(+)}\rho _{i}^{\prime(+)} =\frac{\rho_{1}^{(+)}J^{(+)}}{D^{(+)}(\rho_{1}^{(+)}+\rho_{2}^{(+)}) }, \end{gather}
\begin{gather}\sum_{i}\left( \frac{\partial G_{2}}{\partial\rho_{i}}\right)^{(+)}\rho _{i}^{\prime(+)} =\frac{\rho_{1}^{(+)}J^{(+)}}{D^{(+)}(\rho_{1}^{(+)}+\rho_{2}^{(+)}) }, \end{gather}
where  $D^{(+)}=D(\rho _{1}^{(+)},\rho _{2}^{(+)},T)$ and
$D^{(+)}=D(\rho _{1}^{(+)},\rho _{2}^{(+)},T)$ and
 \begin{equation} \left( \frac{\partial G_{j}}{\partial\rho_{i}}\right)^{(+)}=\frac{\partial G_{j}(\rho_{1}^{(+)},\rho_{2}^{(+)},T)}{\partial\rho_{i}^{(+)}}. \end{equation}
\begin{equation} \left( \frac{\partial G_{j}}{\partial\rho_{i}}\right)^{(+)}=\frac{\partial G_{j}(\rho_{1}^{(+)},\rho_{2}^{(+)},T)}{\partial\rho_{i}^{(+)}}. \end{equation}
Given the smallness of the air-to-liquid and vapour-to-air density ratios (3.32), (3.41)–(3.42) can be simplified. Using the diluted-fluid expressions (3.2) and (3.10) for  $G_{i}$ and
$G_{i}$ and  $D$, respectively, and keeping the leading order only, one obtains
$D$, respectively, and keeping the leading order only, one obtains
 \begin{gather} \rho_{1}^{\prime(+)} =\frac{\rho_{1}^{(l)}\dot{Z}}{\mathcal{D} }, \end{gather}
\begin{gather} \rho_{1}^{\prime(+)} =\frac{\rho_{1}^{(l)}\dot{Z}}{\mathcal{D} }, \end{gather} \begin{gather}\rho_{2}^{\prime(+)} ={-}\rho_{1}^{\prime(+)}\,\frac{R_{1}}{R_{2}}. \end{gather}
\begin{gather}\rho_{2}^{\prime(+)} ={-}\rho_{1}^{\prime(+)}\,\frac{R_{1}}{R_{2}}. \end{gather}
Equality (3.44) is the desired matching condition; it can also be viewed as the relationship of the speed  $\dot {Z}$ of the interface to the vapour flux
$\dot {Z}$ of the interface to the vapour flux  $\mathcal {D}\rho ^{\prime (+)}$, whereas (3.45) is the standard condition of constant pressure for diffusion in diluted fluids. As mentioned before, both conditions are obvious physically, but it is still important to verify that they are intrinsic to the DIM formalism.
$\mathcal {D}\rho ^{\prime (+)}$, whereas (3.45) is the standard condition of constant pressure for diffusion in diluted fluids. As mentioned before, both conditions are obvious physically, but it is still important to verify that they are intrinsic to the DIM formalism.
3.5. The interfacial asymptotic region
Three asymptotic zones exist within the inner (interfacial) region,
 \begin{equation} \left.\begin{array}{ll@{}} \text{Zone 1\ (mostly air):} & \rho_{1}\ll\rho_{2}\ll\rho^{(l)},\\ \text{Zone 2\ (air--vapour mixture):} & \rho_{1}\sim \rho_{2}\ll\rho^{(l)},\\ \text{Zone 3\ (mostly liquid):} & \rho_{2}\ll\rho_{1}\sim \rho^{(l)}. \end{array}\right\} \end{equation}
\begin{equation} \left.\begin{array}{ll@{}} \text{Zone 1\ (mostly air):} & \rho_{1}\ll\rho_{2}\ll\rho^{(l)},\\ \text{Zone 2\ (air--vapour mixture):} & \rho_{1}\sim \rho_{2}\ll\rho^{(l)},\\ \text{Zone 3\ (mostly liquid):} & \rho_{2}\ll\rho_{1}\sim \rho^{(l)}. \end{array}\right\} \end{equation}
Note that Zone 1 is not part of the air region: in the latter, the air density is nearly uniform,  $\rho _{2}\approx \rho _{2}^{(a)}$, whereas in the former, variations of
$\rho _{2}\approx \rho _{2}^{(a)}$, whereas in the former, variations of  $\rho _{2}$ are comparable to
$\rho _{2}$ are comparable to  $\rho _{2}^{(a)}$.
$\rho _{2}^{(a)}$.
A schematic illustrating the asymptotic structure of the problem can be found in figure 3 (which is computed for the equilibrium interface, i.e. that where the relative humidity of air is  $H=1$). Its most counter-intuitive features are the local maximum and minimum of the air density
$H=1$). Its most counter-intuitive features are the local maximum and minimum of the air density  $\rho _{2}(z)$ in Zone 2. Note that they both disappear in the limit
$\rho _{2}(z)$ in Zone 2. Note that they both disappear in the limit  $K_{12}\rightarrow 0$ (see figure 11 of Benilov Reference Benilov2023a). One can thus assume that the maximum of
$K_{12}\rightarrow 0$ (see figure 11 of Benilov Reference Benilov2023a). One can thus assume that the maximum of  $\rho _{2}$ is created by the van der Waals force exerted by the bulk of the liquid by attracting a certain amount of air towards the interface. The existence of the minimum of
$\rho _{2}$ is created by the van der Waals force exerted by the bulk of the liquid by attracting a certain amount of air towards the interface. The existence of the minimum of  $\rho _{2}$ is harder to understand, but it is dynamically unimportant anyway. The air density is extremely small there – even smaller than that of the air dissolved in the liquid – and they both have a minuscule effect on the dynamics of the rest of the fluid.
$\rho _{2}$ is harder to understand, but it is dynamically unimportant anyway. The air density is extremely small there – even smaller than that of the air dissolved in the liquid – and they both have a minuscule effect on the dynamics of the rest of the fluid.
It turns out that only Zone 1 contributes significantly to the evaporation rate, whereas the contributions of Zones 2–3 are negligible. To understand why, recall that the fluid density in the latter two zones exceeds that of the vapour component of air by an order of magnitude. As a result, Zones 2–3 are close to equilibrium (the fact that the ‘outside’ air is undersaturated cannot perturb them too much). Thus Zone 1 is the only region out of equilibrium – hence it is the only place where irreversible processes, such as evaporation, can occur.
The calculations associated with Zones 2–3 can be avoided via a certain workaround developed by Benilov (Reference Benilov2022b, Reference Benilov2023b) for evaporation of pure fluids. The workaround involves the following two steps.
(i) Equations (3.22)–(3.24) can be rearranged in such a way that the velocity
$\dot {Z}$ of the interface is expressed in terms of the fluid's known parameters (relative humidity, etc.) and a certain integral of the solution.(ii) The contribution of Zone 1 to this integral exceeds the contributions of Zones 2–3 by an order of magnitude. Thus to evaluate this integral asymptotically, it is sufficient to find the solution only in Zone 1.

3.5.1. Calculation of $\dot {Z}$

Recall that matching conditions (3.44)–(3.45) emerged from the governing equations at the order of  ${O}(\varepsilon \delta )$. Finding
${O}(\varepsilon \delta )$. Finding  $\dot {Z}$ does not require going that far, as the terms in (3.22)–(3.23) that involve
$\dot {Z}$ does not require going that far, as the terms in (3.22)–(3.23) that involve  $\dot {Z}$ are
$\dot {Z}$ are  ${O} (\varepsilon )$. The terms
${O} (\varepsilon )$. The terms  ${O}(\delta )$ in these equations can also be omitted (as suggested by the estimates in § 3.3).
${O}(\delta )$ in these equations can also be omitted (as suggested by the estimates in § 3.3).
