


“Omics” (genomics, transcriptomics, proteomics, and metabolomics) encompass multiple sets of biological molecules, including DNAs, RNAs, proteins, and metabolites. Advances in the identification and use of omics-based biomarkers have facilitated stratification of patients based on risk. They have allowed improved prognosis, treatment selection, and measurement of response to treatment and outcomes for different diseases, especially in oncology. Among the molecular diagnostic or prognostic tests that have been developed, multi-analyte assays with algorithmic analyses (MAAAs) explore multiple biomarkers in conjunction with clinical data. MAAA applies multi-parametric statistical algorithms to individual biomarkers, thus improving the positive and negative predictive value of a diagnostic or prognostic test. Diagnostic MAAAs help to determine whether a patient has a disease at the time the test is performed, while prognostic MAAAs predict the aggressiveness and the likelihood of relapse to determine the best management for the patient (personalized medicine). Some MAAAs may be used for both diagnostic and prognostic purposes.
MAAAs and other molecular diagnostic algorithms are regulated as in vitro diagnostic (IVD) medical devices (Reference Henschke, Panteli, Perleth and Busse1) in both Europe and the USA Medical device regulation is not formally centralized in Europe, and the IVD rules, under review since 2012 (Reference Garrido, Kristensen, Nielsen and Busse2), have only recently been promulgated (3). As a result, the legal context surrounding MAAAs is not yet well-defined. The situation is also unclear in the United States, where regulatory supervision by the Food and Drug Administration (FDA) depends upon whether or not the test is developed by a laboratory (4).
While the field of molecular diagnostics has grown rapidly, the use of such algorithms in clinical practice remains relatively limited (Reference Peabody, Shimkhada, Tong and Zubiller5). This may be due to a lack of awareness on the part of physicians, unproven utility in real world clinical settings as well as limited availability due to coverage/reimbursement issues. Health technology assessment (HTA) is key to the latter as it evaluates the clinical, economic, organizational, social, ethical, and legal issues of a health intervention to inform policy decision making including reimbursement by health systems (Reference Rosenkötter, Vondeling and Blancquaert6). However, developers of MAAAs may have difficulties in meeting the evidentiary standards used in HTA evaluations, such as demonstrating the existence of a relationship between test results (clinical validity) and improved patient outcomes (clinical utility).
We undertook a review of the literature to explore methods and challenges for the assessment of MAAAs to identify the criteria that could be used for reimbursement purposes. The objectives of this review were to identify existing evaluation frameworks used for MAAAs, identify MAAAs that have undergone HTA evaluations to date, undertake an analysis of the most widely evaluated MAAAs and synthesize the particular challenges that MAAAs present to HTA bodies.
METHODS
We undertook a scoping review of the scientific and gray literature available in English and French (reports in other languages were summarily explored), structured around a five-phase framework for scoping reviews: (i) identifying the research question; (ii) identifying relevant studies; (iii) study selection; (iv) collating, summarizing, and reporting the results; (v) consultation (Reference Armstrong, Hall, Doyle and Waters7).
Three researchers independently searched through the literature to refine the research question and identify key concepts and terminology, key studies, adapted assessment tools, models, and published assessments of MAAA diagnostic/prognostic tests. Keywords such as MAAAs, omics, assays with algorithmic analyses, biomarkers, genetic tests, combined with evaluation, technology assessment were used in the scoping search. A fourth senior researcher then reviewed the outputs, which are summarized in this report.
Identification of MAAAs That Have Been Evaluated by Leading HTA or Insurer/Reimbursement Bodies
A snowball sampling approach was used to identify MAAAs and to expand to additional sources as outlined in Figure 1. Five different starting points were used consisting of tests identified in two review articles analyzing coverage policies (Reference Peabody, Shimkhada, Tong and Zubiller5;Reference Hresko and Haga8), tests listed by the FDA (9) and by the Tufts Evidence-based Practice Center under contract to the USA Agency for Healthcare Research and Quality (Reference Raman, Avendano and Chen10), as well as tests identified by a scoping search performed from February to March 2016 using PubMed and Google.

