



Introduction
The human microbiome is a complex microbial community structure that resides at different body sites, namely skin, oral cavity, gastrointestinal tract (GIT), respiratory tract, and vagina. However, microbial diversity and richness vary across all body sites (Costello et al., Reference Costello, Lauber, Hamady, Fierer, Gordon and Knight2009; Human Microbiome Project Consortium, 2012) The community belongs to several domains of life, that is, bacteria, viruses, fungi, archaea, and protists (Shreiner et al., Reference Shreiner, Kao and Young2015; Sender et al., Reference Sender, Fuchs and Milo2016). Unlike bacterial species, others have been poorly studied for their role in human physiology (Matijašić et al., Reference Matijašić, Meštrović, Paljetak, Perić, Barešić and Verbanac2020). The extensively researched gut bacterial species outnumbers human body cells and genes by 10 and 100 times, respectively (Bull and Plummer, Reference Bull and Plummer2014). Its role in breakdown of complex carbohydrates into short-chain fatty acids (SCFAs) such as acetate, propionate, and butyrate, branched-chain amino acids, hydrolysis of polyphenols, and biosynthesis of Vitamin K and water-soluble B-vitamins is well explored (Magnúsdóttir et al., Reference Magnúsdóttir, Ravcheev, De Crécy-Lagard and Thiele2015; Rowland et al., Reference Rowland, Gibson, Heinken, Scott, Swann, Thiele and Tuohy2018; Sharma et al., Reference Sharma, Rodionov, Leyn, Tran, Iablokov, Ding, Peterson, Osterman and Peterson2019; Chandel et al., Reference Chandel, Somvanshi and Thakur2023).
The microbiome composition varies across different parts of the GIT with distinct community structures along the mucosal-lumen axis (Bäckhed et al., Reference Bäckhed, Fraser, Ringel, Sanders, Sartor, Sherman, Versalovic, Young and Finlay2012; Ruan et al., Reference Ruan, Engevik, Spinler and Versalovic2020), in different development stages of a particular individual (Rinninella et al., Reference Rinninella, Raoul, Cintoni, Franceschi, Miggiano, Gasbarrini and Mele2019), and among individuals (Human Microbiome Project Consortium, 2012; Rinninella et al., Reference Rinninella, Raoul, Cintoni, Franceschi, Miggiano, Gasbarrini and Mele2019). A healthy human gut microbiome is a stable community composed of a defined set of microbial species, which resist change or return to an equilibrium state following perturbation (Bäckhed et al., Reference Bäckhed, Fraser, Ringel, Sanders, Sartor, Sherman, Versalovic, Young and Finlay2012). It consists of a few phyla with a relatively higher abundance (Bacillota, Bacteroidota, Actinomycetota, and Pseudomonadota) as compared to several others (Fusobacteriota, Tenericutes, Spirochaetes, Cyanobacteria, Verrucomicrobia, and TM7) (Human Microbiome Project Consortium, 2012). Some of the highly abundant and/or prevalent genera include Bacteroides, Eubacterium, Faecalibacterium, Alistipes, Ruminococcus, Clostridium, Prevotella, Roseburia, and Blautia, and highly abundant species include Faecalibacterium prausnitzii, Oscillospira guillermondii, and Blautia obeum (Arumugam et al., Reference Arumugam, Raes, Pelletier, Paslier, Yamada, Mende, Fernandes, Tap, Bruls, Batto, Bertalan, Borruel, Casellas, Fernandez, Gautier, Hansen, Hattori, Hayashi, Kleerebezem, Kurokawa, Leclerc, Levenez, Manichanh, Nielsen, Nielsen, Pons, Poulain, Qin, Sicheritz-Ponten, Tims, Torrents, Ugarte, Zoetendal, Wang, Guarner, Pedersen, de Vos, Brunak, Doré, Weissenbach, Ehrlich and Bork2011; Piquer-Esteban et al., Reference Piquer-Esteban, Ruiz-Ruiz, Arnau, Diaz and Moya2022; Qin et al., Reference Qin, Li, Raes, Arumugam, Burgdorf, Manichanh, Nielsen, Pons, Levenez, Yamada, Mende, Li, Xu, Li, Li, Cao, Wang, Liang, Zheng, Xie, Tap, Lepage, Bertalan, Batto, Hansen, Le Paslier, Linneberg, Nielsen, Pelletier, Renault, Sicheritz-Ponten, Turner, Zhu, Yu, Li, Jian, Zhou, Li, Zhang, Li, Qin, Yang, Wang, Brunak, Doré, Guarner, Kristiansen, Pedersen, Parkhill, Weissenbach, Bork, Ehrlich and Wang2010; Ruan et al., Reference Ruan, Engevik, Spinler and Versalovic2020). They are also the core taxa of a healthy individual (Qin et al., Reference Qin, Li, Raes, Arumugam, Burgdorf, Manichanh, Nielsen, Pons, Levenez, Yamada, Mende, Li, Xu, Li, Li, Cao, Wang, Liang, Zheng, Xie, Tap, Lepage, Bertalan, Batto, Hansen, Le Paslier, Linneberg, Nielsen, Pelletier, Renault, Sicheritz-Ponten, Turner, Zhu, Yu, Li, Jian, Zhou, Li, Zhang, Li, Qin, Yang, Wang, Brunak, Doré, Guarner, Kristiansen, Pedersen, Parkhill, Weissenbach, Bork, Ehrlich and Wang2010). However, there is little consensus about how the taxonomic core microbiome should be quantified, as different researchers use different quantification metrics (Neu, Reference Neu2021). For instance, with 90% and 0.01% threshold of prevalence and relative abundance, respectively, only Faecalibacterium prausnitzii was observed as the core microbiome across Indian cohorts from multiple locations (Chandel et al., Reference Chandel, Somvanshi and Thakur2023). Moreover, the studies on inferring core gut microbiome have not fully captured the variability in microbiome composition due to various factors like geographical location, race, diet, lifestyle, and age.
Large-scale studies on human gut microbiomes have largely been from the U.S. and European countries (Human Microbiome Project Consortium, 2012). But if we look at India, it has the largest human population and is spread across six different physiographic regions, and has a huge diversity in habitat, lifestyle, ethnicity, and dietary habits, which makes the Indian gut microbiota an interesting community to study. While population-specific variations in gut microbial composition have earlier been reported (Yatsunenko et al., Reference Yatsunenko, Rey, Manary, Trehan, Dominguez-Bello, Contreras, Magris, Hidalgo, Baldassano, Anokhin, Heath, Warner, Reeder, Kuczynski, Caporaso, Lozupone, Lauber, Clemente, Knights, Knight and Gordon2012), a recent study captured the uniqueness of the Indian gut microbiome (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019). Not only a substantially large number (943,395) of unique genes were observed in Indian samples, but a few species belonging to genera Prevotella, Mitsuokella, Dialister, Megasphaera, and Lactobacillus were also found highly associated with the Indian population (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019).
Pulipati et al. (Reference Pulipati, Sarkar, Jakkampudi, Kaila, Sarkar, Unnisa, Reddy, Khan and Talukdar2020) recently analysed the features, and determinants of Indian gut microbiota and compared it with worldwide data (Pulipati et al., Reference Pulipati, Sarkar, Jakkampudi, Kaila, Sarkar, Unnisa, Reddy, Khan and Talukdar2020). However, the association of gut microbiota with human health and various infectious/noninfectious diseases in the Indian population has not been systematically reviewed. This review provides Indian population-specific characteristics of the gut microbiome at different developmental stages of life, discusses the factors that shape the gut microbiome, and their association with noninfectious and infectious diseases while comparing them with the findings or trends in global populations (Figure 1).
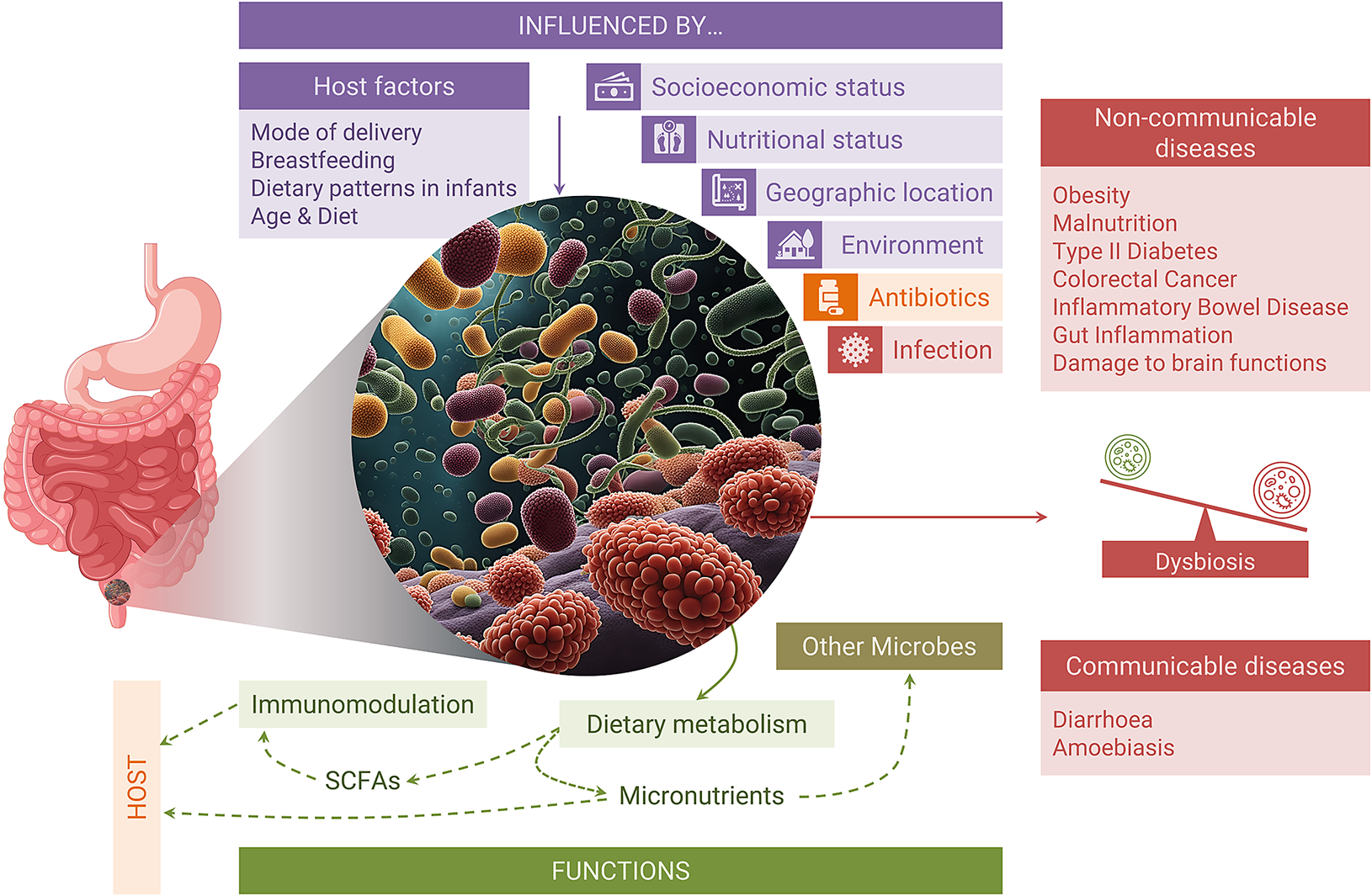
Figure 1. Pictorial representation of the key aspects discussed in this review article.
Establishment of gut microbiome
Pregnancy, birth, and infancy
The sterile womb hypothesis and microbial community acquisition from the external environment (Mackie et al., Reference Mackie, Sghir and Gaskins1999) were challenged when microbes were identified in the placenta, amniotic fluid, and meconium (Perez-Muñoz et al., Reference Perez-Muñoz, Arrieta, Ramer-Tait and Walter2017). It was further supported by the presence of phyla Bacillota, Pseudomonadota, and Bacteroidota and genera Enterococcus and Staphylococcus, in the meconium microbiome, which was majorly affected by maternal rather than perinatal factors (Jiménez et al., Reference Jiménez, Marín, Martín, Odriozola, Olivares, Xaus, Fernández and Rodríguez2008; Perez-Muñoz et al., Reference Perez-Muñoz, Arrieta, Ramer-Tait and Walter2017; Tapiainen et al., Reference Tapiainen, Paalanne, Tejesvi, Koivusaari, Korpela, Pokka, Salo, Kaukola, Pirttilä, Uhari and Renko2018). The similarity of the placental microbial community with the oral (Walker et al., Reference Walker, Clemente, Peter and Loos2017), and a higher dissimilarity with the vaginal and stool microbiome, were highly unlikely the result of contamination (Wassenaar and Panigrahi, Reference Wassenaar and Panigrahi2014; Walker et al., Reference Walker, Clemente, Peter and Loos2017; Cariño et al., Reference Cariño, Takayasu, Suda, Masuoka, Hirayama, Konishi and Umezaki2021).