Substitute expressions (3.38) for  $w$ and
$w$ and  $J$ into (3.22)–(3.23), and, keeping the terms up to
$J$ into (3.22)–(3.23), and, keeping the terms up to  ${O} (\varepsilon )$, obtain
${O} (\varepsilon )$, obtain
 \begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) =\frac{\varepsilon\dot{Z}}{\rho_{1}+\rho_{2} }\,\frac{\partial}{\partial z}\left[ \eta\,\frac{\partial}{\partial z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}\right) \right] , \end{gather}
\begin{gather} \frac{\partial}{\partial z}\left( \sum_{j}K_{1j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{1}\right) =\frac{\varepsilon\dot{Z}}{\rho_{1}+\rho_{2} }\,\frac{\partial}{\partial z}\left[ \eta\,\frac{\partial}{\partial z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}\right) \right] , \end{gather} \begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) =\frac{\varepsilon\dot{Z}}{\rho_{1}+\rho_{2} }\,\frac{\partial}{\partial z}\left[ \eta\,\frac{\partial}{\partial z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}\right) \right] . \end{gather}
\begin{gather}\frac{\partial}{\partial z}\left( \sum_{j}K_{2j}\,\frac{\partial^{2}\rho_{j} }{\partial z^{2}}-G_{2}\right) =\frac{\varepsilon\dot{Z}}{\rho_{1}+\rho_{2} }\,\frac{\partial}{\partial z}\left[ \eta\,\frac{\partial}{\partial z}\left( \frac{\rho_{1}^{(l)}+\rho_{2}^{(l)}}{\rho_{1}+\rho_{2}}\right) \right] . \end{gather}Keeping the same accuracy in boundary condition (3.37), one obtains
 \begin{equation} \rho_{i}\rightarrow\rho_{i}^{(+)}\quad\text{as}\ z\rightarrow +\infty.\end{equation}
\begin{equation} \rho_{i}\rightarrow\rho_{i}^{(+)}\quad\text{as}\ z\rightarrow +\infty.\end{equation}
Now, integrate (3.47) and (3.48) with respect to  $z$ from
$z$ from  $-\infty$ to
$-\infty$ to  $+\infty$. Recalling boundary conditions (3.35) and (3.49), one obtains
$+\infty$. Recalling boundary conditions (3.35) and (3.49), one obtains
 \begin{equation} G_{i}^{(l)}-G_{i}^{(+)}={-}\varepsilon\dot{Z}A,\end{equation}
\begin{equation} G_{i}^{(l)}-G_{i}^{(+)}={-}\varepsilon\dot{Z}A,\end{equation}where
 \begin{equation}
A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{1}+\rho_{2}}\,\frac{\partial}{\partial
z}\left[ \frac{( \rho_{1}^{(l)}+\rho_{2}^{(l)})
\eta}{\left( \rho_{1}+\rho_{2}\right)^{2}}\,\frac{\partial(
\rho_{1}+\rho_{2}) }{\partial z}\right]
\mathrm{d}z.\end{equation}
\begin{equation}
A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{1}+\rho_{2}}\,\frac{\partial}{\partial
z}\left[ \frac{( \rho_{1}^{(l)}+\rho_{2}^{(l)})
\eta}{\left( \rho_{1}+\rho_{2}\right)^{2}}\,\frac{\partial(
\rho_{1}+\rho_{2}) }{\partial z}\right]
\mathrm{d}z.\end{equation}Next, consider the combination
 \begin{equation} \int_{-\infty}^{+\infty}[ \rho_{1}\times \text{(3.47)} +\rho_{2}\times \text{(3.48)} ] \,\mathrm{d}z. \end{equation}
\begin{equation} \int_{-\infty}^{+\infty}[ \rho_{1}\times \text{(3.47)} +\rho_{2}\times \text{(3.48)} ] \,\mathrm{d}z. \end{equation}Integrating by parts the terms involving the third derivatives, take into account boundary conditions (3.35) and (3.49), and recall identity (3.1) to obtain
 \begin{equation} p^{(l)}-p^{(+)}=0.\end{equation}
\begin{equation} p^{(l)}-p^{(+)}=0.\end{equation}Thus the pressure in the liquid coincides with that in the air (as expected physically).
Equalities (3.50) and (3.53) relate the (unknown) liquid densities  $\rho _{i}^{(l)}$ and the velocity
$\rho _{i}^{(l)}$ and the velocity  $\dot {Z}$ of the interface to the air parameters (marked with
$\dot {Z}$ of the interface to the air parameters (marked with  $^{(+)}$). In addition,
$^{(+)}$). In addition,  $\dot {Z}$ is also related to the air parameters by equality (3.44). If two of these three equalities are used to eliminate
$\dot {Z}$ is also related to the air parameters by equality (3.44). If two of these three equalities are used to eliminate  $\rho _{i}^{(l)}$ and
$\rho _{i}^{(l)}$ and  $\dot {Z}$, then the remaining one will inter-relate the air parameters and thus turn into the generalised Maxwell boundary condition (GMBC).
$\dot {Z}$, then the remaining one will inter-relate the air parameters and thus turn into the generalised Maxwell boundary condition (GMBC).
To eliminate  $\rho _{i}^{(l)}$ from equalities (3.50) and (3.53), recall that equations of state of liquids at normal conditions are typically such that even a minuscule change of the density gives rise to a huge change of the pressure. This means effectively that
$\rho _{i}^{(l)}$ from equalities (3.50) and (3.53), recall that equations of state of liquids at normal conditions are typically such that even a minuscule change of the density gives rise to a huge change of the pressure. This means effectively that  $\rho _{i}^{(l)}$ is close to its saturated value,
$\rho _{i}^{(l)}$ is close to its saturated value,  $\rho _{i}^{(l)}\approx \rho _{i}^{(l.sat)}$ – so that (3.50) becomes
$\rho _{i}^{(l)}\approx \rho _{i}^{(l.sat)}$ – so that (3.50) becomes
 \begin{equation} G_{i}^{(l.sat)}-G_{i}^{(+)}={-}\varepsilon\dot{Z}A. \end{equation}
\begin{equation} G_{i}^{(l.sat)}-G_{i}^{(+)}={-}\varepsilon\dot{Z}A. \end{equation}
In the state of saturation, the chemical potentials of the liquid and vapour are equal (see Appendix A.2) – hence one can change in the above equality  $G_{1}^{(l.sat)}\rightarrow G_{1}^{(v.sat)}$. Then, using for both
$G_{1}^{(l.sat)}\rightarrow G_{1}^{(v.sat)}$. Then, using for both  $G_{1}^{(v.sat)}$ and
$G_{1}^{(v.sat)}$ and  $G_{1}^{(+)}$ the diluted-fluid expression (3.2), one obtains
$G_{1}^{(+)}$ the diluted-fluid expression (3.2), one obtains
 \begin{equation} \rho_{1}^{(l.sat)}R_{1}T\ln\frac{\rho_{1}^{(+)}}{\rho_{1}^{(v.sat)} }=\varepsilon\mathcal{D}\rho_{1}^{\prime(+)}A.\end{equation}
\begin{equation} \rho_{1}^{(l.sat)}R_{1}T\ln\frac{\rho_{1}^{(+)}}{\rho_{1}^{(v.sat)} }=\varepsilon\mathcal{D}\rho_{1}^{\prime(+)}A.\end{equation}This is, essentially, the GMBC.
With  $A$ calculated (see the next subsubsection), equality (3.55) inter-relates the near-interface vapour density
$A$ calculated (see the next subsubsection), equality (3.55) inter-relates the near-interface vapour density  $\rho _{1}^{(+)}$ and its gradient
$\rho _{1}^{(+)}$ and its gradient  $\rho _{1}^{\prime (+)}$. If
$\rho _{1}^{\prime (+)}$. If  $\varepsilon =0$, then (3.55) yields
$\varepsilon =0$, then (3.55) yields  $\rho _{1}^{(+)}=\rho _{1}^{(v.sat)}$, which is the classical Maxwell boundary condition. If, however,
$\rho _{1}^{(+)}=\rho _{1}^{(v.sat)}$, which is the classical Maxwell boundary condition. If, however,  $\varepsilon$ is small but not zero, then
$\varepsilon$ is small but not zero, then  $\rho _{1}^{(+)}$ can differ significantly from
$\rho _{1}^{(+)}$ can differ significantly from  $\rho _{1}^{(v.sat)}$ – because
$\rho _{1}^{(v.sat)}$ – because  $A$ can actually be large, so that
$A$ can actually be large, so that  $\varepsilon A\sim 1$. This does not violate the employed asymptotic approach: it requires only
$\varepsilon A\sim 1$. This does not violate the employed asymptotic approach: it requires only  $\varepsilon \ll 1$, which makes the time derivatives in (3.24) small and thus justifies the quasi-steady approximation.
$\varepsilon \ll 1$, which makes the time derivatives in (3.24) small and thus justifies the quasi-steady approximation.
3.5.2. Zone 1 of the interfacial asymptotic region
The coefficient  $A$ arises in all problems where the DIM is applied to evaporation or condensation – but, so far, it has been calculated only for pure fluids and the case where the relative humidity is close to unity (Benilov Reference Benilov2020, Reference Benilov2022a,Reference Benilovb, Reference Benilov2023b). The calculation of
$A$ arises in all problems where the DIM is applied to evaporation or condensation – but, so far, it has been calculated only for pure fluids and the case where the relative humidity is close to unity (Benilov Reference Benilov2020, Reference Benilov2022a,Reference Benilovb, Reference Benilov2023b). The calculation of  $A$ for a binary mixture and arbitrary humidity is more complex technically, but the underlying idea is the same.
$A$ for a binary mixture and arbitrary humidity is more complex technically, but the underlying idea is the same.
It is based on the observation that the main contribution to (3.51) comes from the zone where  $\rho _{1}+\rho _{2}$ is small, but its derivatives are relatively large – i.e. near the air region, but not in it. This means effectively that (3.51) can be calculated asymptotically by considering Zone 1 only (see its definition at the beginning of § 3.5).
$\rho _{1}+\rho _{2}$ is small, but its derivatives are relatively large – i.e. near the air region, but not in it. This means effectively that (3.51) can be calculated asymptotically by considering Zone 1 only (see its definition at the beginning of § 3.5).