Figure 1. Identification of multi-analyte assays with algorithmic analyses (MAAAs) that have been evaluated by at least one health technology assessment (HTA) or insurer/reimbursement body. ODX, Oncotype DX.
Tests Inclusion/Exclusion Criteria
Only multi-analyte, non-companion (stand-alone) diagnostic or prognostic tests that (i) provide actionable results (i.e., can lead to changes in the clinical management of patients), (ii) concern a chronic disease, and (iii) have been evaluated by leading HTA bodies (up to September 2017) were included. We excluded companion diagnostics because they are developed in the context of Phase II or Phase III drug trials and are evaluated under the regulatory scheme for drugs rather than in vitro medical devices.
Identification of Evaluated MAAAs
Preliminary searches revealed that the most widely MAAA assessed was Oncotype DX (ODX). As a result, we initially identified institutions performing reimbursement and coverage decisions for ODX. The websites of these institutions were searched for reports on other MAAAs. To be as inclusive as possible, the websites of the HTA bodies listed in the directory of the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) were searched (11). The assumption was that, in this way, we would not only identify existing reports, but also we would be able to obtain more documentation on the assessments performed. Only reports in English and French were included, although reports in other languages were also summarily explored. This search was completed by examining reports generated by USA and Canadian healthcare providers’ organizations and insurance companies.
RESULTS
Scoping Review
We identified seventy-two potentially relevant reports/articles in our scoping search. We excluded forty-seven items that did not meet our inclusion criteria. This left twenty-five HTA reports evaluating seventeen MAAAs (Figure 2).
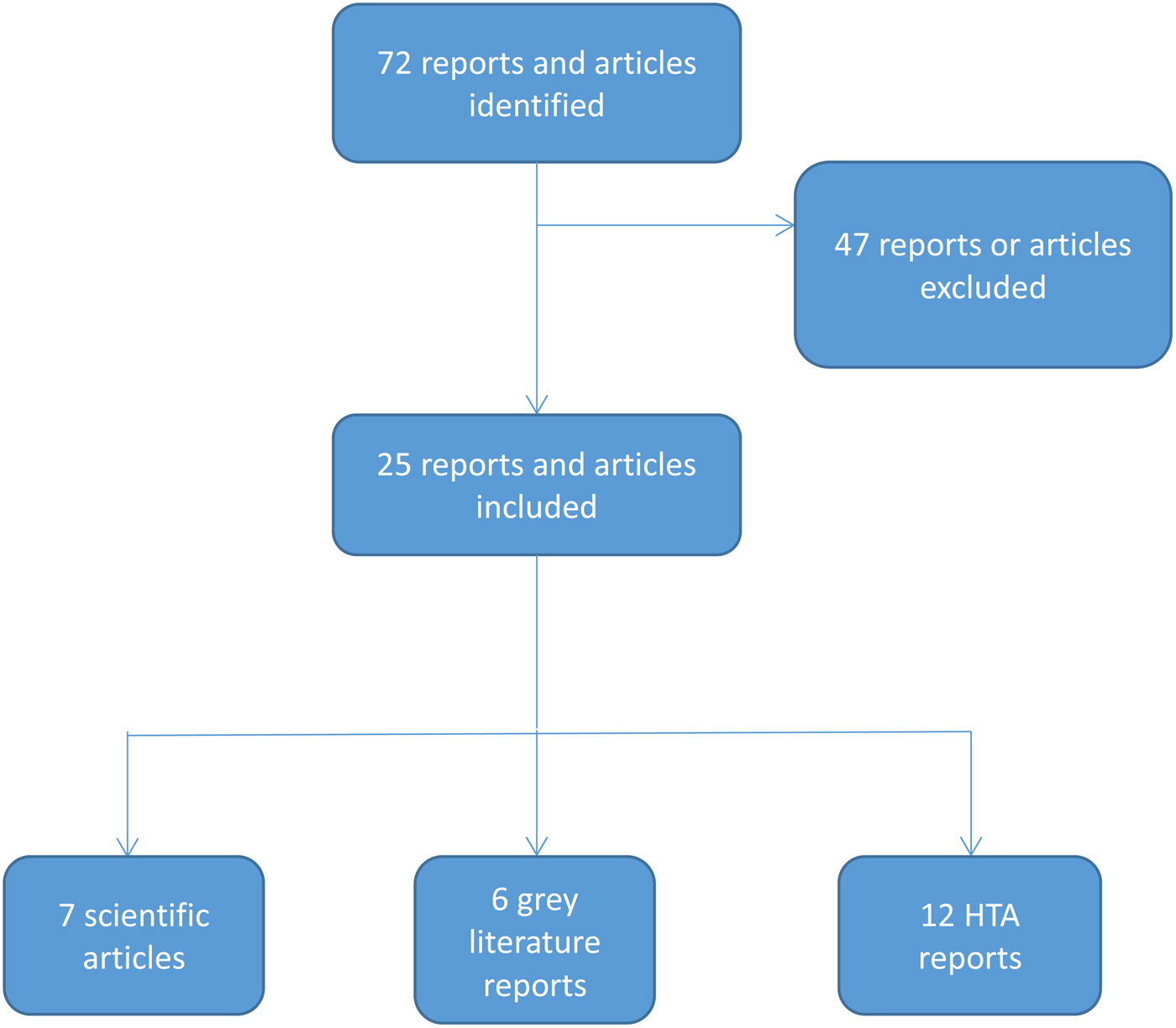
Figure 2. Flowchart of the scoping review. FDA, United States Food and Drug Administration; HTA, health technology assessment; ISPOR, International Society for Pharmacoeconomics and Outcomes Research; ODX, Oncotype DX.
Frameworks for Evaluating Molecular Diagnostic Tests
We identified two main frameworks used in HTA evaluations of diagnostic tests including MAAAs: the EUnetHTA Core Model® for diagnostics and the EGAPP's ACCE framework. Other organizations and initiatives developed guidelines and models, such as the PHGEN II European network (Reference Brand and Lal12). The Canadian Agency for Drugs and Technologies in Health (CADTH) also published a report in 2012 identifying the main evaluation frameworks that were used internationally and outlining some of the most commonly used criteria (Reference Morrison and Boudreau13).