A Finland-based study reported highly variable gut microbiota in T3 (third trimester of pregnancy) as compared to T1, resembling a rather disease-associated dysbiosis. The T3 stage also had a lower abundance of Faecalibacterium (butyrate producer) and a higher abundance of phyla Actinomycetota and Pseudomonadota. The Pseudomonadota has often been associated with inflammation-associated dysbiosis (Koren et al., Reference Koren, Goodrich, Cullender, Spor, Laitinen, Kling Bäckhed, Gonzalez, Werner, Angenent, Knight, Bäckhed, Isolauri, Salminen and Ley2012) (Figure 2). In contrast, there were no significant changes in the gut community structure of the Indian population between T1 and T3; although Pseudomonadota showed a higher abundance during T3, however, this difference was not statistically significant (Kumbhare et al., Reference Kumbhare, Patangia, Mongad, Bora, Bavdekar and Shouche2020). There were no reported adverse effects of higher Pseudomonadota in T3 on infants’ health. The difference in the findings was attributed to either a difference in data analysis or a smaller sample size of the Indian cohort (Kumbhare et al., Reference Kumbhare, Patangia, Mongad, Bora, Bavdekar and Shouche2020).
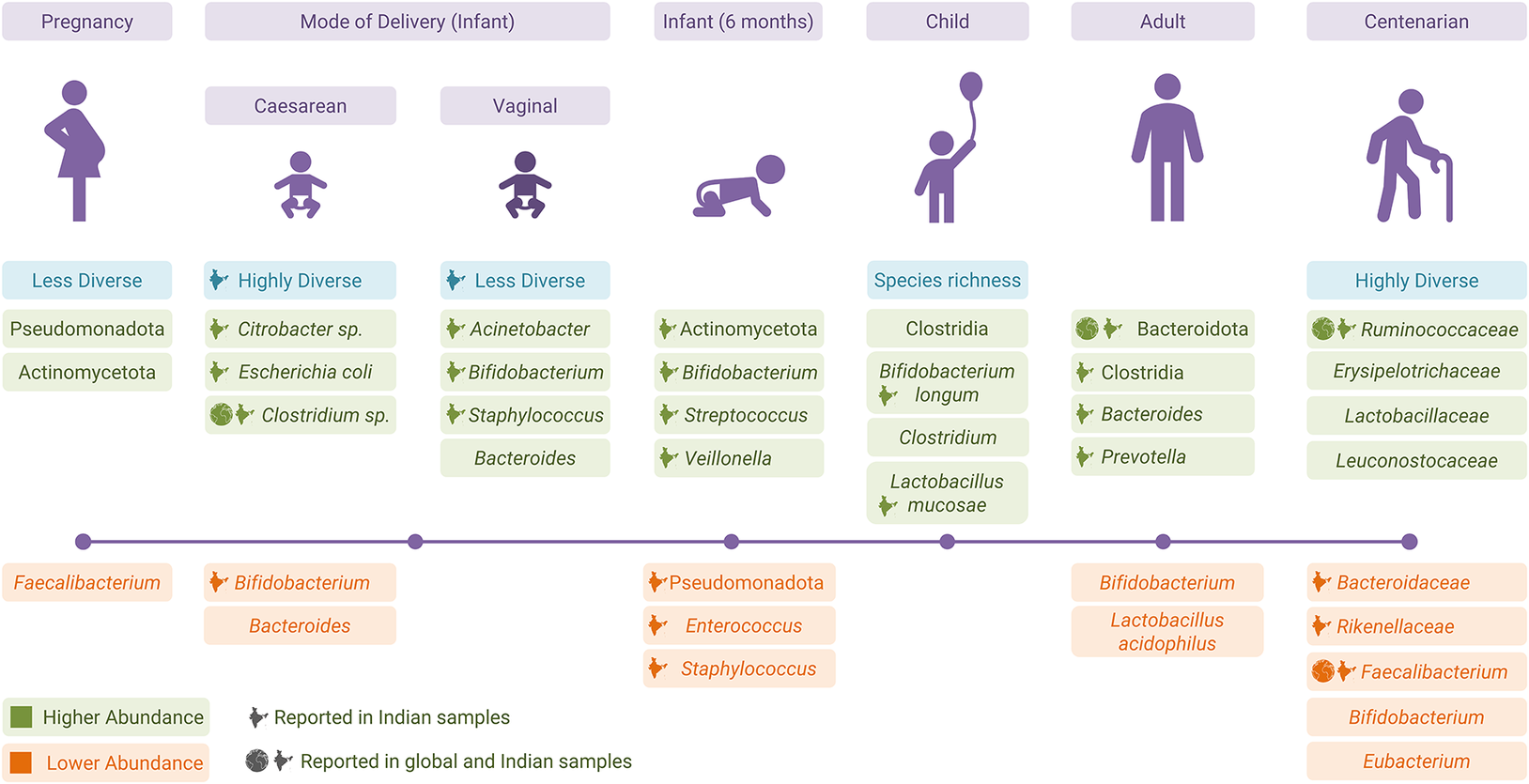
Figure 2. Changes in the gut microbiota from pregnancy to old age.
Mode of delivery, that is, caesarean section delivery (CS) and vaginal delivery (VD), has a strong influence on infants’ gut community. CS infants from Finland and the United States showed a delay in gut microbial community colonisation and reported a lower Bacteroides abundance as compared to VD infants (Grönlund et al., Reference Grönlund, Lehtonen, Eerola and Kero1999; Mitchell et al., Reference Mitchell, Mazzoni, Hogstrom, Bryant, Bergerat, Cher, Pochan, Herman, Carrigan, Sharp, Huttenhower, Lander, Vlamakis, Xavier and Yassour2020). The inverse correlation of Bacteroides with Streptococcus or Haemophilus in CS was the result of direct competition between the two species (Mitchell et al., Reference Mitchell, Mazzoni, Hogstrom, Bryant, Bergerat, Cher, Pochan, Herman, Carrigan, Sharp, Huttenhower, Lander, Vlamakis, Xavier and Yassour2020). Early colonisation of Bifidobacterium-like and Lactobacillus-like beneficial bacteria was seen in the VD children (Grönlund et al., Reference Grönlund, Lehtonen, Eerola and Kero1999). Corroborating the findings from Western countries, an Indian study reported higher Bifidobacterium – a primary coloniser in VD children along with Acinetobacter sp., Staphylococcus sp. (Pandey et al., Reference Pandey, Verma, Kumar, Bavdekar, Patole and Shouche2012). The absence of Bifidobacterium and a higher abundance of opportunistic bacteria (Citrobacter, Clostridium difficile, and E. coli) were seen in Indian CS infants (Pandey et al., Reference Pandey, Verma, Kumar, Bavdekar, Patole and Shouche2012) (Figure 2). The exposure of CS infants to environmental microbes makes them susceptible to colonisation of undesired microbes, which results in higher microbiome diversity (Pandey et al., Reference Pandey, Verma, Kumar, Bavdekar, Patole and Shouche2012).
Studies from Italy and the United States showed that the maternal microbiome from all body sites was the main source of the infant’s gut microbiome; however, the gut microbiome was more persistent compared to other body sites (Ferretti et al., Reference Ferretti, Pasolli, Tett, Asnicar, Gorfer, Fedi, Armanini, Truong, Manara, Zolfo, Beghini, Bertorelli, De Sanctis, Bariletti, Canto, Clementi, Cologna, Crifò, Cusumano, Gottardi, Innamorati, Masè, Postai, Savoi, Duranti, Lugli, Mancabelli, Turroni, Ferrario, Milani, Mangifesta, Anzalone, Viappiani, Yassour, Vlamakis, Xavier, Collado, Koren, Tateo, Soffiati, Pedrotti, Ventura, Huttenhower, Bork and Segata2018; Mitchell et al., Reference Mitchell, Mazzoni, Hogstrom, Bryant, Bergerat, Cher, Pochan, Herman, Carrigan, Sharp, Huttenhower, Lander, Vlamakis, Xavier and Yassour2020). Indian infants at 6 months of age had a higher abundance of phylum Actinomycetota, genera Bifidobacterium, Streptococcus, and Veillonella, and a lower abundance of phylum Pseudomonadota, genera Staphylococcus, and Enterococcus as compared to the birth stage (Kumbhare et al., Reference Kumbhare, Patangia, Mongad, Bora, Bavdekar and Shouche2020). Bifidobacterium and Streptococcus are one of the most abundant and core bacterial members, respectively, of an infant’s gut (Jost et al., Reference Jost, Lacroix, Braegger and Chassard2013; Underwood et al., Reference Underwood, German, Lebrilla and Mills2015). The role of Veillonella in infancy is poorly understood (Ferretti et al., Reference Ferretti, Pasolli, Tett, Asnicar, Gorfer, Fedi, Armanini, Truong, Manara, Zolfo, Beghini, Bertorelli, De Sanctis, Bariletti, Canto, Clementi, Cologna, Crifò, Cusumano, Gottardi, Innamorati, Masè, Postai, Savoi, Duranti, Lugli, Mancabelli, Turroni, Ferrario, Milani, Mangifesta, Anzalone, Viappiani, Yassour, Vlamakis, Xavier, Collado, Koren, Tateo, Soffiati, Pedrotti, Ventura, Huttenhower, Bork and Segata2018; Kumbhare et al., Reference Kumbhare, Patangia, Mongad, Bora, Bavdekar and Shouche2020) (Figure 2). There was a similarity between Indian infants’ and their mothers’ microbiomes, but the results were not significant.
Childhood
Three studies from Norway, Sweden, and Finland were compared with the ones available for Indian cohorts. A Norwegian study showed that a certain bacterial species pool is shared between mother and infant. Mother-associated operational taxonomic units start depleting after 3 months of age. Over the period, microbiota gets enriched with class Bacteroidia and Clostridia (Avershina et al., Reference Avershina, Lundgård, Sekelja, Dotterud, Storrø, Øien, Johnsen and Rudi2016) and species Bifidobacterium breve (Agans et al., Reference Agans, Rigsbee, Kenche, Michail, Khamis and Paliy2011; Avershina et al., Reference Avershina, Lundgård, Sekelja, Dotterud, Storrø, Øien, Johnsen and Rudi2016; Roswall et al., Reference Roswall, Olsson, Kovatcheva-Datchary, Nilsson, Tremaroli, Simon, Kiilerich, Akrami, Krämer, Uhlén, Gummesson, Kristiansen, Dahlgren and Bäckhed2021). Bifidobacterium breve acts as an inhibitor or is negatively associated with late-appearing microbes (Avershina et al., Reference Avershina, Lundgård, Sekelja, Dotterud, Storrø, Øien, Johnsen and Rudi2016). The first 5 years of the developmental trajectory in the Swedish population showed a higher abundance of lactic acid bacteria (Enterococcus, Streptococcus, and Lactobacillus) and gamma-Proteobacteria (Enterobacteriaceae, Citrobacter, and Serratia) along with Bifidobacterium in the first few months. At the age of 1 year, adult-associated genera such as Akkermansia, Faecalibacterium, Prevotella, Roseburia (Roswall et al., Reference Roswall, Olsson, Kovatcheva-Datchary, Nilsson, Tremaroli, Simon, Kiilerich, Akrami, Krämer, Uhlén, Gummesson, Kristiansen, Dahlgren and Bäckhed2021) and Ruminococcus (Agans et al., Reference Agans, Rigsbee, Kenche, Michail, Khamis and Paliy2011) become highly prevalent, and their abundance increases as they grow older (Roswall et al., Reference Roswall, Olsson, Kovatcheva-Datchary, Nilsson, Tremaroli, Simon, Kiilerich, Akrami, Krämer, Uhlén, Gummesson, Kristiansen, Dahlgren and Bäckhed2021).