Before presenting the scaling of Zone 1, it is instructive to apply the ‘obvious’ approximations – i.e. take advantage of the diluted-fluid expressions (3.2), (3.10) and (3.40), plus neglect  $\rho _{1}$ and
$\rho _{1}$ and  $\rho _{2}^{(l)}$ if they appear next to
$\rho _{2}^{(l)}$ if they appear next to  $\rho _{2}$ and
$\rho _{2}$ and  $\rho _{1}^{(l)}$, respectively. As a result, (3.47)–(3.48) and (3.51) become
$\rho _{1}^{(l)}$, respectively. As a result, (3.47)–(3.48) and (3.51) become
 \begin{gather} \frac{\partial}{\partial z}\left( K_{12}\,\frac{\partial^{2}\rho_{2}}{\partial z^{2}}-R_{1}T\ln\rho_{1}\right) =\varepsilon\rho_{1}^{(l)}\dot{Z}\,\frac {\eta_{0}}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1} {\rho_{2}}\right) , \end{gather}
\begin{gather} \frac{\partial}{\partial z}\left( K_{12}\,\frac{\partial^{2}\rho_{2}}{\partial z^{2}}-R_{1}T\ln\rho_{1}\right) =\varepsilon\rho_{1}^{(l)}\dot{Z}\,\frac {\eta_{0}}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1} {\rho_{2}}\right) , \end{gather} \begin{gather}\frac{\partial}{\partial z}\left( K_{22}\,\frac{\partial^{2}\rho_{2}}{\partial z^{2}}-R_{2}T\ln\rho_{2}\right) =\varepsilon\rho_{1}^{(l)}\dot{Z}\,\frac {\eta_{0}}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1} {\rho_{2}}\right) , \end{gather}
\begin{gather}\frac{\partial}{\partial z}\left( K_{22}\,\frac{\partial^{2}\rho_{2}}{\partial z^{2}}-R_{2}T\ln\rho_{2}\right) =\varepsilon\rho_{1}^{(l)}\dot{Z}\,\frac {\eta_{0}}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1} {\rho_{2}}\right) , \end{gather} \begin{gather}A=\eta_{0}\rho_{1}^{(l)}\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}}\,\frac{\partial}{\partial z}\left( \frac{1}{\rho_{2}^{2}}\,\frac{\partial \rho_{2}}{\partial z}\right) \mathrm{d}z. \end{gather}
\begin{gather}A=\eta_{0}\rho_{1}^{(l)}\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}}\,\frac{\partial}{\partial z}\left( \frac{1}{\rho_{2}^{2}}\,\frac{\partial \rho_{2}}{\partial z}\right) \mathrm{d}z. \end{gather}The most general asymptotic limit is when the three terms in (3.57) are of the same order, which implies the rescaling
 \begin{gather} z=z_{0}+\left( \frac{K_{22}\rho_{2}^{(+)}}{R_{2}T}\right)^{1/2} z_{new},\quad \rho_{i}=\rho_{i}^{(+)}\left( \rho_{i}\right)_{new} ,\quad \dot{Z}={-}\frac{E_{0}}{\varepsilon\rho_{1}^{(l)}}\,\xi, \end{gather}
\begin{gather} z=z_{0}+\left( \frac{K_{22}\rho_{2}^{(+)}}{R_{2}T}\right)^{1/2} z_{new},\quad \rho_{i}=\rho_{i}^{(+)}\left( \rho_{i}\right)_{new} ,\quad \dot{Z}={-}\frac{E_{0}}{\varepsilon\rho_{1}^{(l)}}\,\xi, \end{gather} \begin{gather}A=\frac{\rho_{1}^{(l)}R_{2}T}{E_{0}}\,A_{new}, \end{gather}
\begin{gather}A=\frac{\rho_{1}^{(l)}R_{2}T}{E_{0}}\,A_{new}, \end{gather}
where  $z_{0}$ is the ‘location’ of Zone 1,
$z_{0}$ is the ‘location’ of Zone 1,  $E_{0}$ is given by (1.3) of the Introduction, and the minus in the expression for
$E_{0}$ is given by (1.3) of the Introduction, and the minus in the expression for  $\dot {Z}$ is introduced to make
$\dot {Z}$ is introduced to make  $\xi$ positive (recall that evaporation corresponds to
$\xi$ positive (recall that evaporation corresponds to  $\dot {Z}<0$). Since the problem under consideration is translationally invariant, the actual value of
$\dot {Z}<0$). Since the problem under consideration is translationally invariant, the actual value of  $z_{0}$ does not feature in, and is not needed for, any leading-order results. One does not need (3.56) either, because now
$z_{0}$ does not feature in, and is not needed for, any leading-order results. One does not need (3.56) either, because now  $A$ depends only on
$A$ depends only on  $\rho _{2}$.
$\rho _{2}$.
Substitution of (3.59)–(3.60) into (3.57), boundary condition (3.49), and (3.58) yield (with the subscript  $_{new}$ omitted)
$_{new}$ omitted)
 \begin{gather} \frac{\partial}{\partial z}\left( \frac{\partial^{2}\rho_{2}}{\partial z^{2} }-\ln\rho_{2}\right) ={-}\frac{\xi}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1}{\rho_{2}}\right) , \end{gather}
\begin{gather} \frac{\partial}{\partial z}\left( \frac{\partial^{2}\rho_{2}}{\partial z^{2} }-\ln\rho_{2}\right) ={-}\frac{\xi}{\rho_{2}}\,\frac{\partial^{2}}{\partial z^{2}}\left( \frac{1}{\rho_{2}}\right) , \end{gather} \begin{gather}\rho_{2}\rightarrow1\quad\text{as}\ z\rightarrow+\infty, \end{gather}
\begin{gather}\rho_{2}\rightarrow1\quad\text{as}\ z\rightarrow+\infty, \end{gather} \begin{gather}A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}}\,\frac{\partial}{\partial z}\left( \frac{1}{\rho_{2}^{2}}\,\frac{\partial\rho_{2}}{\partial z}\right) \mathrm{d}z. \end{gather}
\begin{gather}A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}}\,\frac{\partial}{\partial z}\left( \frac{1}{\rho_{2}^{2}}\,\frac{\partial\rho_{2}}{\partial z}\right) \mathrm{d}z. \end{gather}As shown in Appendix C, boundary condition (3.62) is sufficient to uniquely fix the solution of (3.61), making it unnecessary to examine the other asymptotic zones of the interfacial region.
It can be derived readily from (3.61) that
 \begin{equation} \rho_{2}\sim z^{2}\ln({-}z) \quad\text{as}\ z\rightarrow -\infty. \end{equation}
\begin{equation} \rho_{2}\sim z^{2}\ln({-}z) \quad\text{as}\ z\rightarrow -\infty. \end{equation}
The growth of  $\rho _{2}(z)$ as
$\rho _{2}(z)$ as  $z\rightarrow -\infty$ in Zone 1 agrees with the fact that this function has a local maximum in Zone 2 (see figure 3). Asymptotics (3.64) also guarantees that (3.63) converges at the lower limit.
$z\rightarrow -\infty$ in Zone 1 agrees with the fact that this function has a local maximum in Zone 2 (see figure 3). Asymptotics (3.64) also guarantees that (3.63) converges at the lower limit.
The function  $A(\xi )$ was computed numerically using the algorithm described in § C.2; the result is plotted in figure 4. For practical use, however, it is more convenient to use the approximation formula (1.4) (for more details, see § C.3).
$A(\xi )$ was computed numerically using the algorithm described in § C.2; the result is plotted in figure 4. For practical use, however, it is more convenient to use the approximation formula (1.4) (for more details, see § C.3).

Figure 4. The function  $A(\xi )$. Note that in the right-hand half of the figure, the scale of
$A(\xi )$. Note that in the right-hand half of the figure, the scale of  $\xi$ is logarithmic.
$\xi$ is logarithmic.
3.5.3. Summarising the GMBC for flat interfaces
Recall that the dimensional variables can be recovered by setting  $\varepsilon =\delta =1$. Then equalities (3.44) and (3.59) yield a dimensional relationship of the near-interface air density
$\varepsilon =\delta =1$. Then equalities (3.44) and (3.59) yield a dimensional relationship of the near-interface air density  $\rho _{1}^{(+)}$ and its gradient
$\rho _{1}^{(+)}$ and its gradient  $\rho _{1}^{\prime (+)}$,
$\rho _{1}^{\prime (+)}$,
 \begin{equation} \xi=\frac{-\mathcal{D}\rho_{1}^{\prime(+)}}{E_{0}},\end{equation}
\begin{equation} \xi=\frac{-\mathcal{D}\rho_{1}^{\prime(+)}}{E_{0}},\end{equation}and (3.55) yields
 \begin{equation} \rho_{1}^{(+)}=\rho_{1}^{(v.sat)}\exp\left[ -\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)\right] .\end{equation}
\begin{equation} \rho_{1}^{(+)}=\rho_{1}^{(v.sat)}\exp\left[ -\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)\right] .\end{equation}
Equalities (3.65) and (3.66) inter-relate the near-surface vapour density  $\rho _{1}^{(+)}$ and its normal derivative
$\rho _{1}^{(+)}$ and its normal derivative  $\rho _{1}^{\prime (+)}$, hence they constitute the generalised Maxwell boundary condition (for flat interfaces), as required.
$\rho _{1}^{\prime (+)}$, hence they constitute the generalised Maxwell boundary condition (for flat interfaces), as required.
3.5.4. Curved interfaces
Interfacial curvature influences evaporation via the so-called Kelvin effect, i.e. by changing the effective saturated vapour pressure near the liquid's (curved) surface. This effect can be incorporated readily into the analysis under the assumption that the curvature radius is much larger than the interfacial thickness. This has been done, and the result has turned out to be physically obvious: the Kelvin effect adds a new term to the pressure condition (3.53),
 \begin{equation} p^{(l)}-p^{(+)}=\varepsilon C\sigma, \end{equation}
\begin{equation} p^{(l)}-p^{(+)}=\varepsilon C\sigma, \end{equation}
where  $C$ is the interfacial curvature, and
$C$ is the interfacial curvature, and  $\sigma$ is the surface tension. (The DIM expression for
$\sigma$ is the surface tension. (The DIM expression for  $\sigma$ is given by (B8) of Appendix B.)
$\sigma$ is given by (B8) of Appendix B.)
The extra term in the pressure equation entails an extra term in the GMBC: instead of (3.66), it becomes (1.1) of the Introduction, whereas definition (3.65) of the relative evaporation rate  $\xi$ coincides with (1.2) if one sets
$\xi$ coincides with (1.2) if one sets  $\rho _{1}^{\prime (+)}=( \boldsymbol {n} \boldsymbol {\cdot }\boldsymbol {\nabla }\rho _{1})^{(+)}$.
$\rho _{1}^{\prime (+)}=( \boldsymbol {n} \boldsymbol {\cdot }\boldsymbol {\nabla }\rho _{1})^{(+)}$.