Among the concepts used, analytic validity corresponds to the capacity of a test to precisely and reliably measure a genotype in the laboratory (in vitro) to predict a clinical disorder or phenotype of interest, such as overall survival. It includes sensibility, specificity, reliability, and reproducibility. It also encompasses positive and negative predictive values and disease prevalence. Clinical utility measures the ability of a test to improve patient care using clinical outcomes to assess the value for patient care. Finally, the ethical, legal, and social implications consider what impediments exist and what safeguards are required.
EUnetHTA Core Model®
The main aim of the EUnetHTA Core Model® is to facilitate collaboration between European and international HTA agencies by means of an HTA ontology containing an extensive list of generic questions, methodological guidance to help researchers find answers to these questions, and a common reporting structure providing a standardized format for the output of HTA projects. The third revision of the EUnetHTA Core Model® published in 2016 (14) is structured according to domains, topics, and issues.
Initially developed for medical and surgical therapeutic applications, it has been adapted for diagnostic and screening purposes. An evaluation of breast cancer MAAAs was undertaken by EUnetHTA using the Core Model framework (see section 3.3). By its own admission, the EUnetHTA Core Model® is not well adapted to prognostic tests, which include several MAAAs currently available. A study produced by EUnetHTA in 2015 discusses the study design that could be applied to personalized medicine technologies to ensure that the evidence available is at the same level as that of other health technologies (15).
The ACCE Framework
In 2000, the ACCE framework—Analytical validity; Clinical validity; Clinical utility; and Ethical, legal, and social aspects—was developed by the USA Office of Public Health Genomics (OHPG). Between 2005 and 2013, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP™) working group undertook evidence reviews and published eight recommendations based on the ACCE framework (16).
Since 2013, the USA Centers for Medicare and Medicaid Services (CMS) has outsourced the assessment of molecular diagnostic tests, including MAAAs. Assessments under the MolDx program developed in 2011 by Palmetto GBA, a CMS Medicare Administrative Contractor, are based on the ACCE framework (Reference Palmetto17). The technology assessment framework summarizes all evidence of clinical validity and clinical utility when seeking CMS coverage and reimbursement (Reference Peabody, Shimkhada, Tong and Zubiller5).