Healthy children from the south Indian slum had a higher abundance of the genera Prevotella, Bifidobacterium, and Escherichia-Shigella (Shivakumar et al., Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021). Partially in line with the Swedish population, children from southern India showed a higher abundance of Lactobacillus, Bifidobacterium, Eubacterium rectale, and Faecalibacterium prausnitzii (Balamurugan et al., Reference Balamurugan, Janardhan, George, Chittaranjan and Ramakrishna2008). A comparison of Indian and Finnish children’s microbiomes showed enrichment of Prevotella and Megasphaera in Indian children (Kumbhare et al., Reference Kumbhare, Kumar, Chowdhury, Dhotre, Endo, Mättö, Ouwehand, Rautava, Joshi, Patil, Patil, Isolauri, Bavdekar, Salminen and Shouche2017) (Figure 2). A higher prevalence of Prevotella indicates enterotype 2 in the Indian population, which is well established in other studies as well (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019; Kaur et al., Reference Kaur, Khatri, Akhtar, Subramanian and Ramya2020)
Adult
The Norwegian data showed that Bifidobacterium breve had a higher prevalence in the first year of life and was negatively associated with a range of adult-like species. Its disappearance suggestively drives (at least partially) the transition from infant to adult-associated gut microbiome (Avershina et al., Reference Avershina, Lundgård, Sekelja, Dotterud, Storrø, Øien, Johnsen and Rudi2016). According to a study from the Netherlands, the adult gut microbiome is stable and highly diverse compared to children, with the dominance of Blautia and Bacteroides in the former and latter groups, respectively (Radjabzadeh et al., Reference Radjabzadeh, Boer, Beth, van der Wal, Kiefte-De Jong, Jansen, Konstantinov, Peppelenbosch, Hays, Jaddoe, Ikram, Rivadeneira, van Meurs, Uitterlinden, Medina-Gomez, Moll and Kraaij2020). On the contrary, data from Ohio, USA showed that it was relative abundance, not the presence–absence of specific genera that differentiated the two groups (Agans et al., Reference Agans, Rigsbee, Kenche, Michail, Khamis and Paliy2011). The western adult gut microbiome is dominated by phyla Bacillota, Bacteroidota, Actinomycetota, and Pseudomonadota with carbohydrate metabolism remaining the dominant pathway (Human Microbiome Project Consortium, 2012).
Comparison of the Indian with Chinese populations showed no difference in diversity; however, composition and relative abundance differed (Jain et al., Reference Jain, Li and Chen2018). Both the populations were enriched with Bacillota and Actinomycetota, with fewer Bacteroides. Differences in dietary patterns led to a significantly higher abundance of Bacteroidota and Prevotella in Indians in contrast to Chinese (Jain et al., Reference Jain, Li and Chen2018). Bacterial succession from childhood to adulthood in Indians showed a decline in Bifidobacterium and Lactobacillus. Contrary to Radjabzadeh et al. (Reference Radjabzadeh, Boer, Beth, van der Wal, Kiefte-De Jong, Jansen, Konstantinov, Peppelenbosch, Hays, Jaddoe, Ikram, Rivadeneira, van Meurs, Uitterlinden, Medina-Gomez, Moll and Kraaij2020) and Jain et al. (Reference Jain, Li and Chen2018), a higher abundance of Bacteroides during late adolescence and adulthood, and a sharp decline of Eubacterium rectale and F. prausnitzii in Indian adults were reported (Balamurugan et al., Reference Balamurugan, Janardhan, George, Raghava, Muliyil and Ramakrishna2008; Jain et al., Reference Jain, Li and Chen2018; Radjabzadeh et al., Reference Radjabzadeh, Boer, Beth, van der Wal, Kiefte-De Jong, Jansen, Konstantinov, Peppelenbosch, Hays, Jaddoe, Ikram, Rivadeneira, van Meurs, Uitterlinden, Medina-Gomez, Moll and Kraaij2020). Similar to the western microbial profile at the phylum level, Indian communities are also dominated by Bacillota, Bacteroidota, Actinomycetota, and Pseudomonadota (Figure 2) (Ramakrishna, Reference Ramakrishna2013; Das et al., Reference Das, Ghosh, Kedia, Rampal, Saxena, Bag, Mitra, Dayal, Mehta, Surendranath, Travis, Tripathi, Nair and Ahuja2018).
Elderly
The transition from a stable and diverse bacterial community in adults to a less diverse one in the elderly population was compared between four global studies (China, Italy, Ireland, and Japan) and available Indian studies. An increase in Pseudomonadota species was reported in several studies (Rampelli et al., Reference Rampelli, Candela, Turroni, Biagi, Collino, Franceschi, O’Toole and Brigidi2013; Kumar et al., Reference Kumar, Babaei, Ji and Nielsen2016; Kong et al., Reference Kong, Deng, Li and Zhao2018). An Ireland-based study reported significantly higher dominance of Prevotella and Ruminococcus in the adults and Alistipes and Oscillibacter in the elderly group (Claesson et al., Reference Claesson, Jeffery, Conde, Power, O’Connor, Cusack, Harris, Coakley, Lakshminarayanan, ’sullivan, Fitzgerald, Deane, O’Connor, Harnedy, O’Connor, O’Mahony, Van Sinderen, Wallace, Brennan, Stanton, Marchesi, Fitzgerald, Shanahan, Hill, Ross and O’Toole2012). The study done on the same cohort showed Bacteroides, Alistipes, Parabacteroides, Faecalibacterium, and Ruminococcus as the core genera in the elderly population (Jeffery et al., Reference Jeffery, Lynch and O’Toole2015). An overall decrease in SCFAs production, shift from proteolytic to saccharolytic fermentation, loss of organisms such as Eubacterium, Bifidobacterium, and Faecalibacterium, and increased abundance of pathogens such as Escherichia-Shigella were considered as functions of the ageing process (Kumar et al., Reference Kumar, Babaei, Ji and Nielsen2016; Kong et al., Reference Kong, Deng, Li and Zhao2018).
In line with the results from other countries, an Indian study done by Tuikhar et al. (Reference Tuikhar, Keisam, Labala, Imrat, Ramakrishnan, Arunkumar, Ahmed, Biagi and Jeyaram2019) also reported a higher diversity in the Ruminococcaceae family in centenarians (~100 years old). Direct comparison with samples from Italy, Japan, and China in the same study also showed similar results. A decrease in the abundance of Faecalibacterium was also observed in the Indian population. Species from genera Akkermansia, Alistipes, and Ruminococcoaceae D16 were reported as signatures of longevity in all four populations. Akkermansia was reported to be associated with health and anti-inflammatory activity. The unclassified species Ruminococcoaceae D16 was reported to be a butyrate producer in herbivorous and omnivorous animals (Figure 2) (Tuikhar et al., Reference Tuikhar, Keisam, Labala, Imrat, Ramakrishnan, Arunkumar, Ahmed, Biagi and Jeyaram2019; Badal et al., Reference Badal, Vaccariello, Murray, Yu, Knight, Jeste and Nguyen2020).
Factors affecting gut microbiome composition
Diet
Trends from three studies done on global cohorts (the United States, Japan, Europe, and Africa) were compared with available data on Indian cohorts. The long-term effect of diet has a huge impact on microbial community structure; however, short-term (5 days) consumption of entirely plant-based or animal-based foods has also rapidly changed the gut community structure (David et al., Reference David, Maurice, Carmody, Gootenberg, Button, Wolfe, Ling, Devlin, Varma, Fischbach, Biddinger, Dutton and Turnbaugh2013). Animal-based diet showed a higher abundance of bile-tolerant bacteria such as Bacteroides, Alistipes, and Bilophila (David et al., Reference David, Maurice, Carmody, Gootenberg, Button, Wolfe, Ling, Devlin, Varma, Fischbach, Biddinger, Dutton and Turnbaugh2013; Pareek et al., Reference Pareek, Kurakawa, Das, Motooka, Nakaya, Rongsen-Chandola, Goyal, Kayama, Dodd, Okumura, Maeda, Fujimoto, Nii, Ogawa, Iida, Bhandari, Kida, Nakamura, Nair and Takeda2019), whereas the higher abundance of Bacillota that metabolise plant polysaccharides such as Roseburia, Eubacterium rectale, and Ruminococcus bromii reported in plant-based diet consuming individuals (David et al., Reference David, Maurice, Carmody, Gootenberg, Button, Wolfe, Ling, Devlin, Varma, Fischbach, Biddinger, Dutton and Turnbaugh2013). Another study done by De Filippo et al. (Reference De Filippo, Cavalieri, Di Paola, Ramazzotti, Poullet, Massart, Collini, Pieraccini and Lionetti2010) on European and African children, consuming western and rural diets, respectively, showed partial overlapping patterns. A higher abundance of phylum Bacteroidota (Prevotella) and SCFAs, and depletion of phylum Bacillota and family Enterobacteriaceae (Shigella and Escherichia) reported in Africans (De Filippo et al., Reference De Filippo, Cavalieri, Di Paola, Ramazzotti, Poullet, Massart, Collini, Pieraccini and Lionetti2010). In line with the above results, the Indian population consuming a plant-based diet had a higher abundance of Prevotella (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019; Jain et al., Reference Jain, Li and Chen2018; Kaur et al., Reference Kaur, Khatri, Akhtar, Subramanian and Ramya2020). It was also reported to have higher lipopolysaccharide pathway genes and serum BCAA levels; Latter is because of the presence of fewer in-ward transporters in bacteria; hence; they get absorbed in serum (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019). In contrast, the omnivorous group showed higher bacterial BCAA transporters and hence their high abundance in faecal matter (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019). Partially overlapping results on the association of omnivorous diet with butyrate-producing bacteria such as Roseburia–E. Rectale (Kabeerdoss et al., Reference Kabeerdoss, Shobana Devi, Regina Mary and Ramakrishna2012), Bacteroides, Ruminococcus, and Faecalibacterium, and enrichment of SCFAs biosynthesis pathways were also observed (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019). Another Indian study by Bamola et al. (Reference Bamola, Ghosh, Kapardar, Lal, Cheema, Sarma and Chaudhry2017), however, presented a completely different picture, reporting a higher Bacteroidota to Bacillota ratio in the non-vegetarian group as compared to vegetarians. It was not clearly explained if the abundance profile comparison of taxa between the vegetarian and omnivorous groups was statistically significant (sequence data involved just 96 sequences per group) (Bamola et al., Reference Bamola, Ghosh, Kapardar, Lal, Cheema, Sarma and Chaudhry2017).
Lifestyle
Despite being crucial in maintaining health, little is known to what extent modernisation has impacted gut microbiota structure. Less affected tribal populations still use traditional ways to survive (Shetty et al., Reference Shetty, Marathe and Shouche2013). Here, the comparison of Indian studies was made with data from Tanzania, America, Malawi, Mongolia, and Italy. Yanomami, who live a hunter-gatherer lifestyle similar to human ancestors, not exposed to antibiotics, were first contacted in ~1960 in Venezuela. Their gut composition showed significantly huge diversity than the U.S. population, with high Prevotella and low Bacteroides abundance, similar to that in African hunter-gatherers, Guahibo Amerindians, and Malawians (Clemente et al., Reference Clemente, Pehrsson, Blaser, Sandhu, Gao, Wang, Magris, Hidalgo, Contreras, Noya-Alarcón, Lander, McDonald, Cox, Walter, Oh, Ruiz, Rodriguez, Shen, Song, Metcalf, Knight, Dantas and Dominguez-Bello2015). They also showed high functional diversity, gene prevalence, and less intragroup variation as compared to the United States (Clemente et al., Reference Clemente, Pehrsson, Blaser, Sandhu, Gao, Wang, Magris, Hidalgo, Contreras, Noya-Alarcón, Lander, McDonald, Cox, Walter, Oh, Ruiz, Rodriguez, Shen, Song, Metcalf, Knight, Dantas and Dominguez-Bello2015). An interesting pattern of seasonal variation in community structure emerged in Hadza hunter-gatherers of Tanzania. This seasonal variation was based on food acquisition activities which were affected by the local environment and type of food availability in two different seasons. Bacillota, for instance, remained stable in both dry (May–October) and wet (November–April) seasons; however, the abundance of family Prevotellace significantly declined during the wet season compared to the dry season (Smits et al., Reference Smits, Leach, Sonnenburg, Gonzalez, Lichtman, Reid, Knight, Manjurano, Changalucha, Elias, Dominguez-Bello and Sonnenburg2017). Surprisingly, seasonally volatile taxa in Hadza differentiated this traditional population from the industrialised one, indicating a decrease in the prevalence and abundance of some taxa in modernised populations (Smits et al., Reference Smits, Leach, Sonnenburg, Gonzalez, Lichtman, Reid, Knight, Manjurano, Changalucha, Elias, Dominguez-Bello and Sonnenburg2017). Prevotella was the dominant genus in Mongolian, Amerindian, and Malawian groups, while Faecalibacterium was in the American, Italian, and Hadza populations (Dehingia et al., Reference Dehingia, Devi, Talukdar, Talukdar, Reddy, Mande, Deka and Khan2015). India, with six major physiographic divisions, namely The Himalayan mountains, Northern plains, Peninsular plateau, Indian desert, Coastal plains, and Islands along with multiple ethnic groups living in each division, have many distinct dietary habits and lifestyles (urban, rural, tribals from forests, hills, hot deserts, cold deserts, remote islands, mangroves, etc.). While there are multiple studies on tribal populations, no proper study has been done on Indian ethnic groups. Similar to the trends mentioned above, gut bacterial profiles of tribal populations from four different geographical locations, namely Assam, Telangana, Manipur, and Sikkim, showed the dominance of Prevotella. Likewise, a comparison of three different tribes from Mongoloid (Ladakh), Caucasoid (Jaisalmer), and Australoid (Khargone) ancestry revealed that despite the differences in ethnicity and geographical locations, genera Prevotella, Bifidobacterium, Bacteroides, Eubacterium, and Faecalibacterium were abundant in overall populations (Kaur et al., Reference Kaur, Khatri, Akhtar, Subramanian and Ramya2020; Hazarika et al., Reference Hazarika, Chattopadhyay, Umpo, Choudhury and Sharma2022). A small cohort size study in Tamil Nadu, India, revealed a higher Bacillota/Bacteroidota ratio and higher Actinomycetota abundance in the rural population than in tribal (Ramadass et al., Reference Ramadass, Rani, Pugazhendhi, John and Ramakrishna2017). A study on the Nicobarese community, one of the six tribal communities of Andaman and Nicobar Islands, revealed that their lifestyle has a profound impact on the gut bacterial composition, where the remote subset of the community had Bacteroides–Prevotella–Porphyromonas as the dominant bacterial group, while the rural and urban subsets had Clostridium coccoides, Eubacterium rectale, and Bifidobacterium as the predominant bacterial groups, respectively (Anwesh et al., Reference Anwesh, Kumar, Nagarajan, Chander, Kartick and Paluru2016).