4. Evaporation of drops as described by the generalised model
To compare the generalised model to the classical one, the former will be applied to evaporation of floating spherical drops. To do so, one needs to replace the classical boundary condition (2.10) with its generalised version (1.1), and leave the rest of the governing set (2.3)–(2.13) the same as before. The ensuing algebra is also the same as before, and one eventually obtains the following differential equation for  $r_{d}(t)$:
$r_{d}(t)$:
 \begin{equation} \underset{\text{diffusion}}{\underbrace{\frac{\rho_{1}^{(l.sat)}} {\mathcal{D}\rho_{1}^{(v.sat)(a)}}\,\dot{r}_{d}r_{d}}}+\exp\left[ \underset{\text{non-isothermality}}{\underbrace{\dfrac{T_{A}\rho_{1}^{(l.sat)}\varDelta}{T^{(a)2}\kappa^{(a)}}\,\dot{r}_{d}r_{d}} }-\underset{\text{interfacial effects}}{\underbrace{\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)}}+\underset{\text{Kelvin effect}}{\underbrace{\frac{2\sigma}{\rho_{1}^{(l.sat)}R_{1}T^{(a)}r_{d}}}}\right] =H,\end{equation}
\begin{equation} \underset{\text{diffusion}}{\underbrace{\frac{\rho_{1}^{(l.sat)}} {\mathcal{D}\rho_{1}^{(v.sat)(a)}}\,\dot{r}_{d}r_{d}}}+\exp\left[ \underset{\text{non-isothermality}}{\underbrace{\dfrac{T_{A}\rho_{1}^{(l.sat)}\varDelta}{T^{(a)2}\kappa^{(a)}}\,\dot{r}_{d}r_{d}} }-\underset{\text{interfacial effects}}{\underbrace{\frac{R_{2}}{R_{1}}\,\xi\, A(\xi)}}+\underset{\text{Kelvin effect}}{\underbrace{\frac{2\sigma}{\rho_{1}^{(l.sat)}R_{1}T^{(a)}r_{d}}}}\right] =H,\end{equation}where
 \begin{equation} \xi=\frac{-\rho_{1}^{(l.sat)}\dot{r}_{d}}{E_{0}}\end{equation}
\begin{equation} \xi=\frac{-\rho_{1}^{(l.sat)}\dot{r}_{d}}{E_{0}}\end{equation}is the ratio of the evaporative flux to the interfacial emission capacity (the latter given by (1.3)).
Equations (4.1)–(4.2) generalise the corresponding classical result (2.21). The term involving  $A(\xi )$ in (4.1) describes emission of vapour by the interface, and the term involving
$A(\xi )$ in (4.1) describes emission of vapour by the interface, and the term involving  $\sigma$ gives the shift of the effective saturation pressure near a curved surface. The remaining two terms on the left-hand side of (4.1) are the same as those in its classical counterpart (2.21).
$\sigma$ gives the shift of the effective saturation pressure near a curved surface. The remaining two terms on the left-hand side of (4.1) are the same as those in its classical counterpart (2.21).
The classical and modified models – described by (2.21) and (4.1), respectively – are compared in figures 5(a,b). For figure 5(a), both were treated as algebraic equations for the non-dimensional evaporation flux  $\xi$ with the drop's radius
$\xi$ with the drop's radius  $r_{d}$ being a parameter – so that the result is the dependence
$r_{d}$ being a parameter – so that the result is the dependence  $\xi$ versus
$\xi$ versus  $r_{d}$. Figure 5(b), in turn, shows the dependence of the time of full evaporation on the drop's initial radius.
$r_{d}$. Figure 5(b), in turn, shows the dependence of the time of full evaporation on the drop's initial radius.

Figure 5. Characteristics of an evaporating spherical drop versus its radius, for  $T = 25\,^{\circ }\mathrm {C}$ and relative humidity
$T = 25\,^{\circ }\mathrm {C}$ and relative humidity  $H = 50\,\%$. Curves (M) and (B) are computed using Maxwell's classical model and its generalised version, respectively. The numbers in the ellipses show the relative deviations of curve (M) from curve (B). (a) The evaporative flux normalised by the emission capacity of the interface (where
$H = 50\,\%$. Curves (M) and (B) are computed using Maxwell's classical model and its generalised version, respectively. The numbers in the ellipses show the relative deviations of curve (M) from curve (B). (a) The evaporative flux normalised by the emission capacity of the interface (where  $\xi$ is defined by (4.2)). (b) The time of evaporation. The shaded areas of (a) and (b) correspond to
$\xi$ is defined by (4.2)). (b) The time of evaporation. The shaded areas of (a) and (b) correspond to  $\xi <1$, which is where the classical model is supposed to work. (c) The four terms (effects) in (4.1).
$\xi <1$, which is where the classical model is supposed to work. (c) The four terms (effects) in (4.1).
The following features of these figures should be observed.
(i) The (classical)
$d^{2}$ law states that $t_{e}\sim r_{d}^{2}$, which is why curve (M) in figure 5(b) is a straight line with slope $2$. In figure 5(a), curve (M) is also a straight line, but its slope is $-1$ (which agrees with the corresponding result of the classical model).(ii) The shaded areas in figures 5(a,b) mark the region
$\xi <1$, where the two models are supposed to yield similar results – and indeed, they do. The value of $r_{d}$ corresponding to $\xi =1$ is $r_{d}\approx 2\ \mathrm {\mu }\mathrm {m}$.(iii) For smaller
$r_{d}$, curves (M) and (B) begin to diverge. The difference between the classical and generalised results peaks when the drop's radius is close to, and slightly less than, $r_{d}=10\ \mathrm {nm}$.A similar effect is observed in models resolving or parametrising the Knudsen layer (Long et al. Reference Long, Micci and Wong1996; Haut & Colinet Reference Haut and Colinet2005; Landry et al. Reference Landry, Mikkilineni, Paharia and McGaughey2007; Jakubczyk et al. Reference Jakubczyk, Kolwas, Derkachov, Kolwas and Zientara2012; Hołyst et al. Reference Hołyst, Litniewski, Jakubczyk, Kolwas, Kolwas, Kowalski, Migacz, Palesa and Zientara2013, Reference Hołyst, Litniewski and Jakubczyk2017; Rana et al. Reference Rana, Lockerby and Sprittles2018, Reference Rana, Lockerby and Sprittles2019; Zhao & Nadal Reference Zhao and Nadal2023).
(iv) For drops with
$r_{d}\lesssim 10\ \mathrm {nm}$, the Kelvin effect ‘kicks in’: it increases the effective saturated pressure and thus accelerates the vapour emission by the interface – as a result, curves (M) and (B) re-converge.To verify this explanation, the four terms of (4.1) have been computed individually and plotted in figure 5(c). Evidently, evaporation of small drops is governed by the balance of interfacial effects and the Kelvin effect. Non-isothermality is less important, whereas the diffusion of vapour in the air is unimportant.
(v) A general criterion of importance of the van der Waals force relative to diffusion can be obtained by estimating the characteristic ratio
$B$ of the second term in the square brackets in (4.1) to the first term. Recalling (4.2), one obtains
(4.3)where\begin{equation} B=\frac{R_{2}\bar{A}T^{(a)2}\kappa^{(a)}}{R_{1}r_{d}E_{0}T_{A}\varDelta}, \end{equation}$\bar {A}$ is a characteristic value of the function $A(\xi )$. The van der Waals force is important when $B\gtrsim 1$, which amounts to
(4.4)For the parameters of water at\begin{equation} r_{d}\lesssim\frac{R_{2}\bar{A}T^{(a)2}\kappa^{(a)}}{R_{1}E_{0}T_{A}\varDelta}. \end{equation}$25\,^{\circ }\mathrm {C}$ and with $\bar {A}=A(0)\approx 0.14219$, one obtains $r_{d}\lesssim 0.4\ \mathrm {\mu }\mathrm {m}$, which agrees qualitatively with the fact that curves (d) and (i) in figure 5(c) intersect at $r_{d}\approx 1\ \mathrm {\mu } \mathrm {m}$.(vi) Figure 5(c) also suggests that for large drops (say,
$r_{d}\gtrsim 0.1\ \mathrm {\mu } \mathrm {m}$), the Kelvin effect is negligible, which agrees broadly with the estimate of Sobac et al. (Reference Sobac, Talbot, Haut, Rednikov and Colinet2015) .It is worth emphasising, however, that this conclusion applies only to large floating drops, whereas large sessile drops can be still influenced by the Kelvin effect via the highly curved part of their surface near the contact line (e.g. Deegan et al. Reference Deegan, Bakajin, Dupont, Huber, Nagel and Witten2000; Dunn et al. Reference Dunn, Wilson, Duffy, David and Sefiane2009; Eggers & Pismen Reference Eggers and Pismen2010; Colinet & Rednikov Reference Colinet and Rednikov2011; Rednikov & Colinet Reference Rednikov and Colinet2013, Reference Rednikov and Colinet2019; Morris Reference Morris2014; Stauber et al. Reference Stauber, Wilson, Duffy and Sefiane2014, Reference Stauber, Wilson, Duffy and Sefiane2015; Janeček et al. Reference Janeček, Doumenc, Guerrier and Nikolayev2015; Sáenz et al. Reference Sáenz, Sefiane, Kim, Matar and Valluri2015; Saxton et al. Reference Saxton, Whiteley, Vella and Oliver2016, Reference Saxton, Vella, Whiteley and Oliver2017; Wray, Duffy & Wilson Reference Wray, Duffy and Wilson2019; Williams et al. Reference Williams, Karapetsas, Mamalis, Sefiane, Matar and Valluri2020).
(vii) Overall, figures 5(a,b) should be viewed as an argument in favour of the GMBC at scales
$\lesssim 1\ \mathrm {\mu }$m, whereas figure 5(c) provides a physical explanation as to why.























5. Concluding remarks
Since the main result of this paper has been summarised in the Introduction, it only remains to comment on further applications of the proposed model and its connections with other models of evaporation.