The EGAPP working group based the ACCE framework on forty-four questions related to five main criteria: analytical validity, clinical validity and clinical utility, as well as ethical, legal, and social aspects (Reference Teutsch, Bradley and Palomaki18).
In 2013, the working group initiative undertook a review of its work (16), examining the quality of evidence and the particular challenges in evaluating molecular tests. They found that evidence on analytic validity and clinical validity was sparse, that reviews were time-consuming and expensive compared with the rapid evolution of genomic technologies, that modeling could be useful in estimating the benefits and harms of genomic technologies, and that greater stakeholder involvement was necessary in developing evaluation recommendations. The group, therefore, proposed that rapid ACCE reviews taking less than 8 weeks and costing less than $20,000, using panels of experts and independent reviewers, may be suitable for topics lacking a large evidence base (Reference Kroese, Elles and Zimmern19).
The ACCE model has been adopted and adapted by several European bodies, including the UK Genetic Testing Network (GTN) (20), the European Commission project EuroGentest (21), and the British PHG Foundation (22;Reference Burke and Zimmern23).
MAAA Tests That Have Been Evaluated by HTA or Insurer/Reimbursement Bodies
Seventeen MAAAs found in twenty-five reports met our criteria (Table 1). Thirteen of the seventeen tests were for cancer indications, most frequently breast cancer: ODX (HTA assessments found in fourteen countries) and MammaPrint (assessed in six countries). MAAAs targeting four noncancer indications, diabetes, obstructive coronary artery disease (CAD), heart transplant, and rheumatoid arthritis (RA), were also identified. Assessments for these noncancer MAAAs were only found in the United States. Overall, ten of the seventeen tests have been assessed only by HTA bodies in the United States.
Table 1. MAAAs Evaluated by at Least One HTA or Insurer/Reimbursement Body

AU, Australia; BE, Belgium; CA, Canada; CH, Switzerland; DE, Denmark; ES, Spain; FISH, fluorescence in situ hybridization; FR, France; GR, Germany; IE, Ireland; IHC, immunohistochemistrty; IL, Israel; IS, Iceland; IT, Italy; NL, The Netherlands; NZ, New Zealand; RT-PCR, reverse transcriptase polymerase chain reaction; UK, United Kingdom; USA, United States.
To further explore the critical elements considered by HTA bodies in assessing MAAAs, we undertook a closer analysis of the most widely evaluated MAAA: ODX for breast cancer.
Evaluations of ODX/MammaPrint for Breast Cancer Carried on by HTA or Insurer/Reimbursement Bodies
The ODX algorithm generates a recurrence score (RS) that identifies the likelihood of distant recurrence of breast cancer as low risk (RS <18), intermediate risk (RS 18–30), and high risk (RS ≥31). It is designed to better target chemotherapy to higher risk patients by estimating baseline risk and response to chemotherapy.
We examined HTA evaluations of ODX by nine HTA bodies in five countries, identifying the methods and criteria applied, with a particular focus on clinical utility and economic analysis (Table 2).
Table 2. Criteria Used by HTA or Insurer/Reimbursement Bodies in Evaluating Oncotype DX (Breast Cancer)

EGAPP, Evaluation of Genomic Applications in Practice and Prevention; HTA, health technology assessment; NCPE, National Centre for Pharmacoeconomics; NICE, National Institute for Health and Care Excellence; UK, United Kingdom; USA, United States.
In terms of clinical utility, the greatest challenge identified was the lack of direct evidence of improved health outcomes. While the impact on clinical decision making was emphasized, with moderate evidence showing that ODX led to changes in treatment decisions, none of the assessments examined both physician decision-making and downstream health outcomes for patients. Indirect evidence of clinical utility was based on the correlation between the risk of disease recurrence and thus the likelihood of chemotherapy benefit.
Cost-effectiveness of this MAAA test was assessed by five HTA bodies, each of which relied upon Markov models adopting the payer's perspective.
