


1. Introduction
DNA-based transposable elements, also called terminal-inverted-repeat elements, are powerful tools for genomic manipulations, such as gene transfer, mutagenesis and gene/promoter/enhancer trapping. Elements of this type currently used for vertebrates include the Tol1 (Koga et al., Reference Koga, Inagaki, Bessho and Hori1995) and Tol2 (Koga et al., Reference Koga, Suzuki, Inagaki, Bessho and Hori1996) elements naturally occurring in the medaka fish Oryzias latipes, as well as the Sleeping Beauty (Ivics et al., Reference Ivics, Hackett, Plasterk and Izsvak1997) and Frog Prince (Miskey et al., Reference Miskey, Izsvak, Plasterk and Ivics2003) elements reconstructed from sequence information of defective elements in the genomes of salmonid fishes and the frog Rana pipiens, respectively. The piggyBac element (Cary et al., Reference Cary, Goebel, Corsaro, Wang, Rosen and Fraser1989), identified in the cabbage looper moth, is also used because this element is efficiently transposed in vertebrate genomes (Wu et al., Reference Wu, Meir, Coates, Handler, Pelczar, Moisyadi and Kaminski2006). Among these elements, Tol1 has the shortest history as a genomic manipulation tool because its transposase gene was only recently identified (Koga et al., Reference Koga, Shimada, Kuroki, Hori, Kusumi, Kyono-Hamaguchi and Hamaguchi2007). However, it is a superior genetic tool in that it can deliver large DNA fragments into the chromosomes: a Tol1 element as long as 22·1 kb can be efficiently transposed and Tol1 arms with a total length of only 263 bp are sufficient as vector portions (Koga et al., in press). The transposition activity of this element has been demonstrated in the human and mouse (Koga et al., Reference Koga, Shimada, Kuroki, Hori, Kusumi, Kyono-Hamaguchi and Hamaguchi2007), in addition to the medaka fish (Tsutsumi et al., Reference Tsutsumi, Imai, Kyono-Hamaguchi, Hamaguchi, Koga and Hori2006). It can therefore be inferred that this element is mobile in a wide range of vertebrates, including the African clawed frog Xenopus laevis.
DNA-based transposable elements move mainly in a cut-and-paste fashion. ‘Cut’ is the process of excision of the element from the chromosome or other DNA molecules where the element resides, and ‘paste’ is the reintegration of the excised element into the same or other DNA molecules. The former, if it occurs, can be easily detected because a PCR analysis of a specific element provides sufficient information. Demonstration of reintegration requires a suitable marker gene and a complex detection system because it is impossible to tell in advance where the element will come to rest.
The aim of the present study was to discover whether excision of the Tol1 element occurs in X. laevis. For this purpose, we prepared an indicator plasmid in which a Tol1 element was embedded, and a helper plasmid that played the role of providing the transposase enzyme in cells. These plasmids were injected into early-stage embryos of the frog and, after a period of cell divisions, recovered from the embryos. Subsequent analyses of the indicator plasmid by PCR, cloning and sequencing indicated that the Tol1 element had been excised from the plasmid, with various footprint sequences left at breakpoints. These results suggest that the Tol1 element is mobile and, therefore, can be used as genomic manipulation tools in this model organism.
The footprint sequences were similar to those observed in fish and mammals. However, a tendency to substitute or insert a particular residue at the very breakpoint was observed. This might reflect some DNA repair mechanism that is specific to this frog species or amphibians. This is another finding of the present study.
2. Materials and methods
(i) Plasmids
Two kinds of plasmids were used in this study: indicator plasmids and helper plasmids. The former contained a non-autonomous Tol1 element and the latter the transposase gene driven by the CMV promoter. It is expected that, in cells, the helper plasmid provides the transposase enzyme and the enzyme catalyses transposition of the Tol1 element embedded in the indicator plasmid.
As a negative control of transposase function, we also prepared a defective helper plasmid, in which two internal codons for the transposase had been changed to stop codons.