(i) Apart from drops, the generalised Maxwell boundary condition can be applied to menisci – and if their radii are small, then the GMBC should produce results more accurate than those of the classical model. This should be important for diffusion of fluids in porous media with small pores.
Note also that interfacial effects are particularly strong for concave menisci, in which case the Kelvin effect works with the interfacial effects, not against them. This should make the difference between the predictions of the classical and generalised models larger than that for convex menisci and drops.
(ii) For flat and cylindrical interfaces, the results obtained via the two models converge with time. In both cases, the diffusion equation does not have a one-dimensional solution describing a steady flux of vapour towards infinity, which effectively means that the throughput of air is zero. As a result, after a sufficiently long time, the air near the interface gets almost saturated.
At finite times, however, the classical and generalised models yield different results even for cylindrical and flat interfaces.
(iii) A sessile or pendant drop – especially that with moving contact lines – evaporates differently from floating ones. Its surface is highly curved near the contact lines, changing the local evaporation rate. To account for this effect is not a straightforward task, however, as the flow inside the drop is not one-dimensional in this case.
One might hope that the generalised model might explain the problem of abnormally small Navier slip arising for several liquids, including water (Podgorski, Flesselles & Limat Reference Podgorski, Flesselles and Limat2001; Winkels et al. Reference Winkels, Peters, Evangelista, Riepen, Daerr, Limat and Snoeijer2011; Puthenveettil, Senthilkumar & Hopfinger Reference Puthenveettil, Senthilkumar and Hopfinger2013; Benilov & Benilov Reference Benilov and Benilov2015).
(iv) All of the problems listed above are easier to solve using the quasi-static approximation. One should keep in mind, however, that Finneran et al. (Reference Finneran, Garner and Nadal2021) compared that with the transient solution of the governing equations (for the classical model) and observed that at high pressure, the quasi-steady approximation overpredicts the droplet lifetime by up to 80 %.
One might conjecture that the difference between the transient and quasi-steady approaches is due to the fact that the latter implies that the mass of evaporated liquid is infinite. Indeed, the steady solution of the spherically symmetric diffusion equation for vapour is
${\sim }1/r$ – hence the integral of this solution over the region between the drop's surface and infinity diverges.Thus for the quasi-steady solution to establish itself around the drop, a significant part of the drop should have evaporated already. This applies to both classical and generalised models.
(v) If the drop's radius is small, but the Kelvin effect is nevertheless neglected, the generalised model – i.e. (4.1) with
$\sigma =0$ – predicts that the time of a drop's evaporation is proportional to its radius. A similar behaviour occurs due to the temperature jump associated with the Knudsen layer (e.g. Rana et al. Reference Rana, Lockerby and Sprittles2019; Zhao & Nadal Reference Zhao and Nadal2023), but this seems to be just a coincidence. Indeed, the DIM does not describe temperature jumps (which are a purely kinetic effect (Bond & Struchtrup Reference Bond and Struchtrup2004; Persad & Ward Reference Persad and Ward2016; Rana et al. Reference Rana, Lockerby and Sprittles2018)), whereas the existing kinetic models do not include the van der Waals force (which is vital for Zone 1 of the present analysis).Yet the Knudsen layer and Zone 1 are both located at the outskirts of the interface, out of equilibrium and affected by irreversible processes: the former by collisions, and the latter by viscosity (which is the hydrodynamic analogue of collisions). All this suggests that Zone 1 is the DIM equivalent of the Knudsen layer. If this is indeed the case, then the main conclusion of this paper is that the van der Waals force is one of the driving effects in the Knudsen layer and thus should be accounted for.
This conjecture can be verified using the Enskog–Vlasov kinetic equation; it includes the van der Waals force (just like the DIM) and can handle temperature jumps (like the Boltzmann equation). Since both effects work the same way – to slow evaporation down – their joint effect should be stronger than predicted by the DIM and existing models of the Knudsen layer separately.
(vi) Despite the fact that the DIM cannot handle temperature jumps, it is still closely related to the Enskog–Vlasov equation, with the former being the hydrodynamic approximation of the latter (Giovangigli Reference Giovangigli2020, Reference Giovangigli2021). The asymptotic structure of the solution of the two models should be similar, and the results obtained in the present paper should make the analysis of the Enskog–Vlasov equation much easier.
(vii) Note that, strictly speaking, the surface tension depends on the drop's curvature (Tolman's effect), which can have a profound influence on, say, cavitation (Menzl et al. Reference Menzl, Gonzalez, Geiger, Caupin, Abascal, Valeriani and Dellago2016; Magaletti, Gallo & Casciola Reference Magaletti, Gallo and Casciola2021; Aasen et al. Reference Aasen, Wilhelmsen, Hammer and Reguera2023; Lamas et al. Reference Lamas, Vega, Noya and Sanz2023). Since cavitating bubbles are created with zero radii, Tolman's length – no matter how small – may affect the whole ‘trajectory’ of their growth. Evaporation appears to be different, however; since Tolman's length for, say, water under normal conditions is less than 1 Å (e.g. Wilhelmsen, Bedeaux & Reguera Reference Wilhelmsen, Bedeaux and Reguera2015; Burian et al. Reference Burian, Isaiev, Termentzidis, Sysoev and Bulavin2017), it is much smaller than typical radii of evaporating drops, and the whole effect is likely to be negligible.


Declaration of interests
The author reports no conflict of interests.
Appendix A. The thermodynamics incorporated into the DIM
A.1. The definition of the chemical potential $G_{i}$

Consider an  $N$-component compressible non-ideal fluid, characterised by the temperature
$N$-component compressible non-ideal fluid, characterised by the temperature  $T$ and partial densities
$T$ and partial densities  $\rho _{i}$, where
$\rho _{i}$, where  $i=1,\ldots, N$. The fluid's thermodynamic properties are fully described by the internal energy
$i=1,\ldots, N$. The fluid's thermodynamic properties are fully described by the internal energy  $e(\rho _{1},\ldots,\rho _{N},T)$ and entropy
$e(\rho _{1},\ldots,\rho _{N},T)$ and entropy  $s(\rho _{1},\ldots,\rho _{N},T)$ – both specific, i.e. per unit mass. Note that
$s(\rho _{1},\ldots,\rho _{N},T)$ – both specific, i.e. per unit mass. Note that  $e$ and
$e$ and  $s$ are not fully arbitrary, but should satisfy the fundamental thermodynamic relation, which can be written in the form (see Appendix A of Benilov Reference Benilov2023a)
$s$ are not fully arbitrary, but should satisfy the fundamental thermodynamic relation, which can be written in the form (see Appendix A of Benilov Reference Benilov2023a)
 \begin{equation} \frac{\partial e}{\partial T}=T\,\frac{\partial s}{\partial T}.\end{equation}
\begin{equation} \frac{\partial e}{\partial T}=T\,\frac{\partial s}{\partial T}.\end{equation}
The dependence of the fluid pressure  $p$ on
$p$ on  $(\rho _{1},\ldots,\rho _{N},T)$, or the equation of state, is given by (e.g. Giovangigli & Matuszewski Reference Giovangigli and Matuszewski2013)
$(\rho _{1},\ldots,\rho _{N},T)$, or the equation of state, is given by (e.g. Giovangigli & Matuszewski Reference Giovangigli and Matuszewski2013)
 \begin{equation} p=\rho\sum_{i}\rho_{i}\left( \frac{\partial e}{\partial\rho_{i}} -T\,\frac{\partial s}{\partial\rho_{i}}\right) ,\end{equation}
\begin{equation} p=\rho\sum_{i}\rho_{i}\left( \frac{\partial e}{\partial\rho_{i}} -T\,\frac{\partial s}{\partial\rho_{i}}\right) ,\end{equation}where
 \begin{equation} \rho=\sum_{i}\rho_{i}\end{equation}
\begin{equation} \rho=\sum_{i}\rho_{i}\end{equation}is the total density. The partial chemical potentials, in turn, are given by
 \begin{equation} G_{i}=\frac{\partial( \rho e) }{\partial\rho_{i}}-T\,\frac {\partial( \rho s) }{\partial\rho_{i}}.\end{equation}
\begin{equation} G_{i}=\frac{\partial( \rho e) }{\partial\rho_{i}}-T\,\frac {\partial( \rho s) }{\partial\rho_{i}}.\end{equation}A.2. The Maxwell construction (conditions of saturation)
Consider a flat interface in a multicomponent fluid in the state of equilibrium. This implies that there should be no flow and fluxes ( $w=J_{i} =0$), and no dependence on
$w=J_{i} =0$), and no dependence on  $t$, so that (3.14)–(3.15) yield
$t$, so that (3.14)–(3.15) yield
 \begin{equation} \frac{\mathrm{d}}{\mathrm{d}z}\left( \sum_{j}K_{ij}\,\frac{\mathrm{d}^{2} \rho_{j}}{\mathrm{d}z^{2}}-G_{i}\right) =0.\end{equation}
\begin{equation} \frac{\mathrm{d}}{\mathrm{d}z}\left( \sum_{j}K_{ij}\,\frac{\mathrm{d}^{2} \rho_{j}}{\mathrm{d}z^{2}}-G_{i}\right) =0.\end{equation}One should also impose the following boundary conditions:
 \begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l.sat)}\quad\text{as}\ z\rightarrow -\infty, \end{gather}
\begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l.sat)}\quad\text{as}\ z\rightarrow -\infty, \end{gather} \begin{gather}\rho_{i}\rightarrow\rho_{i}^{(v.sat)}\quad\text{as}\ z\rightarrow +\infty, \end{gather}
\begin{gather}\rho_{i}\rightarrow\rho_{i}^{(v.sat)}\quad\text{as}\ z\rightarrow +\infty, \end{gather}
where  $\rho _{i}^{(l.sat)}$ and
$\rho _{i}^{(l.sat)}$ and  $\rho _{i}^{(v.sat)}$ are the densities of saturated liquid and vapour, respectively.