Three HTA bodies used Budget Impact Analysis (BIA) as part of their economic analyses. Overall, the budget impact depended on whether the test was made available to all breast cancer patients or to a risk-specific subgroup.
The studies consistently showed an overall shift to less-intensive treatment recommendations as a result of ODX. While lower use of chemotherapy would result not only in reduced costs but also presumably in lower exposure to harms, none of the studies followed patients to assess the overall balance of clinical benefits and harms. However, some evaluations considered the effect of ODX on decisional conflict (i.e., whether use of the test increased patients’ confidence in the treatment decision).
In 2012, EUnetHTA assessed prognostic tests for breast cancer recurrence and stressed the fundamental differences between diagnostic and prognostic tests. The first version of the EUnetHTA Core Model was considered insufficient and not suited to prognostic technologies (24).
In January 2018, the National Institute for Health and Care Excellence (NICE) (25) released for public consultation an evidence overview regarding the tumor profiling tests for breast cancer (EndoPredict, MammaPrint, Oncotype DX Breast Recurrence Score, Prosigna, and IHC4+C). The outcomes used for the assessment of clinical and cost-effectiveness were: (i) Intermediate measures: time to test results, analytical validity, prognostic ability, ability to predict benefit from chemotherapy, impact of test results on decision making. (ii) Clinical outcomes: disease-free survival, overall survival, distant recurrence, disease-related morbidity and mortality, chemotherapy-related morbidity and mortality. (iii) Patient-reported outcomes: health-related quality of life, anxiety. (iv) Costs were considered from an NHS and Personal Social Services perspective and included: costs of treating breast cancer, including drug cost, administration cost, outpatient appointments, and treatment of adverse events, costs of the tests, including equipment costs and reagents when relevant, costs of staff and associated training. (v) The cost-effectiveness measure by incremental cost per quality-adjusted life year.
In February 2018, EUnetHTA released an assessment report regarding the added value of using MammaPrint for adjuvant chemotherapy in early breast cancer (26). The authors determined that MammaPrint testing has not yet demonstrated improved patient outcomes due to withholding adjuvant chemotherapy and thus its clinical utility has not been proven.
Analysis of Assessments of Non-cancer MAAAs Conducted by HTA or Insurer/Reimbursement Bodies
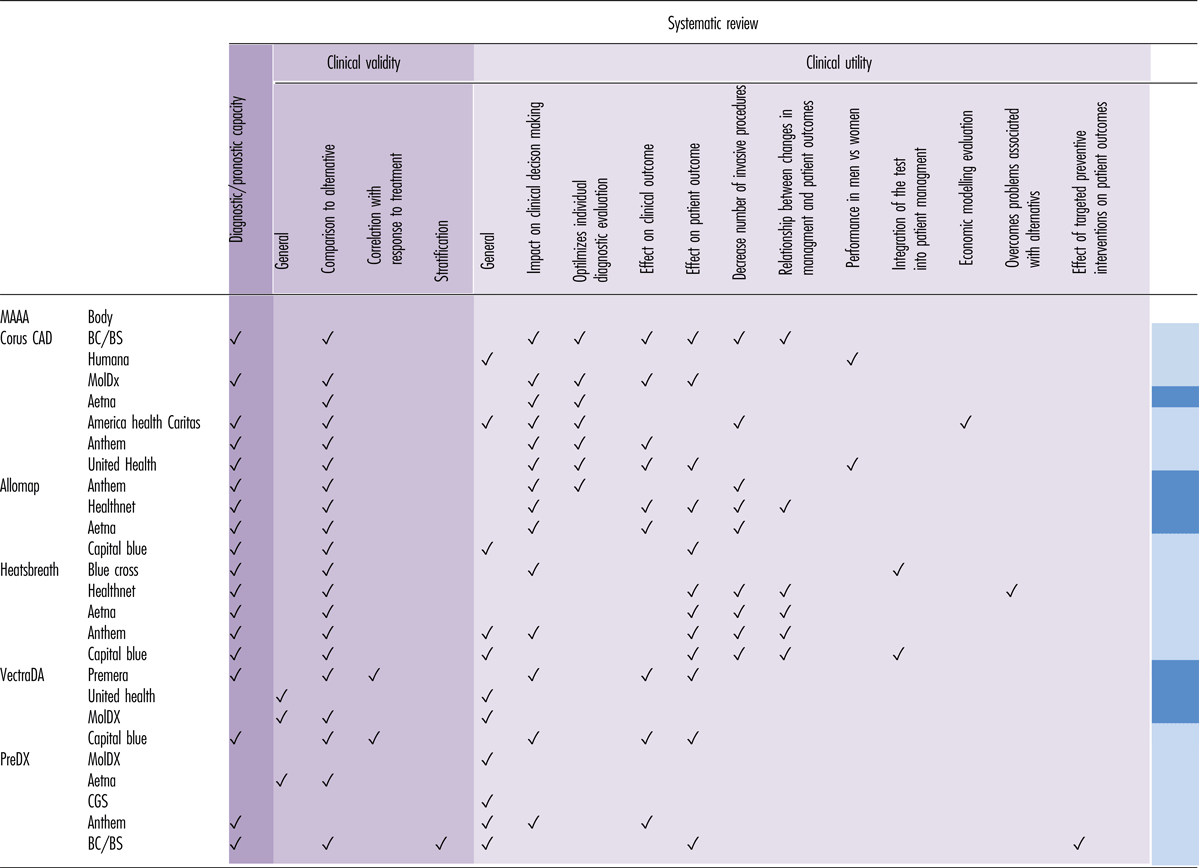