Antibiotic usage
The benefits of antibiotic usage in humans as well as livestock come at a cost with the inevitable evolution of antibiotic-resistant variants and the collateral damaging effect of antibiotics on commensal bacteria (Blaser, Reference Blaser2016). A longitudinal study conducted on 12 individuals in Denmark observed that antibiotic usage reduces microbial diversity, especially that of butyrate-producing species with a restoration period of 1.5 months to obtain the baseline composition (Palleja et al., Reference Palleja, Mikkelsen, Forslund, Kashani, Allin, Nielsen, Hansen, Liang, Feng, Zhang, Pyl, Coelho, Yang, Wang, Typas, Nielsen, Nielsen, Bork, Wang, Vilsbøll, Hansen, Knop, Arumugam and Pedersen2018) A similar restoration period of 1 month was observed in a study which included 39 children from Finland (Yassour et al., Reference Yassour, Vatanen, Siljander, Hämäläinen, Härkönen, Ryhänen, Franzosa, Vlamakis, Huttenhower, Gevers, Lander, Knip and Xavier2016). However, Palleja et al. (Reference Palleja, Mikkelsen, Forslund, Kashani, Allin, Nielsen, Hansen, Liang, Feng, Zhang, Pyl, Coelho, Yang, Wang, Typas, Nielsen, Nielsen, Bork, Wang, Vilsbøll, Hansen, Knop, Arumugam and Pedersen2018) observed that several common species were not restored even after 1.5 months and until the end of their study period which was 180 days. Moreover, disruptions in the balance of gut microbial species lead to an increase in pathobionts such as Clostridium difficile (Buffie and Pamer Reference Buffie and Pamer2013). Another study conducted on 21 participants from Spain, who were treated with broad-spectrum antibiotics indicated a reduction in bacterial diversity due to the elimination of antibiotic-susceptible bacteria and an increase in the overall microbial load due to the replacement and rapid multiplication of antibiotic-resistant bacterial species (Panda et al., Reference Panda, El Khader, Casellas, López Vivancos, García Cors, Santiago, Cuenca, Guarner and Manichanh2014). Studies conducted across Canada and the United States provide increasing evidence that early antibiotic exposure in life is associated with obesity, diabetes, inflammatory bowel diseases (IBDs), allergies, and asthma (Arrieta et al., Reference Arrieta, Stiemsma, Dimitriu, Thorson, Russell, Yurist-Doutsch, Kuzeljevic, Gold, Britton, Lefebvre, Subbarao, Mandhane, Becker, McNagny, Sears, Kollmann, Mohn, Turvey and Finlay2015; Azad et al., Reference Azad, Bridgman, Becker and Kozyrskyj2014; Bokulich et al., Reference Bokulich, Chung, Battaglia, Henderson, Jay, Li, Lieber, Wu, Perez-Perez, Chen, Schweizer, Zheng, Contreras, Dominguez-Bello and Blaser2016) in the later stages of life. Whereas, the short-term and medium-term consequences include antibiotic-associated diarrhoea, C. difficile infections, and H. pylori-related gut dysbiosis (Ramirez et al., Reference Ramirez, Guarner, Bustos Fernandez, Maruy, Sdepanian and Cohen2020).
In the Indian context, a study from southern India, which included 120 infants, revealed that azithromycin has a moderate impact on their gut microbiota (Parker et al., Reference Parker, Praharaj, John, Kaliappan, Kampmann, Kang and Grassly2017). This study indicated a decrease in the microbial diversity and abundance during antibiotic intake; however, no effect was observed on the maturity of the microbiota. Although studies depicting the direct effect of antibiotic usage on the gut microbiota may be rare in India, the other major concern of gut microbiota acting as a reservoir for antibiotic resistance genes has been reported in various studies. Antibiotic abuse is a common phenomenon in low- and middle-income countries. In India, the usage of antibiotics has increased from 3.2 billion defined daily doses in 2000 to 6.5 billion in 2015, an increase of 103% (Klein et al., Reference Klein, Van Boeckel, Martinez, Pant, Gandra, Levin, Goossens and Laxminarayan2018). In such situations, the human gut microbiome acts as a reservoir of antibiotic-resistance genes, capable of transferring the genes rapidly to transient pathogens within the holobiont through horizontal gene transfer (Sitaraman Reference Sitaraman2018; Groussin et al., Reference Groussin, Poyet, Sistiaga, Kearney, Moniz, Noel, Hooker, Gibbons, Segurel, Froment, Mohamed, Fezeu, Juimo, Lafosse, Tabe, Girard, Iqaluk, Nguyen, Shapiro, Lehtimäki, Ruokolainen, Kettunen, Vatanen, Sigwazi, Mabulla, Domínguez-Rodrigo, Nartey, Agyei-Nkansah, Duah, Awuku, Valles, Asibey, Afihene, Roberts, Plymoth, Onyekwere, Summons, Xavier and Alm2021). An insightful gut microbiome study among 18 Swedish students who travelled to India on an exchange programme showed that 12 of the students acquired ESBL-producing E. coli, even without taking antibiotics (Bengtsson-Palme et al., Reference Bengtsson-Palme, Angelin, Huss, Kjellqvist, Kristiansson, Palmgren, Joakim Larsson and Johansson2015). Another study on 122 travellers from the Netherlands to India revealed increased acquisition rates of beta-lactam and quinolone resistance genes (von Wintersdorff et al., Reference von Wintersdorff, Penders, Stobberingh, Oude Lashof, Hoebe, Savelkoul and Wolffs2014). This emphasises the potential for antibiotic resistance transmission in regions with heightened antibiotic use. Furthermore, a study conducted in 2019 among 207 healthy individuals from Chandigarh, India, reported that 70.5% of the stool samples had antibiotic-resistant isolates of which 2.4% were multi-drug resistant and the most common genes identified were β-lactamases (Gupta et al. Reference Gupta, Didwal, Bansal, Kaushal, Batra, Gautam and Ray2019b). Similarly, a high prevalence of β-lactamases was observed in the rectal swabs collected from neonates and mothers in India (Carvalho et al. Reference Carvalho, Sands, Thomson, Portal, Mathias, Milton, Gillespie, Dyer, Akpulu, Boostrom and Hogan2022). A study on 25 healthy individuals from Kolkata, India, reported that all the samples carried aminoglycoside resistance markers and most of them showed resistance to tetC and sul-2 genes (De et al. Reference De, Kanungo, Mukhopadhyay and Dutta2023).
Gut microbiome association with health and diseases
Gut microbiota has a crucial role in regulating gut homeostasis, maintaining intestinal barrier and immunity by metabolising complex dietary substrates, and synthesising micronutrients. The microbial community dysbiosis or modulation could lead to or associate with various noncommunicable and communicable diseases. Studies across the globe and from India have suggested their role/association in malnourishment, diabetes, obesity, inflammatory diseases, neurological disorders, diarrhoea, amoebiasis, and so forth.
Noncommunicable diseases
Malnourishment
Excess, deficiency, and/or imbalanced micronutrients and energy intake lead to malnutrition. The various forms of malnutrition include undernutrition, micronutrient-related malnutrition, overweight, obesity, and other diet-related diseases. Around 45% of children’s deaths are caused by malnutrition globally (Fact Sheets – Malnutrition, n.d.).
A comparison of four global studies from Indonesia, Mexico, Bangladesh, South Africa, Guatemala, and Malawi with Indian studies provides evidence that gut microbiota dysbiosis could also predispose to various forms of malnutrition. A study from Indonesia reported low Bacteroidota and high Bacillota in stunted children of 3–5 years (Surono et al., Reference Surono, Widiyanti, Kusumo and Venema2021), which was also true in undernourished and obese children from Mexico (Méndez-Salazar et al., Reference Méndez-Salazar, Ortiz-López, Granados-Silvestre, Palacios-González and Menjivar2018). High species richness and diversity along with significant enrichment of Prevotella 9 in healthy children correlated with their height and high dietary fibre intake (Méndez-Salazar et al., Reference Méndez-Salazar, Ortiz-López, Granados-Silvestre, Palacios-González and Menjivar2018; Surono et al., Reference Surono, Widiyanti, Kusumo and Venema2021). However, it has not been confirmed if this species could revert the malnutrition. Malnourished and poorly growing Bangladeshi children had a higher abundance of Pseudomonadota species such as Klebsiella, Escherichia/Shigella, and a lower abundance of Prevotella, compared to healthy controls (Monira et al., Reference Monira, Nakamura, Gotoh, Izutsu, Watanabe, Alam, Endtz, Cravioto, Ali, Nakaya, Horii, Iida and Alam2011, Perin et al., Reference Perin, Burrowes, Almeida, Ahmed, Haque, Parvin, Biswas, Azmi, Bhuyian, Talukder, Faruque, Stine and George2020) (Table 1). The gastrointestinal infection caused by these pathogenic species could lead to nutrient malabsorption (Monira et al., Reference Monira, Nakamura, Gotoh, Izutsu, Watanabe, Alam, Endtz, Cravioto, Ali, Nakaya, Horii, Iida and Alam2011), likely by dissolution of the brush border membrane and loss of microvilli structure due to lesions induced by adherence of pathogens to the intestine (Neto and Scaletsky, Reference Fagundes Neto and Affonso Scaletsky2000). These pathogens are also associated with poor growth, and inflammation and can also detoxify nitric oxide, which is produced by colonic epithelial cells as an inflammatory response (Perin et al., Reference Perin, Burrowes, Almeida, Ahmed, Haque, Parvin, Biswas, Azmi, Bhuyian, Talukder, Faruque, Stine and George2020). Million et al., Reference Million, Diallo and Raoult2017 also reviewed the link between malnutrition and gut microbiota in studies from countries including South Africa, Guatemala, Bangladesh, Malawi, and India, and reported early depletion of Bifidobacterium longum as the first step in severe acute malnutrition.