$\rho _{i}^{(v.sat)}$ are the densities of saturated liquid and vapour, respectively.
Integrating (A5) with respect to  $z$, and taking into account conditions (A6)–(A7), one obtains
$z$, and taking into account conditions (A6)–(A7), one obtains
 \begin{equation} G_{i}^{(l.sat)}=G_{i}^{(v.sat)}. \end{equation}
\begin{equation} G_{i}^{(l.sat)}=G_{i}^{(v.sat)}. \end{equation}Then consider
 \begin{equation} \int_{-\infty}^{+\infty}\sum_{i}\rho_{i}\times\text{(A5)}\, \mathrm{d}z. \end{equation}
\begin{equation} \int_{-\infty}^{+\infty}\sum_{i}\rho_{i}\times\text{(A5)}\, \mathrm{d}z. \end{equation}Integrating the resulting equality by parts, and keeping in mind that
 \begin{equation} \sum_{i,j}K_{ij}\,\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\frac{\mathrm{d}^{2} \rho_{j}}{\mathrm{d}z^{2}}\,\mathrm{d}z=\frac{1}{2}\,\frac{\mathrm{d}} {\mathrm{d}z}\sum_{i,j}K_{ij}\,\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\, \frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}\,\mathrm{d}z, \end{equation}
\begin{equation} \sum_{i,j}K_{ij}\,\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\frac{\mathrm{d}^{2} \rho_{j}}{\mathrm{d}z^{2}}\,\mathrm{d}z=\frac{1}{2}\,\frac{\mathrm{d}} {\mathrm{d}z}\sum_{i,j}K_{ij}\,\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\, \frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}\,\mathrm{d}z, \end{equation}one can use boundary conditions (A6)–(A7) to obtain
 \begin{equation} -\sum_{i}\rho_{i}^{(v.sat)}G_{i}^{(v.sat)}+\sum_{i}\rho_{i}^{(l.sat)} G_{i}^{(l.sat)}+\int_{-\infty}^{+\infty}\sum_{i}\frac{\mathrm{d}\rho_{i} }{\mathrm{d}z}\,G_{i}\,\mathrm{d}z=0. \end{equation}
\begin{equation} -\sum_{i}\rho_{i}^{(v.sat)}G_{i}^{(v.sat)}+\sum_{i}\rho_{i}^{(l.sat)} G_{i}^{(l.sat)}+\int_{-\infty}^{+\infty}\sum_{i}\frac{\mathrm{d}\rho_{i} }{\mathrm{d}z}\,G_{i}\,\mathrm{d}z=0. \end{equation}Observe that identity (3.1) implies that
 \begin{equation} G_{i}=\frac{\partial}{\partial\rho_{i}}\left( \sum_{j}\rho_{j}G_{j}-p\right). \end{equation}
\begin{equation} G_{i}=\frac{\partial}{\partial\rho_{i}}\left( \sum_{j}\rho_{j}G_{j}-p\right). \end{equation}This equality helps one to integrate the last term of (A11) and obtain
 \begin{equation} p^{(l.sat)}=p^{(v.sat)}. \end{equation}
\begin{equation} p^{(l.sat)}=p^{(v.sat)}. \end{equation}Physically, equalities (A8) and (A13) reflect the famous Maxwell construction (Maxwell Reference Maxwell1875), which inter-relates the partial densities of the components in vapour and liquid. One should also require
 \begin{equation} p^{(v.sat)}=p_{A}, \end{equation}
\begin{equation} p^{(v.sat)}=p_{A}, \end{equation}
where  $p_{A}$ is the atmospheric pressure.
$p_{A}$ is the atmospheric pressure.
An example of a numerical solution of boundary-value problem (A5)–(A7) in application to a water/air interface (with the chemical potentials  $G_{i}$ described in the next appendix) is shown in figure 3.
$G_{i}$ described in the next appendix) is shown in figure 3.
Appendix B. The parameters of the DIM
Benilov (Reference Benilov2023a) proposed to use the DIM with the so-called Enskog–Vlasov equation of state. In application to a binary fluid with one diluted component, it amounts to the following expressions for the chemical potentials:
 \begin{align} G_{1}&=T\left[ R_{1}\ln\rho_{1}+\varTheta(\rho_{1})+( \rho_{1}+\rho _{2})\,\frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right] \nonumber\\ &\quad -2( a_{11}\rho_{1}+a_{12}\rho_{2}) +T[ R_{1}+3R_{1}( 1-\ln T)] , \end{align}
\begin{align} G_{1}&=T\left[ R_{1}\ln\rho_{1}+\varTheta(\rho_{1})+( \rho_{1}+\rho _{2})\,\frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right] \nonumber\\ &\quad -2( a_{11}\rho_{1}+a_{12}\rho_{2}) +T[ R_{1}+3R_{1}( 1-\ln T)] , \end{align} \begin{align} G_{2}&=T[ R_{2}\ln\rho_{2}+\varTheta(\rho_{1})] -2a_{12}\rho _{1}+T[ R_{2}+\tfrac{5}{2}R_{2}( 1-\ln T) ] , \end{align}
\begin{align} G_{2}&=T[ R_{2}\ln\rho_{2}+\varTheta(\rho_{1})] -2a_{12}\rho _{1}+T[ R_{2}+\tfrac{5}{2}R_{2}( 1-\ln T) ] , \end{align}
where  $a_{11}$ and
$a_{11}$ and  $a_{12}$ are adjustable parameters, and
$a_{12}$ are adjustable parameters, and  $\varTheta (\rho _{1})$ is an adjustable function. For water and air, Benilov (Reference Benilov2023a) proposed
$\varTheta (\rho _{1})$ is an adjustable function. For water and air, Benilov (Reference Benilov2023a) proposed
 \begin{gather} a_{11}=2112.1\ \mathrm{m}^{5}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1},\quad a_{12}=208\ \mathrm{m}^{5} \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}, \end{gather}
\begin{gather} a_{11}=2112.1\ \mathrm{m}^{5}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1},\quad a_{12}=208\ \mathrm{m}^{5} \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}, \end{gather} \begin{gather}\varTheta(\rho_{1})=R\left[{-}q^{(0)}\ln\left( 1-0.99\,\frac{\rho_{1}}{\rho_{tp} }\right) +\sum_{n=1}^{4}q^{(n)}\left( \frac{\rho_{1}}{\rho_{tp}}\right) ^{n}\right] , \end{gather}
\begin{gather}\varTheta(\rho_{1})=R\left[{-}q^{(0)}\ln\left( 1-0.99\,\frac{\rho_{1}}{\rho_{tp} }\right) +\sum_{n=1}^{4}q^{(n)}\left( \frac{\rho_{1}}{\rho_{tp}}\right) ^{n}\right] , \end{gather}where
 \begin{gather} \rho_{tp}=999.79\ \mathrm{kg\ m}^{{-}3}, \end{gather}
\begin{gather} \rho_{tp}=999.79\ \mathrm{kg\ m}^{{-}3}, \end{gather} \begin{gather}q^{(0)}=0.071894,\quad q^{(1)}=1.4139,\quad q^{(2)}=8.1126, \end{gather}
\begin{gather}q^{(0)}=0.071894,\quad q^{(1)}=1.4139,\quad q^{(2)}=8.1126, \end{gather} \begin{gather}q^{(3)}={-}8.3669,\quad q^{(4)}=4.0238. \end{gather}
\begin{gather}q^{(3)}={-}8.3669,\quad q^{(4)}=4.0238. \end{gather}
When using the DIM, one also needs the Korteweg matrix  $K_{ij}$. The value of
$K_{ij}$. The value of  $K_{11}$ can be deduced from the surface tension of the liquid–water/vapour–water interface, and
$K_{11}$ can be deduced from the surface tension of the liquid–water/vapour–water interface, and  $K_{22}$ from that of the liquid–air/vapour–air interface. The resulting values, computed by Benilov (Reference Benilov2023a), are listed in equality (3.12).
$K_{22}$ from that of the liquid–air/vapour–air interface. The resulting values, computed by Benilov (Reference Benilov2023a), are listed in equality (3.12).
The coefficient  $K_{12}$ is harder to calculate. One might think that it can be determined by comparing the liquid–water/vapour–water interface with the liquid–water/air interface – but at normal conditions, their characteristics are almost indistinguishable. Instead,
$K_{12}$ is harder to calculate. One might think that it can be determined by comparing the liquid–water/vapour–water interface with the liquid–water/air interface – but at normal conditions, their characteristics are almost indistinguishable. Instead,  $K_{12}$ was deduced from the surface tension of the liquid–water/air interface at high pressure, measured by Hinton & Alvarez (Reference Hinton and Alvarez2021). The following approach was used.
$K_{12}$ was deduced from the surface tension of the liquid–water/air interface at high pressure, measured by Hinton & Alvarez (Reference Hinton and Alvarez2021). The following approach was used.
Within the framework of the DIM, the surface tension is given by
 \begin{equation} \sigma=\sum_{i,j}K_{ij} {\displaystyle\int_{-\infty}^{\infty}} \dfrac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\dfrac{\mathrm{d}\rho_{j}}{\mathrm{d} z}\,\mathrm{d}z, \end{equation}
\begin{equation} \sigma=\sum_{i,j}K_{ij} {\displaystyle\int_{-\infty}^{\infty}} \dfrac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\dfrac{\mathrm{d}\rho_{j}}{\mathrm{d} z}\,\mathrm{d}z, \end{equation}
where  $\rho _{i}(z)$ is the solution of the boundary-value problem (A5)–(A7). It was solved numerically with different values of
$\rho _{i}(z)$ is the solution of the boundary-value problem (A5)–(A7). It was solved numerically with different values of  $K_{12}$ and the rest of the parameters given by (B1)–(B7) and (3.12). For each value of
$K_{12}$ and the rest of the parameters given by (B1)–(B7) and (3.12). For each value of  $K_{12}$, problem (A5)–(A7) was solved for various values of the atmospheric pressure
$K_{12}$, problem (A5)–(A7) was solved for various values of the atmospheric pressure  $p_{A}$, and this computation was repeated until the best fit with the experimental dependence
$p_{A}$, and this computation was repeated until the best fit with the experimental dependence  $\sigma$ versus
$\sigma$ versus  $p_{A}$ is achieved. The results are shown in figure 6.