The evaluations of non-cancer MAAAs (ncMAAAs) focused on clinical utility, clinical validity, or both (Table 3). The ncMAAAs related to very different diseases and had disparate objectives: Corus CAD was used in the diagnosis of CAD, Allomap in the diagnosis and prognosis of acute cellular heart transplant rejection, Vectra DA as a measurement for RA disease progression, and PreDX to stratify patients into low and high risk for developing diabetes.
Table 3. Summary of the General Characteristics of the Studies Cited by HTAs or Insurer/Reimbursement Bodies in Their Evaluation of MAAAs
![]() Investigational
Investigational ![]() Medical necessary
Medical necessary
BC/BS, Blue Cross/Blue Shield; CGS, ; HTA, health technology assessment; MAAA, multi-analyte assays with algorithmic analyses.
HTA bodies evaluated the clinical utility of the ncMAAAs by the impact on clinical decision making/management and on patient outcomes, as well as the relationship between changes in management and patient outcomes, such as net reclassification improvement or stratification. All HTA bodies considered the ability of ncMAAAs to diagnose or predict future outcomes.
Clinical validity was most often evaluated by comparing the performance of ncMAAAs with alternative tests in current clinical practice. Comparators of Corus CAD included clinical predictors and imaging technologies. Allomap and Heartsbreath were compared with the current diagnostic gold standard; the disease activity measure Vectra DA was compared with patient-reported and activity measures of disease progression, while PreDX was compared with current diabetes risk scores.
The HTA bodies identified several limitations regarding the studies supporting the ncMAAAs. They were critical of industry sponsorship of studies, small sample sizes, lack of controls and the nature of the control groups used (e.g., in the case of Corus CAD, lack of paired anatomic studies or the use of historical controls was criticized), limited follow-up times and thus evaluation of short-term outcomes, as well as the inclusion/exclusion criteria that resulted in study populations potentially having biased disease prevalence or pretest probability (e.g., in the case of Allomap, only patients who had received a cardiac transplant more than 6 months previously, and who were at a lower risk of rejection compared with the first 6 months following transplantation, were eligible).
Upon comparison with alternatives, other limitations were identified: the reported sensitivity of a comparator of Corus CAD in some studies was considerably lower than generally reported in the literature, and in the case of VectraDA, only moderate level of agreement above chance with alternative scores of disease progression was cited. In terms of the impact on clinical decision making, the lack of standardized use of PreDx for clinical decision-making and the fact that the choices of interventions were left up to physician and/or patient discretion, meant that it could not be determined whether observed changes would have occurred in the absence of any intervention or with only routine medical care.