Table 1. Common and/or unique trends observed between gut microbiome of Indian and global populations in noncommunicable and communicable diseases
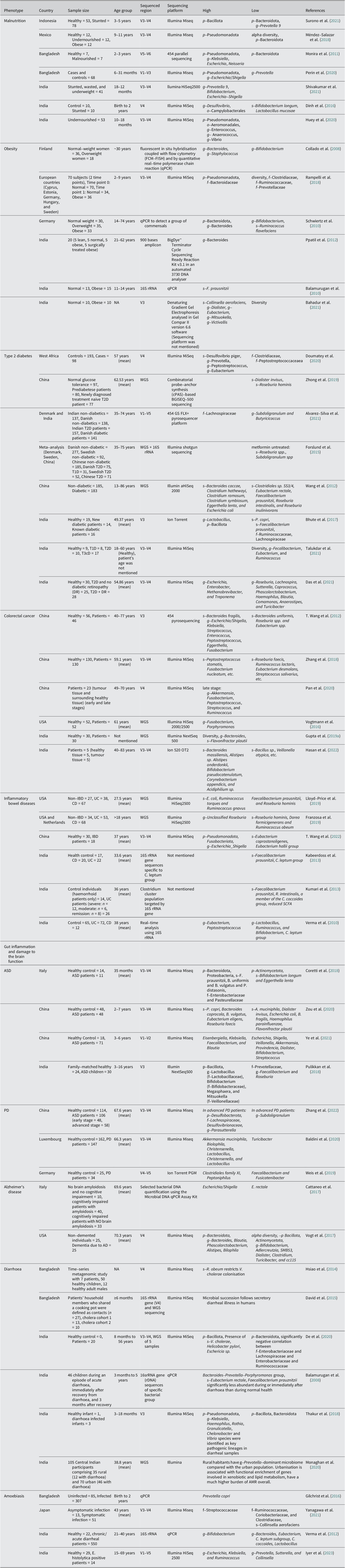
An Indian study showed enrichment of bacterial genera Prevotella 7, Prevotella 9, and Sutterella, and depletion of Clostridiaceae 1 family, Intestinibacter and Fusicatenibacter genera and Bifidobacterium longum subsp longum species in stunted children compared to non-stunted children (Shivakumar et al., Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021). This conflicting trend (of Prevotella genera in malnourished children) in Shivakumar et al. (Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021), which was also observed in Kristensen et al. (Reference Kristensen, Wiese, Rytter, Özçam, Hansen, Namusoke, Friis and Nielsen2016), could be either due to the difference in the age group of children being compared (<2 years vs. 3–5 years) or due to dietary differences between the cohorts, which needs further examination (Kristensen et al. (Reference Kristensen, Wiese, Rytter, Özçam, Hansen, Namusoke, Friis and Nielsen2016); Shivakumar et al., Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021). However, a higher abundance of pathogenic genera Escherichia/Shigella was in sync with the global trend (Shivakumar et al., Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021; Surono et al., Reference Surono, Widiyanti, Kusumo and Venema2021). A longitudinal study on persistently stunted children from south India showed an increase in diversity in both groups (stunted and healthy controls) with age. Partially in line with Shivakumar et al. (Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021), stunted children at 12 months of age showed a higher abundance of Bacteroidota. Enrichment of inflammogenic taxa, that is, genus Desulfovibrio and order Campylobacterales, and lower abundance of probiotic species Bifidobacterium longum and Lactobacillus mucosae in stunted children were also observed (Dinh et al., Reference Dinh, Ramadass, Kattula, Sarkar, Braunstein, Tai, Wanke, Hassoun, Kane, Naumova, Kang and Ward2016; Shivakumar et al., Reference Shivakumar, Sivadas, Devi, Jahoor, McLaughlin, Smith, Kurpad and Mukhopadhyay2021). The gut microbiota of children living in Mumbai slums was enriched with Pseudomonadota and less Actinomycetota, representing the immaturity of the gut (Huey et al., Reference Huey, Jiang, Fedarko, McDonald, Martino, Ali, Russell, Udipi, Thorat, Thakker, Ghugre, Potdar, Chopra, Rajagopalan, Haas, Finkelstein, Knight and Mehta2020) (Table 1).
The majority of the microbiota-associated malnutrition reports are coming from countries with low socioeconomic status. Increasing poverty, poor hygiene, altered dietary habits, exposure to pollutants, and accumulation of environmental pathogens could make them more prone to long-term health problems such as malnutrition (Leocádio et al., Reference Leocádio, Lopes, Dias, Alvarez-Leite, Guerrant, Malva and Oriá2021). Association of a higher abundance of pathogenic genera from phylum Pseudomonadota with malnutrition, and depletion of Bifidobacterium longum emerged as a common trend in both Indian and Global populations. However, the sample size, age group, and sequenced region of the 16S rRNA gene were different in the above comparisons.
Obesity
Excessive or abnormal accumulation of fat in the body that could impair health is termed obesity or overweight (Obesity and Overweight, n.d.). Nearly 650 million people around the globe and 135 million in India are affected by obesity. Changes in gut microbial composition also lead to excessive energy storage and a high risk of obesity. Four studies from Germany, Finland, the United States, and other European countries were compared with Indian studies. The gut bacterial-regulated low-grade inflammation was associated with obesity. For instance, inflammation associated Staphylococcus aureus was enriched in overweight mothers (Collado et al., Reference Collado, Isolauri, Laitinen and Salminen2008). The onset of obesity was associated with an increase in the Pseudomonadota phylum and a decrease in the family Clostridiaceae and Ruminococcaceae, as reported in a longitudinal study from Europe (Rampelli et al., Reference Rampelli, Guenther, Turroni, Wolters, Veidebaum, Kourides, Molnár, Lissner, Benitez-Paez, Sanz, Fraterman, Michels, Brigidi, Candela and Ahrens2018). The gut microbiota of obese individuals was reported to exhibit a lower abundance of the genus Bifidobacterium (Collado et al., Reference Collado, Isolauri, Laitinen and Salminen2008), Clostridium leptum group of phylum Bacillota (Schwiertz et al., Reference Schwiertz, Taras, Schäfer, Beijer, Bos, Donus and Hardt2010), and family Prevotellaceae (Rampelli et al., Reference Rampelli, Guenther, Turroni, Wolters, Veidebaum, Kourides, Molnár, Lissner, Benitez-Paez, Sanz, Fraterman, Michels, Brigidi, Candela and Ahrens2018). Additionally, enrichment of Bacteroides (Collado et al., Reference Collado, Isolauri, Laitinen and Salminen2008; Schwiertz et al., Reference Schwiertz, Taras, Schäfer, Beijer, Bos, Donus and Hardt2010; Rampelli et al., Reference Rampelli, Guenther, Turroni, Wolters, Veidebaum, Kourides, Molnár, Lissner, Benitez-Paez, Sanz, Fraterman, Michels, Brigidi, Candela and Ahrens2018) and faecal SCFAs concentrations, particularly propionate and butyrate, were also observed. The latter could be a result of factors like higher microbial production, changes in microbial cross-feeding patterns, and low absorption (Schwiertz et al., Reference Schwiertz, Taras, Schäfer, Beijer, Bos, Donus and Hardt2010). (Table 1).
A consistent pattern was observed while comparing the global (the United States, Germany, Finland, and six other European countries) results to the Indian gut microbiota, for instance, a higher abundance of Bacteroides and a higher level of faecal SCFAs in obese as compared to lean/normal individuals was reported. However, no difference in the distribution of Bacillota and Bacteroidota was observed (Ppatil et al., Reference Ppatil, Pdhotre, Gchavan, Sultan, Jain, Lanjekar, Gangawani, Sshah, Stodkar, Shah, Ranade, Patole and Shouche2012). Faecalibacterium prausnitzii from the Clostridium leptum group was higher in obese south Indian children suggesting an increase in energy salvage from undigested/unabsorbed carbohydrates, which otherwise would be unavailable (Balamurugan et al., Reference Balamurugan, George, Kabeerdoss, Hepsiba, Chandragunasekaran and Ramakrishna2010) (Table 1). Inconsistent with both global as well as other Indian studies, Bahadur et al., Reference Bahadur, Chaudhry, Bamola, Chutani, Verma and Paul2021 reported bacterial composition with denaturing gradient gel electrophoresis technique. They detected Collinsella aerofaciens, Dialister, Eubacterium, Mitsuokella, Victivallis in obese, and Paraclostridium bifermentans in lean individuals (Bahadur et al., Reference Bahadur, Chaudhry, Bamola, Chutani, Verma and Paul2021). Obesity-related microbiota differences strongly influenced by geographical location, lifestyle, and diet as western individuals follow a low fibre and saturated fat-rich diet (Ecklu-Mensah et al., Reference Ecklu-Mensah, Choo-Kang, Maseng, Donato, Bovet, Viswanathan, Bedu-Addo, Plange-Rhule, Boateng, Forrester, Williams, Lambert, Rae, Sinyanya, Luke, Layden, O’Keefe, Gilbert and Dugas2023). These could be the reasons for non-overlapping pattern between global and Indian studies. Inconsistency within Indian studies could be due to different methodologies used for taxonomy identification, different targeted regions of the 16S rRNA gene, and variable age groups (Table 1). However, the association of Bacteroides with obesity has been observed in both Indian and global data.
Type 2 diabetes
The condition of increased blood glucose level due to impaired insulin production by pancreatic beta-cells and the inability of body cells to utilise it (insulin resistance) is termed Type 2 diabetes (T2D). There are about 422 million cases across the globe and India harbours 77 million diabetic cases in adults with a prevalence rate of 8.3% (Members, n.d.). This metabolic disorder is caused by genetic, environmental, or both factors. Here, five studies from global cohorts (Africa, China, and Denmark) were compared with reports from India. A direct link between gut microbiome alteration and T2D comes from clinical studies reporting an increase in the incidence of T2D in total or partial colectomy (Jensen et al., Reference Jensen, Sørensen, Pedersen, Jess, Brunak and Allin2018). The dysbiosis leading to a reduction in the Bacillota phylum, which is otherwise enriched in the healthy subjects, was observed in Africa and Denmark (Zhong et al., Reference Zhong, Ren, Lu, Fang, Hou, Yang, Chen, Yang, Zhao, Shi, Zhou, Wu, Zou, Zi, Chen, Bao, Hu, Gao, Zhang, Xu, Hou, Yang, Wang, Liu, Jia, Madsen, Brix, Kristiansen, Liu and Li2019; Doumatey et al., Reference Doumatey, Adeyemo, Zhou, Lei, Adebamowo, Adebamowo and Rotimi2020). Differences in gut microbial profiles in healthy, pre-diabetic, and treatment-naive T2D were shown in Chinese cohorts. There was an insignificant difference in microbial gene-based diversity and richness among all three groups. However, the butyrate producers from class Clostridia (Dialister invisus and Roseburia hominis) were highly abundant in healthy compared to the other two groups. Treatment-naive T2D group had a higher abundance of Bacteroides spp and lower Akkermansia muciniphila compared to healthy and pre-diabetic groups (Zhong et al., Reference Zhong, Ren, Lu, Fang, Hou, Yang, Chen, Yang, Zhao, Shi, Zhou, Wu, Zou, Zi, Chen, Bao, Hu, Gao, Zhang, Xu, Hou, Yang, Wang, Liu, Jia, Madsen, Brix, Kristiansen, Liu and Li2019). Similarly, African, Danish, and Chinese T2D patients also showed a reduced abundance of butyrate producers (Collinsella, Ruminococcus lactaris, Anaerostipes, and Clostridium) (J. Wang et al., Reference Wang, Qin, Li, Cai, Li, Zhu, Zhang, Liang, Zhang, Guan, Shen, Peng, Zhang, Jie, Wu, Qin, Xue, Li, Han, Lu, Wu, Dai, Sun, Li, Tang, Zhong, Li, Chen, Xu, Wang, Feng, Gong, Yu, Zhang, Zhang, Hansen, Sanchez, Raes, Falony, Okuda, Almeida, LeChatelier, Renault, Pons, Batto, Zhang, Chen, Yang, Zheng, Li, Yang, Wang, Ehrlich, Nielsen, Pedersen, Kristiansen and Wang2012; Forslund et al., Reference Forslund, Hildebrand, Nielsen, Falony, Le Chatelier, Sunagawa, Prifti, Vieira-Silva, Gudmundsdottir, Krogh Pedersen, Arumugam, Kristiansen, Yvonne Voigt, Vestergaard, Hercog, Igor Costea, Roat Kultima, Li, Jørgensen, Levenez, Dore, Nielsen, Brunak, Raes, Hansen, Wang, Ehrlich, Bork and Pedersen2015; Doumatey et al., Reference Doumatey, Adeyemo, Zhou, Lei, Adebamowo, Adebamowo and Rotimi2020; Alvarez-Silva et al., Reference Alvarez-Silva, Kashani, Hansen, Pinna, Anjana, Dutta, Saxena, Støy, Kampmann, Nielsen, Jørgensen, Gnanaprakash, Gnanavadivel, Sukumaran, Rani, Færch, Radha, Balasubramanyam, Nair, Das, Vestergaard, Hansen, Mande, Mohan, Arumugam and Pedersen2021) (Table 1). In contrast to Zhong et al., microbial gene diversity increased upon treatment with metformin (Forslund et al., Reference Forslund, Hildebrand, Nielsen, Falony, Le Chatelier, Sunagawa, Prifti, Vieira-Silva, Gudmundsdottir, Krogh Pedersen, Arumugam, Kristiansen, Yvonne Voigt, Vestergaard, Hercog, Igor Costea, Roat Kultima, Li, Jørgensen, Levenez, Dore, Nielsen, Brunak, Raes, Hansen, Wang, Ehrlich, Bork and Pedersen2015). The high diversity and richness in urban African T2D patients could be due to different lifestyles (Doumatey et al., Reference Doumatey, Adeyemo, Zhou, Lei, Adebamowo, Adebamowo and Rotimi2020).