$p_{A}$ is achieved. The results are shown in figure 6.

Figure 6. The surface tension of the water/air interface versus the pressure, at  $T=22.5\,^{\circ }\mathrm {C}$. The black squares show the experimental data of Hinton & Alvarez (Reference Hinton and Alvarez2021), and the solid line shows the theoretical result (B8) computed using the parameter values described in Appendix B and
$T=22.5\,^{\circ }\mathrm {C}$. The black squares show the experimental data of Hinton & Alvarez (Reference Hinton and Alvarez2021), and the solid line shows the theoretical result (B8) computed using the parameter values described in Appendix B and  $K_{12}=0.84978\times 10^{-17}\ \mathrm {m}^{7}\ \mathrm {s}^{-2}\ \mathrm {kg}^{-1}$ (chosen as the best fit of the experimental results). The dashed curves marked with ‘
$K_{12}=0.84978\times 10^{-17}\ \mathrm {m}^{7}\ \mathrm {s}^{-2}\ \mathrm {kg}^{-1}$ (chosen as the best fit of the experimental results). The dashed curves marked with ‘ $\pm$’ are computed for
$\pm$’ are computed for  $K_{12}=(0.84978\pm 0.04810)\times 10^{-17} \ \mathrm {m}^{7}\ \mathrm {s}^{-2}\ \mathrm {kg}^{-1}$, respectively.
$K_{12}=(0.84978\pm 0.04810)\times 10^{-17} \ \mathrm {m}^{7}\ \mathrm {s}^{-2}\ \mathrm {kg}^{-1}$, respectively.
Unfortunately, Hinton & Alvarez (Reference Hinton and Alvarez2021) provide data for only six values of  $p_{A}$, and the resulting pairs
$p_{A}$, and the resulting pairs  $( p_{A},\sigma )$ do not sit on a smooth curve. Thus these measurements do not seem to merit the use of formal curve-fitting methods; instead, a value of
$( p_{A},\sigma )$ do not sit on a smooth curve. Thus these measurements do not seem to merit the use of formal curve-fitting methods; instead, a value of  $K_{12}$ was chosen ‘manually’. If sought in the form
$K_{12}$ was chosen ‘manually’. If sought in the form
 \begin{equation} K_{12}=b( K_{11}K_{22})^{1/2},\end{equation}
\begin{equation} K_{12}=b( K_{11}K_{22})^{1/2},\end{equation}
the best fit appears to be for  $b=0.53$ – for which (B9) yields the value of
$b=0.53$ – for which (B9) yields the value of  $K_{12}$ given by (3.12). For comparison, figure 6 shows curves with
$K_{12}$ given by (3.12). For comparison, figure 6 shows curves with  $K_{12}$ corresponding to
$K_{12}$ corresponding to  $b=0.50$ and
$b=0.50$ and  $b=0.56$.
$b=0.56$.
Note that in the pure-fluid reduction of (B8), one can replace the coordinate  $z$ with the density
$z$ with the density  $\rho$ and then use the pure-fluid reduction of boundary-value problem (A5)–(A7) to obtain a closed-form integral involving the fluid's thermodynamic functions (see equation (7.12) of Benilov Reference Benilov2023a). As a result,
$\rho$ and then use the pure-fluid reduction of boundary-value problem (A5)–(A7) to obtain a closed-form integral involving the fluid's thermodynamic functions (see equation (7.12) of Benilov Reference Benilov2023a). As a result,  $\sigma$ can be found without solving the equations for the interfacial profile. In the multicomponent case, however, a similar change
$\sigma$ can be found without solving the equations for the interfacial profile. In the multicomponent case, however, a similar change  $z\rightarrow \rho _{1}$ does allow one to bypass solving the equations, as (B8) would involve the dependence of
$z\rightarrow \rho _{1}$ does allow one to bypass solving the equations, as (B8) would involve the dependence of  $\rho _{2}$ on
$\rho _{2}$ on  $\rho _{1}$.
$\rho _{1}$.
Appendix C. Equation (3.61)
The ordinary differential equation (ODE) (3.61) is third order, but it can be reduced to a first-order equation of the Chini kind. To do so, multiply (3.61) by  $\rho _{2}$ and integrate with respect to
$\rho _{2}$ and integrate with respect to  $z$. Fixing the constant of integration via boundary condition (3.62), one obtains
$z$. Fixing the constant of integration via boundary condition (3.62), one obtains
 \begin{equation} \rho_{2}\,\frac{\mathrm{d}^{2}\rho_{2}}{\mathrm{d}z^{2}}-\frac{1}{2}\left( \frac{\mathrm{d}\rho_{2}}{\mathrm{d}z}\right)^{2}-\rho_{2}+1=\frac{\xi} {\rho_{2}^{2}}\,\frac{\mathrm{d}\rho_{2}}{\mathrm{d}z}. \end{equation}
\begin{equation} \rho_{2}\,\frac{\mathrm{d}^{2}\rho_{2}}{\mathrm{d}z^{2}}-\frac{1}{2}\left( \frac{\mathrm{d}\rho_{2}}{\mathrm{d}z}\right)^{2}-\rho_{2}+1=\frac{\xi} {\rho_{2}^{2}}\,\frac{\mathrm{d}\rho_{2}}{\mathrm{d}z}. \end{equation}
Then changing  $( \rho _{2},z) \rightarrow ( q,\rho )$, where
$( \rho _{2},z) \rightarrow ( q,\rho )$, where
 \begin{equation} q=\frac{\mathrm{d}\rho_{2}}{\mathrm{d}z},\quad\rho=\rho_{2}, \end{equation}
\begin{equation} q=\frac{\mathrm{d}\rho_{2}}{\mathrm{d}z},\quad\rho=\rho_{2}, \end{equation}one obtains
 \begin{equation} \rho q\,\frac{\mathrm{d}q}{\mathrm{d}\rho}-\frac{1}{2}\,q^{2}-\rho+1=\frac{\xi }{\rho^{2}}\,q. \end{equation}
\begin{equation} \rho q\,\frac{\mathrm{d}q}{\mathrm{d}\rho}-\frac{1}{2}\,q^{2}-\rho+1=\frac{\xi }{\rho^{2}}\,q. \end{equation}
In terms of  $( q,\rho )$, boundary condition (3.62) becomes
$( q,\rho )$, boundary condition (3.62) becomes
 \begin{equation} q=0\quad\text{at}\ \rho=1. \end{equation}
\begin{equation} q=0\quad\text{at}\ \rho=1. \end{equation}C.1. Numerical results
Observe that boundary-value problem (C3)–(C4) is singular at the boundary point, hence to solve it numerically, one needs to ‘step away’ from  $\rho =1$. This can be done using the asymptotic expansion
$\rho =1$. This can be done using the asymptotic expansion
 \begin{equation} q\sim\sum_{n=1}^{\infty}a_{n}( \rho-1)^{n}\quad\text{as} \ \rho\rightarrow1.\end{equation}
\begin{equation} q\sim\sum_{n=1}^{\infty}a_{n}( \rho-1)^{n}\quad\text{as} \ \rho\rightarrow1.\end{equation}
Substituting into (C3), one can calculate recursively as many coefficients  $a_{n}$ as one needs, e.g.
$a_{n}$ as one needs, e.g.
 \begin{equation} a_{1}=\frac{\xi-\sqrt{\xi^{2}+4}}{2},\quad a_{2}=\frac{4\xi a_{1}+a_{1}^{2} }{2\xi-6a_{1}},\quad \ldots. \end{equation}
\begin{equation} a_{1}=\frac{\xi-\sqrt{\xi^{2}+4}}{2},\quad a_{2}=\frac{4\xi a_{1}+a_{1}^{2} }{2\xi-6a_{1}},\quad \ldots. \end{equation}
To rewrite (3.63) for  $A(\xi )$ in terms of
$A(\xi )$ in terms of  $( q,\rho )$, one should first integrate it by parts and use boundary conditions (3.62) and (3.64) to obtain
$( q,\rho )$, one should first integrate it by parts and use boundary conditions (3.62) and (3.64) to obtain
 \begin{equation} A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}^{4}}\left( \frac{\mathrm{d} \rho_{2}}{\mathrm{d}z}\right)^{2}\mathrm{d}z. \end{equation}
\begin{equation} A=\int_{-\infty}^{+\infty}\frac{1}{\rho_{2}^{4}}\left( \frac{\mathrm{d} \rho_{2}}{\mathrm{d}z}\right)^{2}\mathrm{d}z. \end{equation}Rewriting this integral in terms of variables (C2), one obtains
 \begin{equation} A=\int_{1}^{+\infty}\frac{q(\rho)}{\rho^{4}}\,\mathrm{d}\rho,\end{equation}
\begin{equation} A=\int_{1}^{+\infty}\frac{q(\rho)}{\rho^{4}}\,\mathrm{d}\rho,\end{equation}
where  $q(\rho )$ is determined by (C3).
$q(\rho )$ is determined by (C3).