The limitations identified by the HTA bodies regarding the different ncMAAAs did not predict whether a particular test was deemed investigational (and thus not reimbursable) or medically necessary. The cost-effectiveness of the ncMAAAs and their effect on preventive interventions and patient outcomes was given little attention. None of the HTA evaluations of the ncMAAAs mentioned any economic evaluation or meta-analysis.
DISCUSSION
We described the assessment processes for MAAAs mostly in Europe and North America. To our knowledge, it is the first review contributing to the discussion on the pertinent criteria for MAAA assessment and providing insights for clinical research and evidence development.
Among the difficulties encountered in identifying which HTA bodies had evaluated the MAAAs was the fact that assessments were not always undertaken by the national HTA body. For example, in France, the assessment of three breast cancer MAAAs was carried out initially by the national cancer institute (INCa), rather than the national HTA authority the Haute Autorité de Santé (HAS) (Reference Luporsi, Bellocq and Barrière27). The evaluation of one breast cancer MAAA was included in the 2016 working program of HAS (28).
Despite multiple search methods, the data may simply not have been available. In a recent study examining the U.S. payer coverage policies (Reference Chambers, Saret and Anderson29), authors found that payers less often cited clinical studies, systematic reviews, technology assessments, and cost-effectiveness analyses in their coverage policies for multi-gene panels than in their policies for other intervention types.
Another reason few HTA assessments of non-companion MAAAs were identified in Europe may be the fact that HTA bodies have not adapted their processes to accommodate such evaluations, although this appears to be changing. In the United Kingdom, the NICE has developed a Diagnostics Assessment Programme (DAP) that mainly focuses on stand-alone molecular diagnostics (30). DAP essentially follows the same approach as applied to drugs and measures patient benefit in quality-adjusted life-years. With the public consultation published in early 2018 (25), the NICE broadened its scope to include MAAAs.
In Germany, the German Institute for Quality and Efficiency in Health Care (IQWiG), expressly referenced MAAAs and recommended that the same evaluation principles as for diagnostic tests be applied (31). In Spain, a guideline for the evaluation of genetic based technologies was elaborated by the Andalusian Agency for Health Technology Assessment (AETSA) (Reference Márquez Calderón and Briones Pérez de la Blanca32). Indeed, these countries concurred in applying their assessment framework to MAAAs, with minor adaptations. Analytic validity is normally established as part of the market access process. However, in the case of stand-alone MAAAs, this may not take place before HTA evaluations, and HTA bodies must, therefore, consider questions related to the analytical validity of MAAAs as well.
Finally, the two frameworks identified in our review, EUnetHTA Core Model® and the ACCE framework, have not been until now adopted by key European national HTA bodies (e.g., NICE, HAS, IQWiG), although these bodies are evaluating clinical utility which “plays a key role in the vast majority of reimbursement decisions” (Reference Akhmetov and Bubnov33).
The concept of clinical utility for diagnostics has evolved in recent years, and the MolDX program used by Medicare in the United States is considered by some to be the de facto measure (Reference Palmetto34). In the assessment process of MolDX, organizational aspects must play an increasing role, especially linked with the external validity of algorithms. This aspect is taken into account in the EUnetHTA model but not in the ACCE framework developed by EGAPP. Other issues that have to be developed by the models are ethical ones, not least because of the genetic data that are communicated to manufacturers.
MAAAs are better-developed in cancer than in other disease areas. Nonetheless, even cancer test developers face challenges in providing evidence of clinical utility. In 2014, the USA Agency for Healthcare Research and Quality (AHQR) undertook a systematic review of molecular pathology tests that estimate the prognosis (recurrence) of common cancers to determine whether there was evidence that test results changed physician decision making and improved clinical outcomes for patients (Reference Meleth, Reeder-Hayes and Ashok35). In terms of clinical utility, no studies directly assessing the impact of a test on both physician decision making and downstream health outcomes were found. In terms of impact of tests in changing treatment decisions, moderate strength of evidence was found only for ODX Breast. This important point must be addressed in future research and must be a requirement of HTA agencies, namely providing strong evidence that links the use of the algorithm with the clinical outcomes for the patient in real life practice. Difficulty in producing final outcomes/endpoints could also be due to the existence of a confounding factor, namely the clinical effectiveness of the treatment proposed in the case of a positive test.