Consistent with the above results, Indian T2D patients also showed a reduction in butyrate producers (family Ruminococcaceae and Lachnospiraceae, genera Prevotella, Fecalibacterium, Ruminococcus, Roseburia) (Bhute et al., Reference Bhute, Suryavanshi, Joshi, Yajnik, Shouche and Ghaskadbi2017; Alvarez-Silva et al., Reference Alvarez-Silva, Kashani, Hansen, Pinna, Anjana, Dutta, Saxena, Støy, Kampmann, Nielsen, Jørgensen, Gnanaprakash, Gnanavadivel, Sukumaran, Rani, Færch, Radha, Balasubramanyam, Nair, Das, Vestergaard, Hansen, Mande, Mohan, Arumugam and Pedersen2021; Talukdar et al., Reference Talukdar, Sarkar, Jakkampudi, Sarkar, Aslam, Jandhyala, Deepika, Unnisa and Reddy2021). Reduction in anti-inflammatory (Roseburia, Lachnospira, Coprococcus, Phascolarctobacterium, Blautia, Anaerostipes), pro-inflammatory (Sutterella), a few pathogens (Haemophilus, Comamonas), and enrichment of pathogenic (Escherichia, Enterobacter, Treponem), Pro-inflammatory (Methanobrevibacter), anti-inflammatory bacteria (Butyricimonas, Acidaminococcus, Weissella) was reported in Indian T2D patients (Das et al., Reference Das, Jayasudha, Chakravarthy, Prashanthi, Bhargava, Tyagi, Rani, Pappuru, Sharma and Shivaji2021), indicating that a balance between anti-inflammatory and pro-inflammatory bacteria is crucial. Global studies were fairly different in their experimental design and sample size (Table 1). Taking together, it has been observed that T2D diseases could be associated with a decreased abundance of butyrate producers; however, butyrate-producing species can be different.
Colorectal cancer
Colorectal cancer (CRC), a digestive tract tumour, is a leading cause of morbidity and mortality in developed countries like Japan and the United States. Mutation in tumour repressor genes (p53, DPC4/Smad4, APC, MSH2, MLH1, and PMS2) and activation of oncogenes (beta-catenin, COX-2, and K-RAS) are the causes of CRC (Hisamuddin & Yang, Reference Hisamuddin and Yang2006). In this section, four studies from China and the United States were compared with all available Indian ones.
Association studies of gut bacterial dysbiosis with CRC revealed the reduced abundance of butyrate producers (Roseburia spp., Eubacterium spp., E. hallii, E. hadrum, E. desmolans, Roseburia faecis, and Coprococcus comes) (T. Wang et al., Reference Wang, Cai, Qiu, Fei, Zhang, Pang, Jia, Cai and Zhao2012; Zhang et al., Reference Zhang, Yu, Yu, Wang, Cai, Shuai, Yan, Jiang, Wang, Liu, Chen, Li and Jiang2018) and a higher abundance of opportunistic pathogens (Enterococcus, Escherichia/Shigella, Klebsiella, Streptococcus, and Peptostreptococcus) in CRC patients of China. Species Bacteroides vulgatus and Bacteroides uniformis were enriched in healthy (T. Wang et al., Reference Wang, Qin, Li, Cai, Li, Zhu, Zhang, Liang, Zhang, Guan, Shen, Peng, Zhang, Jie, Wu, Qin, Xue, Li, Han, Lu, Wu, Dai, Sun, Li, Tang, Zhong, Li, Chen, Xu, Wang, Feng, Gong, Yu, Zhang, Zhang, Hansen, Sanchez, Raes, Falony, Okuda, Almeida, LeChatelier, Renault, Pons, Batto, Zhang, Chen, Yang, Zheng, Li, Yang, Wang, Ehrlich, Nielsen, Pedersen, Kristiansen and Wang2012) (Table 1); however, species Bacteroides fragilis, reported to trigger cell proliferation, was enriched in CRC patients (T. Wang et al., Reference Wang, Qin, Li, Cai, Li, Zhu, Zhang, Liang, Zhang, Guan, Shen, Peng, Zhang, Jie, Wu, Qin, Xue, Li, Han, Lu, Wu, Dai, Sun, Li, Tang, Zhong, Li, Chen, Xu, Wang, Feng, Gong, Yu, Zhang, Zhang, Hansen, Sanchez, Raes, Falony, Okuda, Almeida, LeChatelier, Renault, Pons, Batto, Zhang, Chen, Yang, Zheng, Li, Yang, Wang, Ehrlich, Nielsen, Pedersen, Kristiansen and Wang2012; Pan et al., Reference Pan, Du, Li, Yang, Zhang and Wang2020). The reduced abundance of butyrate producers was possibly due to a higher abundance of pathogens such as Fusobacterium nucleatum (Vogtmann et al., Reference Vogtmann, Hua, Zeller, Sunagawa, Voigt, Hercog, Goedert, Shi, Bork and Sinha2016; Zhang et al., Reference Zhang, Yu, Yu, Wang, Cai, Shuai, Yan, Jiang, Wang, Liu, Chen, Li and Jiang2018; Pan et al., Reference Pan, Du, Li, Yang, Zhang and Wang2020), Porphyromonas asaccharolytica, (Vogtmann et al., Reference Vogtmann, Hua, Zeller, Sunagawa, Voigt, Hercog, Goedert, Shi, Bork and Sinha2016; Zhang et al., Reference Zhang, Yu, Yu, Wang, Cai, Shuai, Yan, Jiang, Wang, Liu, Chen, Li and Jiang2018) Peptostreptococcus stomatis (Zhang et al., Reference Zhang, Yu, Yu, Wang, Cai, Shuai, Yan, Jiang, Wang, Liu, Chen, Li and Jiang2018; Pan et al., Reference Pan, Du, Li, Yang, Zhang and Wang2020), Parvimonas micra etc., which are oral periodontopathic bacteria (Zhang et al., Reference Zhang, Yu, Yu, Wang, Cai, Shuai, Yan, Jiang, Wang, Liu, Chen, Li and Jiang2018). Healthy and CRC tissue microbiota from Chinese showed no difference in diversity; however, a significant difference was observed while comparing different CRC stages. Cancer progression was marked by an increasing abundance of phyla Bacteroidota, Bacillota, Fusobacteriota, genera Fusobacterium, Peptostreptococcus, Streptococcus, and Ruminococcus, Verrucomicrobia, and a decreasing abundance of Pseudomonadota (Pan et al., Reference Pan, Du, Li, Yang, Zhang and Wang2020).
In accordance with global studies, Bacteroides fragilis, Peptostreptococcus stomatis, and Parvimonas micra were associated with Indian CRC patients (Table 1). Apart from them, species Akkermansia muciniphila, Bacteroides eggerthii, Escherichia coli, Odoribacter splanchnicus, and Parabacteroides distasonis were also associated with CRC (Gupta et al., Reference Gupta, Dhakan, Maji, Saxena, Vishnu Prasoodanan, Mahajan, Pulikkan, Kurian, Gomez, Scaria, Amato, Sharma and Sharma2019a). Species Flavonifractor plautii, a degrader of key flavonoids, was differentially abundant in Indian CRC samples and separated Indian from Austrian and Chinese samples (Gupta et al., Reference Gupta, Dhakan, Maji, Saxena, Vishnu Prasoodanan, Mahajan, Pulikkan, Kurian, Gomez, Scaria, Amato, Sharma and Sharma2019a). Differentially higher abundance of phylum Pseudomonadota and species Alistipes onderdonkii, Bacteroides massiliensis, Bifidobacterium pseudocatenulatum, and Corynebacterium appendicis was also reported by Hasan et al. (Reference Hasan, Bose, Roy, Paul, Rawat, Nilwe, Chauhan and Choudhury2022). The above comparisons revealed a common trend of higher abundance of genus Bacteroides in both Indian and Global CRC patients; however, species were different. A higher abundance of Fusobacterium in global and Flavonifractor in Indian CRC patients was the unique trend.
Inflammatory bowel diseases
IBDs consist of Crohn’s disease (CD) and ulcerative colitis (UC). The CD is an inflammatory disease affecting the GIT with abdominal pain, fever, diarrhoea with mucus or blood, or both (Baumgart & Sandborn, Reference Baumgart and Sandborn2012). UC is also a relapsing inflammatory disease mainly affecting the inner linings of the large intestine and rectum (Gajendran et al., Reference Gajendran, Loganathan, Jimenez, Catinella, Ng, Umapathy, Ziade and Hashash2019). Two major hypotheses have emerged for the nature of the pathogenesis of IBDs. One is an excessive immunological response to the normal gut microbiome by dysregulation of the mucosal immune system and the second is dysbiosis in the gut microbiome that evokes an inflammatory response (Strober et al., Reference Strober, Fuss and Mannon2007; Kabeerdoss et al., Reference Kabeerdoss, Sankaran, Pugazhendhi and Ramakrishna2013). As the gut microbiome flourishes on dietary components, an anti-inflammatory microbiota could be nourished by specific food intake. High animal food intake, alcohol, soft drinks, sugar, and processed food could lead to gut inflammation, while plant-based foods are associated with low pathobiont abundance and high SCFA producers (Bolte et al., Reference Bolte, Vich Vila, Imhann, Collij, Gacesa, Peters, Wijmenga, Kurilshikov, Campmans-Kuijpers, Fu, Dijkstra, Zhernakova and Weersma2021). Three studies from the United States, Netherlands, and China were compared with the Indians.
A characteristic feature of IBD deduced in cohorts from the United States was an increase in facultative anaerobes with a decrease in obligate anaerobes (butyrate producers), specifically enrichment of E. coli and depletion of F. prausnitzii and Roseburia hominis in CD. The differential abundance of two prominent species in IBD, Ruminococcus torques and Ruminococcus gnavus in CD and UC, respectively, was also confirmed in this study (Lloyd-Price et al., Reference Lloyd-Price, Arze, Ananthakrishnan, Schirmer, Avila-Pacheco, Poon, Andrews, Ajami, Bonham, Brislawn, Casero, Courtney, Gonzalez, Graeber, Hall, Lake, Landers, Mallick, Plichta, Prasad, Rahnavard, Sauk, Shungin, Vázquez-Baeza, White, Braun, Denson, Jansson, Knight, Kugathasan, DPB, Petrosino, Stappenbeck, Winter, Clish, Franzosa, Vlamakis, Xavier and Huttenhower2019). Partially overlapping results from a study on the United States and Netherlands cohorts showed depletion of Roseburia hominis, Dorea formicigenerans, and Ruminococcus obeum and enrichment of unclassified Roseburia species in IBD patients. Symbiosis of Bifidobacterium breve and Clostridium symbiosum was uniquely abundant in UC, while species R. gnavus, E. coli, and Clostridium clostridioforme were in CD (Franzosa et al., Reference Franzosa, Sirota-Madi, Avila-Pacheco, Fornelos, Haiser, Reinker, Vatanen, Hall, Mallick, McIver, Sauk, Wilson, Stevens, Scott, Pierce, Deik, Bullock, Imhann, Porter, Zhernakova, Fu, Weersma, Wijmenga, Clish, Vlamakis, Huttenhower and Xavier2019). Reduced diversity, low Bacillota, higher Pseudomonadota, and Fusobacteriota, in IBD patients, were also reported (Franzosa et al., Reference Franzosa, Sirota-Madi, Avila-Pacheco, Fornelos, Haiser, Reinker, Vatanen, Hall, Mallick, McIver, Sauk, Wilson, Stevens, Scott, Pierce, Deik, Bullock, Imhann, Porter, Zhernakova, Fu, Weersma, Wijmenga, Clish, Vlamakis, Huttenhower and Xavier2019; T. Wang et al., Reference Wang, Yu, Zhu, Wang and Yang2022) (Table 1).
In comparison with the results from global studies, a higher abundance of Pseudomonadota, depletion of butyrate producers F. prausnitzii and Clostridial cluster IV & XIVa (Roseburia, Clostridium, Eubacterium, and Ruminococcus), was observed in UC and CD patients of India (Kabeerdoss et al., Reference Kabeerdoss, Sankaran, Pugazhendhi and Ramakrishna2013; Kumari et al., Reference Kumari, Ahuja and Paul2013; Das et al., Reference Das, Ghosh, Kedia, Rampal, Saxena, Bag, Mitra, Dayal, Mehta, Surendranath, Travis, Tripathi, Nair and Ahuja2018). In contrast, Verma et al. (Reference Verma, Verma, Ahuja and Paul2010) reported a higher abundance of species from Clostridium cluster XIVa (Eubacterium and Peptostreptococcus) in CD but not in UC indicating their different roles in pathogenesis in both groups (Verma et al., Reference Verma, Verma, Ahuja and Paul2010) (Table 1).