Boundary-value problem (C3)–(C4) was solved numerically by calculating 7 terms of series (C5) to set up a boundary condition at  $\rho =1.1$ (a smaller number of terms was found to be insufficient for large
$\rho =1.1$ (a smaller number of terms was found to be insufficient for large  $\xi$). Then the solution was shot towards
$\xi$). Then the solution was shot towards  $\rho \rightarrow +\infty$ using the MATLAB function ‘ode23t’ (all other MATLAB functions for ODEs were found to be slow and inaccurate for large
$\rho \rightarrow +\infty$ using the MATLAB function ‘ode23t’ (all other MATLAB functions for ODEs were found to be slow and inaccurate for large  $\xi$). Finally, the computed solution was substituted into (C8), which was evaluated via Simpson's rule.
$\xi$). Finally, the computed solution was substituted into (C8), which was evaluated via Simpson's rule.
C.2. Asymptotic results
As discussed in the main body of the paper, the classical and generalised models do not differ much for  $\xi \sim 1$. Thus the asymptotic limit
$\xi \sim 1$. Thus the asymptotic limit  $\xi \rightarrow \infty$ is of particular importance.
$\xi \rightarrow \infty$ is of particular importance.
If  $\xi \gg 1$ and
$\xi \gg 1$ and  $\rho \sim 1$, then the first two terms in (C3) can be omitted, and one obtains
$\rho \sim 1$, then the first two terms in (C3) can be omitted, and one obtains
 \begin{equation} q\approx\xi^{{-}1}\rho^{2}( -\rho+1) .\end{equation}
\begin{equation} q\approx\xi^{{-}1}\rho^{2}( -\rho+1) .\end{equation}The omitted terms become important (and the above solution inapplicable) if
 \begin{equation} \rho\gtrsim\xi^{2/5}. \end{equation}
\begin{equation} \rho\gtrsim\xi^{2/5}. \end{equation}
To find the solution for large  $\rho$, introduce
$\rho$, introduce  $( q^{\prime },\rho ^{\prime })$ such that
$( q^{\prime },\rho ^{\prime })$ such that
 \begin{equation} q=\xi^{1/5}\rho^{\prime},\quad\rho=\xi^{2/5}\rho^{\prime}.\end{equation}
\begin{equation} q=\xi^{1/5}\rho^{\prime},\quad\rho=\xi^{2/5}\rho^{\prime}.\end{equation}In terms of the new variables, (C3) becomes, to leading order,
 \begin{equation} \rho^{\prime}q^{\prime}\,\frac{\mathrm{d}q^{\prime}}{\mathrm{d}\rho^{\prime} }-\frac{1}{2}\,q^{\prime2}-\rho^{\prime}=\frac{q^{\prime}}{\rho^{\prime2} },\end{equation}
\begin{equation} \rho^{\prime}q^{\prime}\,\frac{\mathrm{d}q^{\prime}}{\mathrm{d}\rho^{\prime} }-\frac{1}{2}\,q^{\prime2}-\rho^{\prime}=\frac{q^{\prime}}{\rho^{\prime2} },\end{equation}and its solution matches (C9) only if
 \begin{equation} q^{\prime}\sim{-}\rho^{\prime3}+{O}(\rho^{\prime8})\quad\text{as} \ \rho\rightarrow0.\end{equation}
\begin{equation} q^{\prime}\sim{-}\rho^{\prime3}+{O}(\rho^{\prime8})\quad\text{as} \ \rho\rightarrow0.\end{equation}
The composite solution, valid for all  $\rho$, is
$\rho$, is
 \begin{equation} q=\frac{\rho^{2}( \rho-1) }{\xi}+\xi^{1/5}q^{\prime}(\xi ^{{-}2/5}\rho)-\frac{\rho^{3}}{\xi}, \end{equation}
\begin{equation} q=\frac{\rho^{2}( \rho-1) }{\xi}+\xi^{1/5}q^{\prime}(\xi ^{{-}2/5}\rho)-\frac{\rho^{3}}{\xi}, \end{equation}
where the first term is the finite- $\rho$ solution (C9), the second term reflects scaling (C11) for large
$\rho$ solution (C9), the second term reflects scaling (C11) for large  $\rho$, and the last term is the common part of the two solutions. Substituting the above expression into (C8), one obtains
$\rho$, and the last term is the common part of the two solutions. Substituting the above expression into (C8), one obtains
 \begin{equation} A={-}\frac{1}{\xi}-\frac{1}{\xi}\int_{\xi^{{-}1/5}}^{\infty}\frac{q^{\prime} (\rho^{\prime})}{\rho^{\prime4}}\, \mathrm{d}\rho^{\prime}.\end{equation}
\begin{equation} A={-}\frac{1}{\xi}-\frac{1}{\xi}\int_{\xi^{{-}1/5}}^{\infty}\frac{q^{\prime} (\rho^{\prime})}{\rho^{\prime4}}\, \mathrm{d}\rho^{\prime}.\end{equation}
One might be tempted now to take the limit  $\xi \rightarrow \infty$, but the integral in (C15) is actually divergent (due to asymptotics (C13)). One can still use (C13) to express this integral through a convergent one:
$\xi \rightarrow \infty$, but the integral in (C15) is actually divergent (due to asymptotics (C13)). One can still use (C13) to express this integral through a convergent one:
 \begin{equation} \int_{\xi^{{-}1/5}}^{\infty}\frac{q^{\prime}(\rho^{\prime})}{\rho^{\prime4} }\,\mathrm{d}\rho^{\prime}={-}\frac{2}{5}\ln\xi-\int_{\xi^{{-}1/5}}^{\infty}\ln \rho^{\prime}\,\frac{\mathrm{d}}{\mathrm{d}\rho^{\prime}}\left( \frac{q^{\prime}}{\rho^{\prime3}}\right) \mathrm{d}\rho^{\prime}. \end{equation}
\begin{equation} \int_{\xi^{{-}1/5}}^{\infty}\frac{q^{\prime}(\rho^{\prime})}{\rho^{\prime4} }\,\mathrm{d}\rho^{\prime}={-}\frac{2}{5}\ln\xi-\int_{\xi^{{-}1/5}}^{\infty}\ln \rho^{\prime}\,\frac{\mathrm{d}}{\mathrm{d}\rho^{\prime}}\left( \frac{q^{\prime}}{\rho^{\prime3}}\right) \mathrm{d}\rho^{\prime}. \end{equation}
Now take the limit  $\xi \rightarrow \infty$, so that (C15) becomes
$\xi \rightarrow \infty$, so that (C15) becomes
 \begin{equation} A\approx\frac{1}{\xi}\left[{-}1+\frac{2}{5}\ln\xi+\int_{0}^{\infty}\ln \rho^{\prime}\,\frac{\mathrm{d}}{\mathrm{d}\rho^{\prime}}\left( \frac{q^{\prime}}{\rho^{\prime3}}\right) \mathrm{d}\rho^{\prime}\right].\end{equation}
\begin{equation} A\approx\frac{1}{\xi}\left[{-}1+\frac{2}{5}\ln\xi+\int_{0}^{\infty}\ln \rho^{\prime}\,\frac{\mathrm{d}}{\mathrm{d}\rho^{\prime}}\left( \frac{q^{\prime}}{\rho^{\prime3}}\right) \mathrm{d}\rho^{\prime}\right].\end{equation}
The integral in this expression does not involve  $\xi$, hence it can be evaluated ‘once and for all’. This was done by solving boundary-value problem (C12)–(C13) numerically and substituting the resulting solution into (C17), which yields
$\xi$, hence it can be evaluated ‘once and for all’. This was done by solving boundary-value problem (C12)–(C13) numerically and substituting the resulting solution into (C17), which yields
 \begin{equation} A\approx\frac{\frac{2}{5}\ln\xi-0.81571}{\xi}.\end{equation}
\begin{equation} A\approx\frac{\frac{2}{5}\ln\xi-0.81571}{\xi}.\end{equation}C.3. Curve-fitting results
The large- $\xi$ asymptotics (C18) can be used to derive an approximation of
$\xi$ asymptotics (C18) can be used to derive an approximation of  $A(\xi )$ for all
$A(\xi )$ for all  $\xi$. To do so, assume that
$\xi$. To do so, assume that
 \begin{equation} A\approx\frac{\frac{2}{5}\ln( \xi+\xi_{0}) -0.81571}{\xi+\xi_{0} }+\frac{A_{0}}{(\xi+\xi_{0})^{7/5}},\end{equation}
\begin{equation} A\approx\frac{\frac{2}{5}\ln( \xi+\xi_{0}) -0.81571}{\xi+\xi_{0} }+\frac{A_{0}}{(\xi+\xi_{0})^{7/5}},\end{equation}
where  $\xi _{0}$ and
$\xi _{0}$ and  $A_{0}$ are undetermined constants; the former regularises asymptotics (C18) at
$A_{0}$ are undetermined constants; the former regularises asymptotics (C18) at  $\xi =0$, and the latter improves the accuracy for large
$\xi =0$, and the latter improves the accuracy for large  $\xi$. (Note that a term
$\xi$. (Note that a term  ${\sim }\xi ^{-7/5}$ would appear as a correction to the leading-order solution if (C18) were extended to higher orders.)
${\sim }\xi ^{-7/5}$ would appear as a correction to the leading-order solution if (C18) were extended to higher orders.)
The coefficients  $\xi _{0}$ and
$\xi _{0}$ and  $A_{0}$ were fixed by fitting (C19) to the corresponding numerical result for the range
$A_{0}$ were fixed by fitting (C19) to the corresponding numerical result for the range  $\xi \in [ 0,10^{5}]$, using the MATLAB function ‘fit’. The resulting values are those in (1.4) of the Introduction. The relative error of (1.4) within the range
$\xi \in [ 0,10^{5}]$, using the MATLAB function ‘fit’. The resulting values are those in (1.4) of the Introduction. The relative error of (1.4) within the range  $\xi \in [ 0,10^{5}]$ is approximately
$\xi \in [ 0,10^{5}]$ is approximately  $0.8\,\%$.
$0.8\,\%$.
































 Open access
Open access