The indicator, functional helper and defective helper plasmids were designated as pInd263GFP (Tol1 arms of 263 bp plus the GFP gene), pHel851aa (coding for the entire transposase of 851 amino acids) and pHel316aa (for a protein truncated at 316 amino acids), respectively, and their structures are shown in Fig. 1. The GFP gene in pInd263GFP consists of the CMV promoter, the EGFP coding sequence and a polyadenylation signal, and played the role of a marker gene for successful introduction of DNA into the nucleus after microinjection. It will serve in future studies as a marker of reintegration into the chromosomes.
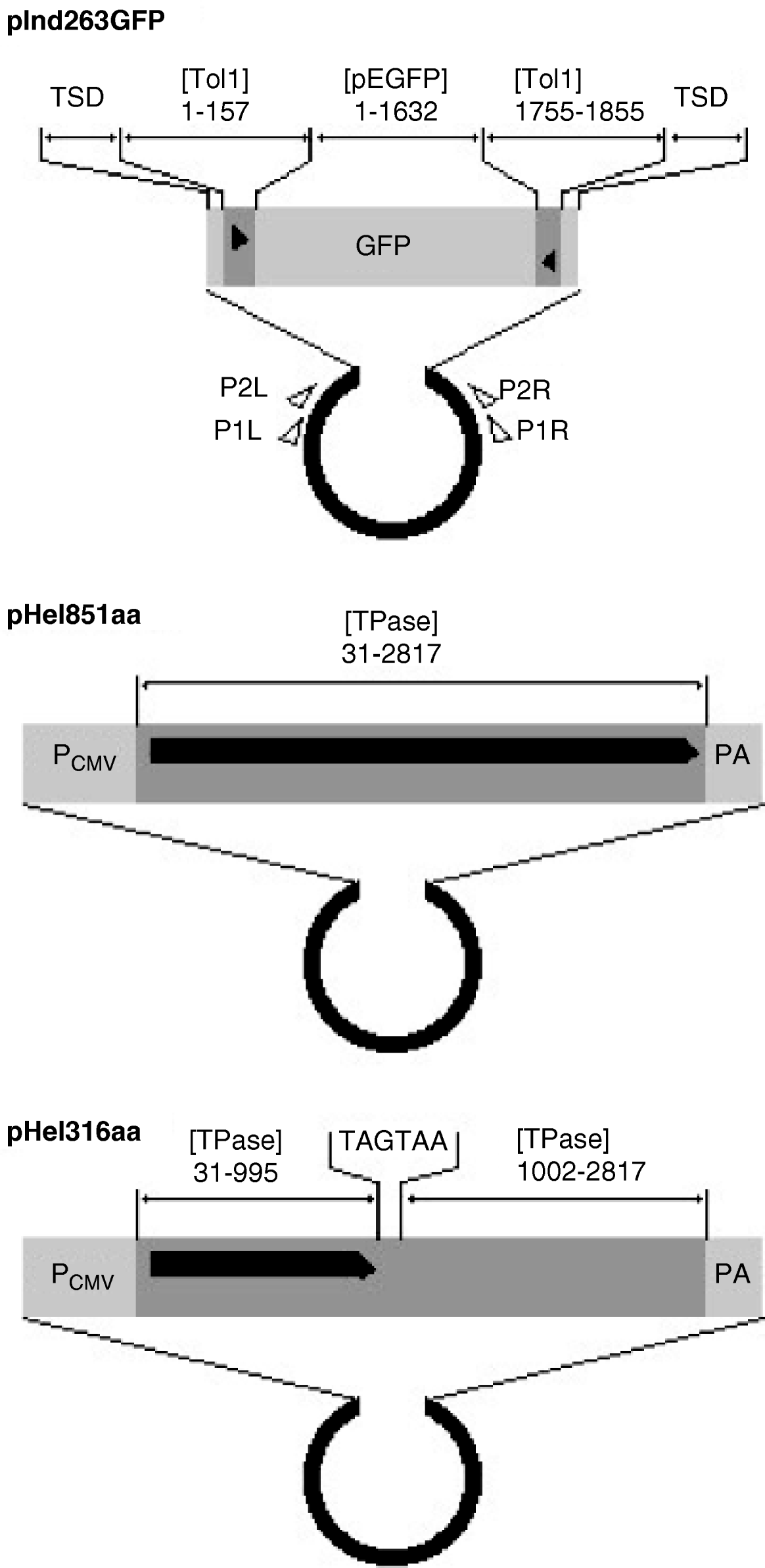
Fig. 1. Organization of indicator and helper plasmids. pInd263GFP is an indicator plasmid that carries the left and right terminal regions of the earliest-discovered copy of the Tol1 element (Tol1-tyr, 1855 bp) and the GFP gene inserted in it, with the plasmid backbone of pUC19. pHel851aa is a functional helper plasmid constructed by inserting the entire coding sequence for the Tol1 transposase (851 amino acids) between the CMV promoter and the poly(A) addition signal regions of the plasmid pCI (Promega, Madison, WI, USA). pHel316aa is a defective helper plasmid created by introducing nucleotide alterations by PCR into the helper plasmid. The sequence for nt 996–1001 is ATGAAA, which corresponds to the amino acids methionine and lysine in the transposase. This sequence was changed to TAGTAA, which resulted in the introduction of two consecutive stop codons in the middle of the transposase ORF. Nucleotide blocks included as components of the plasmids are indicated with abbreviations for GenBank sequence files and the nucleotide positions. The abbreviations are: [Tol1] D84375 (the Tol1-tyr element), [TPase] AB264112 (the transposase gene), [pEGFP] U55763 (plasmid pEGFP-C1 of Clontech Laboratories, Mountain View, CA, USA). TSD indicates the target site duplication whose sequence is CCTTTAGC. Other abbreviations are: PCMV, the CMV promoter; PA, poly(A) addition signal. The thick black horizontal line with a pointed right end indicates the reading frames of the helper and defective helper plasmids. The black triangles are terminal inverted repeats in the Tol1-tyr element. Small open triangles show locations and directions of the PCR primers used in excision assays.