Low gut bacterial diversity and reduction in butyrate producers (Kabeerdoss et al., Reference Kabeerdoss, Sankaran, Pugazhendhi and Ramakrishna2013; Lloyd-Price et al., Reference Lloyd-Price, Arze, Ananthakrishnan, Schirmer, Avila-Pacheco, Poon, Andrews, Ajami, Bonham, Brislawn, Casero, Courtney, Gonzalez, Graeber, Hall, Lake, Landers, Mallick, Plichta, Prasad, Rahnavard, Sauk, Shungin, Vázquez-Baeza, White, Braun, Denson, Jansson, Knight, Kugathasan, DPB, Petrosino, Stappenbeck, Winter, Clish, Franzosa, Vlamakis, Xavier and Huttenhower2019), which inhibit the gut inflammatory response in IBD patients, were observed in both Indian and global samples (Kabeerdoss et al., Reference Kabeerdoss, Sankaran, Pugazhendhi and Ramakrishna2013; Lloyd-Price et al., Reference Lloyd-Price, Arze, Ananthakrishnan, Schirmer, Avila-Pacheco, Poon, Andrews, Ajami, Bonham, Brislawn, Casero, Courtney, Gonzalez, Graeber, Hall, Lake, Landers, Mallick, Plichta, Prasad, Rahnavard, Sauk, Shungin, Vázquez-Baeza, White, Braun, Denson, Jansson, Knight, Kugathasan, DPB, Petrosino, Stappenbeck, Winter, Clish, Franzosa, Vlamakis, Xavier and Huttenhower2019). All these results suggest that the nature of the pathogenesis of IBD could be explained by the second hypothesis, that dysbiosis in the gut microbiome evokes an inflammatory response.
Gut inflammation and damage to the brain function
The bidirectional communication between gut bacterial cells and the brain is called the gut-microbiota brain axis. The bacterial cells produce neurotransmitters, amino acids, and metabolites, which influence host immune systems, gut barrier integrity, and the brain. Gut barrier integrity also gets disturbed during stress, anxiety, autism spectrum disorders (ASDs), and Parkinson’s disease (PD) (Morais et al., Reference Morais, Schreiber and Mazmanian2020). An association study from the United Kingdom revealed a positive correlation of abundant Lactobacillus spp. with positive self-judgement, and an inverse relation of CRP (C-reactive protein), a pro-inflammatory molecule, with cognitive empathy (Heym et al., Reference Heym, Heasman, Hunter, Blanco, Wang, Siegert, Cleare, Gibson, Kumari and Sumich2019).
ASDs are a group of complex neurodevelopmental disorders, and, unfortunately, the cause is still unclear (Geetha et al., Reference Geetha, Sukumar, Dhivyadeepa, Reddy and Balachandar2019). However, an association of socioeconomic and environmental risk factors with ASD has suggested that family history of ASD, paternal age, nutrition during pregnancy, mode of delivery, breastfeeding, and NICU stay were statistically significant factors associated with ASDs (Geetha et al., Reference Geetha, Sukumar, Dhivyadeepa, Reddy and Balachandar2019). Three gut microbial association studies with ASD, from Italy and China, were compared with an Indian study. A Chinese and Italian study reported an increased abundance of Bacteroidota in ASD children (Coretti et al., Reference Coretti, Paparo, Riccio, Amato, Cuomo, Natale, Borrelli, Corrado, Comegna, Buommino, Castaldo, Bravaccio, Chiariotti, Canani and Lembo2018; Zou et al., Reference Zou, Xu, Wang, Duan, Guo, Zhang, Zhao and Zheng2020); however, the opposite trend was reported other Chinese data (Ye et al., Reference Ye, Gao, Wang, Cao, Liang, He, Lv, Wang, Xu and Zhang2021). High bacterial diversity (Zou et al., Reference Zou, Xu, Wang, Duan, Guo, Zhang, Zhao and Zheng2020; Ye et al., Reference Ye, Gao, Wang, Cao, Liang, He, Lv, Wang, Xu and Zhang2021), a significant increase in BCAAs synthesising species (B. vulgatus and P. copri), a reduction in butyrate-producing genera clusters Clostridium clusters IV and XIVa, probiotic bacteria like B. fragilis and A. muciniphila in ASD children compared to normal controls in China (Zou et al., Reference Zou, Xu, Wang, Duan, Guo, Zhang, Zhao and Zheng2020). Depletion of the dominant infant gut bacterium Bifidobacterium longum (Coretti et al., Reference Coretti, Paparo, Riccio, Amato, Cuomo, Natale, Borrelli, Corrado, Comegna, Buommino, Castaldo, Bravaccio, Chiariotti, Canani and Lembo2018; Ye et al., Reference Ye, Gao, Wang, Cao, Liang, He, Lv, Wang, Xu and Zhang2021) an increase in Faecalibacterium prausnitzii, a significant butyrate producer and late coloniser of the healthy gut, was also reported (Coretti et al., Reference Coretti, Paparo, Riccio, Amato, Cuomo, Natale, Borrelli, Corrado, Comegna, Buommino, Castaldo, Bravaccio, Chiariotti, Canani and Lembo2018; Ye et al., Reference Ye, Gao, Wang, Cao, Liang, He, Lv, Wang, Xu and Zhang2021) (Table 1).
The results from Indian studies were not in line with the above global studies. However, a comparison done in the same study with ASD children from the United States showed an overlap. There was no difference in diversity between the control and ASD groups of Indian children. A higher relative abundance of families Lactobacillaceae (Lactobacillus), Bifidobacteraceae (Bifidobacterium), and Veillonellaceae (Megasphaera) was observed in ASD children. Despite the different diets of Indian ASD children (normal native diet) and the United States (gluten-free), the Lactobacillus genus was highly abundant compared to healthy. Support for this finding was also provided in the articles by Coretti et al. (Reference Coretti, Paparo, Riccio, Amato, Cuomo, Natale, Borrelli, Corrado, Comegna, Buommino, Castaldo, Bravaccio, Chiariotti, Canani and Lembo2018) and Zou et al. (Reference Zou, Xu, Wang, Duan, Guo, Zhang, Zhao and Zheng2020). However, it remains obscure whether the higher abundance of Lactobacillus is a cause or an effect of ASD (Pulikkan et al., Reference Pulikkan, Maji, Dhakan, Saxena, Mohan, Anto, Agarwal, Grace and Sharma2018). Further metagenomic and metabolomic studies are needed to confirm this (Table 1).
The other common neurodegenerative disorders are PD and Alzheimer’s disease (AD). The former is caused by dead or impaired dopamine-producing basal ganglia cells, deposition of alpha-synuclein protein in the cells, and genetic or environmental factors (Parkinson’s Disease: Causes, Symptoms, and Treatments | National Institute on Aging, n.d.). The data from two studies from China and Germany were discussed here. Chinese study showed decreased levels of BCAAs (Leu, Ile, and Val) and Tyr in advanced as compared to early PD, which is probably due to increased energy expenditure which further accelerates amino acid consumption in advanced PD. It also showed a negative correlation between plasma BCAAs, aromatic amino acids, and microbial taxa such as Streptococcaceae, Streptococcus, and Lactobacillus, which consume or catabolise them (Zhang et al., Reference Zhang, He, Qian, Xu, Mo, Yan, Yang and Xiao2022). The German study reported a decreased abundance of neuroprotective, health-promoting, anti-inflammatory species such as Faecalibacterium and Fusicatenibacter, enrichment of opportunistic pathogens, that is, Peptoniphilus and Finegoldia, higher level of calprotectin, a faecal inflammation marker in PD patients (Weis et al., Reference Weis, Schwiertz, Unger, Becker, Faßbender, Ratering, Kohl, Schnell, Schäfer and Egert2019). Fang et al. (Reference Fang, Kazmi, Jameson and Hsiao2020) reviewed several articles and revealed a higher abundance of Bifidobacterium, Lactobacillus, Akkermansia, and a lower abundance of Blautia, Coprococcus, and Prevotella in PD patients. The pro-inflammatory Bilophila species were associated with the progression of disease symptoms (Baldini et al., Reference Baldini, Hertel, Sandt, Thinnes, Neuberger-Castillo, Pavelka, Betsou, Krüger and Thiele2020) (Table 1). The burden of noncommunicable neurological disorders is increasing in India. There were 771,000 cases of PD in 2019 and 45,300 deaths reported in PD (Singh et al., Reference Singh, Sharma, Kumar, Rao, Prasad, Mathur, Pandian, Steinmetz, Biswas, Pal, Prakash, Sylaja, Nichols, Dua, Kaur, Alladi, Agarwal, Aggarwal, Ambekar, Bagepally, Banerjee, Bender, Bhagwat, Bhargava, Bhatia, Chakma, Chowdhary, Dey, Dirac, Feigin, Ganguli, Golechha, Gourie-Devi, Goyal, Gupta, Gupta, Gupta, Gururaj, Hemalatha, Jeemon, Johnson, Joshi, Kant, Kataki, Khurana, Krishnankutty, Kyu, Lim, Lodha, Ma, Malhotra, Malhotra, Mathai, Mehrotra, Misra, Mutreja, Naghavi, Naik, Nguyen, Pandey, Parmar, Perianayagam, Prabhakaran, Rath, Reinig, Roth, Sagar, Sankar, Shaji, Sharma, Sharma, Singh, MVP, Stark, Tandon, Thakur, AS, Thomas, Tripathi, Vongpradith, Wunrow, Xavier, Shukla, Reddy, Panda, Dandona, CJL, Vos, Dhaliwal and Dandona2021). The other noncommunicable disease is AD. It is a common type of dementia characterised by extracellular amyloid beta plaque and intracellular tau protein accumulation. In India, there were 3.69 million cases of AD or other dementias in 2019 (Singh et al., Reference Singh, Sharma, Kumar, Rao, Prasad, Mathur, Pandian, Steinmetz, Biswas, Pal, Prakash, Sylaja, Nichols, Dua, Kaur, Alladi, Agarwal, Aggarwal, Ambekar, Bagepally, Banerjee, Bender, Bhagwat, Bhargava, Bhatia, Chakma, Chowdhary, Dey, Dirac, Feigin, Ganguli, Golechha, Gourie-Devi, Goyal, Gupta, Gupta, Gupta, Gururaj, Hemalatha, Jeemon, Johnson, Joshi, Kant, Kataki, Khurana, Krishnankutty, Kyu, Lim, Lodha, Ma, Malhotra, Malhotra, Mathai, Mehrotra, Misra, Mutreja, Naghavi, Naik, Nguyen, Pandey, Parmar, Perianayagam, Prabhakaran, Rath, Reinig, Roth, Sagar, Sankar, Shaji, Sharma, Sharma, Singh, MVP, Stark, Tandon, Thakur, AS, Thomas, Tripathi, Vongpradith, Wunrow, Xavier, Shukla, Reddy, Panda, Dandona, CJL, Vos, Dhaliwal and Dandona2021).
Results from an Italian study showed a lower abundance of anti-inflammatory Eubacterium rectale and anti-inflammatory cytokines (IL-10), and a high abundance of pro-inflammatory Escherichia/Shigella in patients (cognitively impaired with and without brain amyloidosis) (Table 1). Both the studies from the United States and Italy showed more elevated pro-inflammatory cytokines (CXCL2, IL-1Beta, and NLRP3) in cognitively impaired patients with amyloidosis positively correlated with Escherichia/Shigella and negatively correlated with E. rectale (Cattaneo et al., Reference Cattaneo, Cattane, Galluzzi, Provasi, Lopizzo, Festari, Ferrari, Guerra, Paghera, Muscio, Bianchetti, Volta, Turla, Cotelli, Gennuso, Prelle, Zanetti, Lussignoli, Mirabile, Bellandi and Frisoni2017; Vogt et al., Reference Vogt, Kerby, Dill-McFarland, Harding, Merluzzi, Johnson, Carlsson, Asthana, Zetterberg, Blennow, Bendlin and Rey2017) (Table 1). Despite increasing neurodegenerative cases in India, and their evident association with gut health in global studies, there are no studies done in India on gut microbial association with PD and AD.