Not surprisingly, HTA bodies use different criteria in evaluating cancer MAAAs versus non-cancer MAAAs. Targeted use of chemotherapy resulting in reduced costs and side effects to patients was found to be a crucial element in the assessment process for ODX. No such single strong driving criteria was found in any of the evaluations of ncMAAAs. In addition, impact on clinical outcomes was a criterion sought by HTA bodies in their evaluations of ncMAAAs, whereas impact on patient anxiety was a parameter of interest only in evaluations of ODX. Common to evaluations of both cancer and non-cancer MAAAs was their impact on clinical decision making and on patient outcomes; of interest, the relationship between changes in patient management and improvement of patient outcomes was a parameter of interest mainly in the HTA evaluations of ncMAAAs.
Differences between algorithms for diagnostic and prognostic purposes were expected but not found. Indeed, the prognosis of a disease is determined by final results, and the best way to provide evidence is related to the final clinical outcomes. One explanation could be that, at this point in time, clinical research has provided few clinical endpoints to HTA bodies. Another could be that frameworks used are quite wide and differences in the precise criteria used for evaluating clinical utility were not accurately described in the HTA reports (Reference Chambers, Saret and Anderson29).
Neglecting the external validity of these algorithms could in part explain the limitations of their use in clinical practice toward the goal of personalized medicine. Regardless of the nature of the test being evaluated, the main problem encountered by HTA agencies for the assessment of clinical utility of diagnostic and prognostic tests was the lack of available data, which may be due to trial length and proprietary interests.
Economic evidence on MAAAs was scarce, and we found no consensus on whether existing economic evaluation methods were sufficient (Reference Garau, Towse, Garrison, Housman and Ossa36). Some of the methodological challenges pertaining to any diagnostic workup include the choice of comparator, perspective, and timeframe. Costing MAAAs may be difficult because of the need to collect a broad range of costs, often in a data-limited environment. Measuring the impact of MAAAs on clinical outcomes requires information on patients’ and clinicians’ behavior. Alternative metrics (e.g., personal utility) are underdeveloped and alternative approaches (patients reported outcomes and the measure of health and non-health outcomes) are underused (Reference Buchanan, Wordsworth and Schuh37).
In developing trial protocols for MAAAs in Europe, the potential challenges and considerations for future HTA evaluations must be considered (Reference Peabody, Shimkhada, Tong and Zubiller5). They include the ability to measure clinical utility and to estimate long-term costs as well as benefit from the inputs of new categories of stakeholders concerned by precision medicine. The HTA Core Model recently became accessible free of charge to allow development of the model and broaden its future applications (Reference Buchanan, Wordsworth and Schuh37;Reference Kristensen, Lampe and Wild38). MAAA assessments will be reinforced by new European regulations (3) designed to ensure that by May 26, 2022, increased requirements for clinical evidence (including an EU-wide coordinated procedure for authorization of multi-center clinical investigations), general safety, performance, and postmarketing surveillance are met.
The regulation also reinforces the criteria for designation and processes for oversight of Notified Bodies. This action was supported by PACITA and PerMed projects. The PerMed 2015 report (39) estimates that a Europe-wide process to evaluate and validate biomarkers, the development of new clinical trial designs adapted to these new approaches, and the integration of preclinical testing with innovative clinical trials may further improve the effectiveness of interventions.
In conclusion, the assessment of MAAAs that include “omics” is possible with current health technology assessment methods but requires some adjustments.
Stronger evidence is needed, especially on clinical utility, to link these algorithms with the clinical outcomes in real life practice. Existing economic evaluation methods appear to be insufficiently developed to evaluate MAAAs, although they continue to be applied in major systems.
CONFLICTS OF INTEREST
Dr. Barna has nothing to disclose. Ms. Cruz-Sanchez has nothing to disclose. Ms. Berg Brigham reports grants from European Commission, during the conduct of the study. C.T. Thuong has nothing to disclose. Dr. Kristensen has nothing to disclose. Dr. Durand-Zaleski reports personal fees from Roche, outside the submitted work.






 Open access
Open access