Communicable diseases
Diarrhoea
Diarrhoea is one of the leading causes of mortality and is more prevalent in low- and middle-income countries (Naghavi et al., Reference Naghavi, Wang, Lozano, Davis, Liang, Zhou, Vollset, Abbasoglu Ozgoren, Abdalla, Abd-Allah and Abdel Aziz2015). The common causes of diarrhoea are Vibrio cholera, Cryptosporidium sp., enterotoxigenic Escherichia coli, Clostridioides difficile, Rotavirus, and Shigella sp. infection (Guerrant et al., Reference Guerrant, Hughes, Lima and Crane1990; Monaghan et al., Reference Monaghan, Sloan, Stockdale, Blanchard, Emes, Wilcox, Biswas, Nashine, Manke, Gandhi, Jain, Bhotmange, Ambalkar, Satav, Draper, Hill and Kashyap2020). All the diarroeal studies compared with Indian ones were from Bangladesh.
Recovery from V. cholerae infection was characterised by the accumulation of a healthy gut microbial profile. For instance; upon infecting mice with the pathogen, the species Ruminococcus obeum consistently increased, which in turn restricted pathogens’ growth. The increased expression of autoinducer-2 synthase (luxS) in R. obeum repressed several colonisation factors of the pathogen (Table 1) (Hsiao et al., Reference Hsiao, Ahmed, Subramanian, Griffin, Drewry, Petri, Haque, Ahmed and Gordon2014). The recovery mechanism showed that infection or antibiotic treatment cleared both obligate and facultative anaerobes from the gut, followed by the accumulation of oxygen and dietary substrates in the gut. Recolonising facultative anaerobes majorly from dietary resources lowered the oxygen stress that enabled obligate anaerobes to colonise and utilise accumulated carbohydrates. Competition for the dietary substrates returned to the original state community (David et al., Reference David, Weil, Ryan, Calderwood, Harris, Chowdhury, Begum, Qadri, LaRocque and Turnbaugh2015). The disease-specific associations or changes in microbial composition revealed in a meta-analysis, where a higher abundance of Pseudomonadota and a low abundance of Bacteroidota and a few Bacillota, in particular, a reduction of butyrate producers from family Ruminococcaceae and Lachnospiraceae in diarrhoeal patients (Duvallet et al., Reference Duvallet, Gibbons, Gurry, Irizarry and Alm2017).
Similar to the above trends, Indian infants with acute and persistent diarrhoea showed the proliferation of facultative anaerobes of phylum Pseudomonadota (Chelonobacter, Granulicatella, Haemophilus, Klebsiella, Rothia, and Vibrio) and collapse of anaerobic bacteria (Bacillota, Bacteroides) (Thakur et al., Reference Thakur, Changotra, Grover and Vashistt2018). However, the sample size was quite small in this study population. A high Bacillota to Bacteroidota ratio was associated with V. cholera infection (Thakur et al., Reference Thakur, Changotra, Grover and Vashistt2018; De et al., Reference De, Mukhopadhyay and Dutta2020). A negative correlation between commensals of the family Bifidobacteriaceae and Lachnospiraceae and pathogenic families Enterobacteriaceae and Vibrionaceae, implying the obvious trend in diarrheal dysbiosis (De et al., Reference De, Mukhopadhyay and Dutta2020) (Table 1). The gut microbiome of acute diarrheal children from India showed a lower abundance of butyrate producers (E. rectale, F. prauznitzii, L. acidophilus), compared to after recovery microbiome (Balamurugan et al., Reference Balamurugan, Janardhan, George, Chittaranjan and Ramakrishna2008). Antibiotic-exposed urban diarrheal samples from central India were positive for Clostridioides difficile infection and were enriched with cephalosporins and carbapenem resistance genes (Monaghan et al., Reference Monaghan, Sloan, Stockdale, Blanchard, Emes, Wilcox, Biswas, Nashine, Manke, Gandhi, Jain, Bhotmange, Ambalkar, Satav, Draper, Hill and Kashyap2020). The observed differences between Indian and global studies are possible due to the experiment design, age of participants, and targeted region for the taxonomy profiling (Table 1).
Amoebiasis
Amoebiasis is caused by Entamoeba histolytica, and is the second most prevalent protozoan disease, especially in infants in developing countries (Gilchrist et al., Reference Gilchrist, Petri, Schneider, Reichman, Jiang, Begum, Watanabe, Jansen, Elliott, Burgess, Ma, Alam, Kabir, Haque and Petri2016). Upon perturbation or host immune response compromisation, this can become virulent, and cause diarrhoea, and bloody stools. It can also invade other organs if left untreated (Sarjapuram et al., Reference Sarjapuram, Mekala, Singh and Tatu2017; Yanagawa et al., Reference Yanagawa, Nagata, Yagita, Watanabe, Okubo, Kikuchi, Gatanaga, Oka and Watanabe2021). Two studies on gut microbial association with amoebiasis from Bangladesh and Japan were compared with the Indian ones.
A report from Bangladesh showed a significantly higher parasitic load (E. histolytica) during the first year of life in symptomatic as compared to asymptomatics diarrheal infants and association of diarrheal onset with P. copri (Gilchrist et al., Reference Gilchrist, Petri, Schneider, Reichman, Jiang, Begum, Watanabe, Jansen, Elliott, Burgess, Ma, Alam, Kabir, Haque and Petri2016). Japanese asymptomatic and symptomatic diarrheal children differed with significantly lower Streptococcaceae (Streptococcus salivarius and Streptococcus sinensis) and higher protective bacteria from Ruminococcaceae, Coriobacteriaceae, and Clostridiaceae families in former as compared to latter. However, there was no significant difference in the diversity (Yanagawa et al., Reference Yanagawa, Nagata, Yagita, Watanabe, Okubo, Kikuchi, Gatanaga, Oka and Watanabe2021).
Real-time PCR quantification of E. histolytica infected gut microbiota of North Indians showed a significant decrease of predominant gut microbiome members (Bacteroides, Clostridium coccoides subgroup, Clostridium leptum subgroup, Campylobacter, Eubacterium, and Lactobacillus). An unusual rise in the Bifidobacterium population (SCFAs producer), which could also ferment mucin, in E. histolytica infected patients was reported (Verma et al., Reference Verma, Verma, Ahuja and Paul2012). E. histolytica infection induces hypersecretion of mucus from goblet cells to counter adherence of pathogens, which in turn promotes Bifidobacterium growth (Verma et al., Reference Verma, Verma, Ahuja and Paul2012; Cornick et al., Reference Cornick, Moreau, Gaisano and Chadee2017). Another study by Iyer et al. (Reference Iyer, Chandel, Verma, Thakur, Paul, Mandal and Bhattacharya2023) revealed a decreased abundance of Faecalibacterium, Prevotella, Sutterella, Subdoligranulum, and Colinsella and a higher abundance of Escherichia, Klebsiella, and Ruminococcus in the E. histolytica positive patients from Delhi, India. Association of high P. copri levels with diarrhoea was already reported; however, an opposite trend was observed in India (Gilchrist et al., Reference Gilchrist, Petri, Schneider, Reichman, Jiang, Begum, Watanabe, Jansen, Elliott, Burgess, Ma, Alam, Kabir, Haque and Petri2016; Iyer et al., Reference Iyer, Chandel, Verma, Thakur, Paul, Mandal and Bhattacharya2023) (Table 1). Another interesting finding was the preferential phagocytosis of beneficial bacteria from order Bifidobacteriales, Clostridales, Erysipelotrichales, and Lactobacillales cause dysbiosis which could help in the proliferation of pathogens (Iyer et al., Reference Iyer, Verma, Paul and Bhattacharya2019). Treatment of this protozoal disease with antiprotozoal drugs like Metronidazole could give rise to resistant E. histolytica. So efforts have been made to use LAB as probiotics to prevent this disease. The use of Saccharomyces boulardii strain and metronidazole in the clinical trial significantly reduced the duration of diarrhoea (Dinleyici et al. Reference Dinleyici, Eren, Yargic, Dogan and Vandenplas2009). Co-culturing Lactobacillus casei and Enterococcus faecium with E. histolytica showed a significant reduction in parasite survival (Sarjapuram et al., Reference Sarjapuram, Mekala, Singh and Tatu2017). The use of these probiotic strains could lead to amoebiasis treatment without using antibiotics.
Conclusion
This review provides insight into the establishment of the gut microbiome from pregnancy to birth, up till old age, and highlights the dynamics of gut microbiota upon perturbation during communicable and noncommunicable diseases. Gut metagenomic studies from diverse populations of Europe, North and South America, South Africa, and Asia were reviewed and the emerging global pattern of community composition, diversity, and abundance was compared with the Indian population. The differences start appearing right from the mode of delivery, where early colonisation of beneficial bacteria (Bifidobacterium and Lactobacillus) was seen in VD infants. The developmental trajectory from infant, child, and adult to elderly individuals from Indian and global studies showed overlapping as well as unique Indian-specific patterns. For instance, high diversity in the Ruminococcaceae family, and decreased abundance of Faecalibacterium in centenarians were reported in both global as well as Indian studies. On the other hand, a higher abundance of Bacteroides during late adolescence and adulthood, and a sharp decline of Eubacterium rectale and F. prausnitzii in adults were the unique features reported in Indians.
Among key factors influencing gut microbial composition, diet, lifestyle, antibiotic usage, and various diseased conditions have been discussed in depth. To the question of whether population affects these trends, both overlapping as well as unique trends were found, based on a limited number of populations. Since it was earlier reported that the major enterotypes are associated more with the diet rather than with the populations (Arumugam et al., Reference Arumugam, Raes, Pelletier, Paslier, Yamada, Mende, Fernandes, Tap, Bruls, Batto, Bertalan, Borruel, Casellas, Fernandez, Gautier, Hansen, Hattori, Hayashi, Kleerebezem, Kurokawa, Leclerc, Levenez, Manichanh, Nielsen, Nielsen, Pons, Poulain, Qin, Sicheritz-Ponten, Tims, Torrents, Ugarte, Zoetendal, Wang, Guarner, Pedersen, de Vos, Brunak, Doré, Weissenbach, Ehrlich and Bork2011), so from where do the unique trends appear? Populations are known to have (a small set of) unique taxa (Dhakan et al., Reference Dhakan, Maji, Sharma, Saxena, Pulikkan, Grace, Gomez, Scaria, Amato and Sharma2019), which may (at least partially) explain the observed unique trends. This review also highlighted that although reports on core gut microbiomes exist, they are highly limited in terms of capturing the variation present in populations across the globe. This hints towards the need for a systematic study that will prevent any bias associated with meta-analyses.
Studies within India and their comparison with global data also revealed contradictory/inconsistent patterns, which reflects the variability and complexity of metagenomic data. Apart from the various factors mentioned in the article, sampling, storage, DNA isolation methods, library preparation kits, sequencing techniques, and bioinformatic analysis could also influence the outcome of the metagenomic study (Szóstak et al., Reference Szóstak, Szymanek, Havránek, Tomela, Rakoczy, Samelak-Czajka, Schmidt, Figlerowicz, Majta, Milanowska-Zabel, Handschuh and Philips2022). The majority of the Indian studies used amplicon-based different sequencing techniques such as Illumina, pyrosequencing, Ion-torrent, PCR quantification of specific anaerobes, denaturing gradient gel electrophoresis (DGGE), and only a few had used whole genome shotgun sequencing, suggesting a possible explanation for higher-level taxonomy resolution in most cases. Small sample size and lack of controls in comparative studies are other aspects that emerged while reviewing Indian studies. A smaller sample size does not represent a general population-based outcome and influences the significance of the results. As an example, a study done by De et al. (Reference De, Mukhopadhyay and Dutta2020) on gut microbial signatures in diarrheal conditions has inferred the results without comparing them with healthy control. Another important limitation of several studies was their analysis’s ignorance of confounding factors, which might have added bias to the findings.
Lastly, dysbiosis linked with neurodevelopment and neurodegenerative disorders is an active area of research, yet there is only one study on ASD and none on AD and PD in the Indian population. Taken together, a large sample size across multiple geographical locations, analysed through the same robust pipeline, could give the true picture of the gut metagenome in healthy as well as diseased conditions.
Acknowledgements
We would like to thank Mr. Priyansh Patel, a JRF, and Mr. Angeo Saji, a PhD student, for their critical remarks on the manuscript.
Disclosure statement
The authors declare that there are no competing interests.
Author contribution
Conceptualisation: N.C., A.K.V., S.S., V.T.; Data curation: N.C.; Formal analysis: N.C., V.T.; Funding acquisition: V.T.; Supervision: V.T.; Writing – original draft: N.C., A.M.; Writing – review and editing: N.C., A.K.V., V.T.
Funding statement
N.C. was financially supported by funding from the University of Hyderabad’s Institution of Eminence (Grant No. UoH-IoE-RC2-21-023).




 Open access
Open access