(ii) Xenopus embryos and microinjection
For natural mating, X. laevis females and males were injected with 600 and 300 units of chorionic gonadotropin (Aska Pharmaceutical, Tokyo, Japan), respectively. Fertilized eggs were collected and dejellied in 3% cystein (pH 7·9), washed with 0·1× Steinberg's solution (Peng, Reference Peng, Kay and Peng1991), and transferred to [3% Ficoll, 0·1× Steinberg's solution]. Next, 2- to 4-cell-stage embryos were injected with approximately 5 nl of plasmid DNA. Its solvent was [88 mM NaCl, 15 mM Tris-HCl (pH 8·0)]. The final DNA concentrations of the indicator and helper plasmids were 5 μg/ml and 50 μg/ml, respectively. The 1:10 ratio of the indicator and helper plasmid was selected because this ratio is close to the ratio (1:9) that gave the highest transposition frequency among several ratios tested with mammalian cells (Koga et al., Reference Koga, Shimada, Kuroki, Hori, Kusumi, Kyono-Hamaguchi and Hamaguchi2007). The injected embryos were cultured in 0·1× Steinberg's solution at 20°C.
(iii) PCR analysis
GFP-positive embryos were harvested at the tailbud stage. Each embryo was homogenized in 100 μl of [10 mM Tris-HCl, 10 mM EDTA (pH 8·0), 200 μg/ml proteinase K] and incubated at 50°C for more than 12 h, and then 2 μl of the homogenate was used for PCR to detect excision. PCR was performed using KOD Plus polymerase (Toyobo, Osaka, Japan). The primers used, representing parts of pUC19, were P1L (nt 208–237 of GenBank file L09137) and P1R (nt 770–741). Note that the Tol1 element was inserted in the indicator plasmid in place of nt 400–441. The concentrations of dNTPs, MgSO4 and the primers were 0·2 mM, 2 mM and 0·5 μM, respectively. The PCR conditions are described in each case.
(iv) Cloning and sequencing analyses
The PCR reaction mixture was diluted with water to 1:500, and 1 μl of the diluted solution was used for another round of PCR. The primers used for this PCR were P2L (nt 338–367 of L01937) and P2R (nt 650–621). This nested PCR was to ease cloning of the products of the first-round PCR. PCR products obtained were cloned into pBluescript II KS(–) (Stratagene, La Jolla, CA, USA) at its EcoRV site and sequenced with the T3 and T7 primers using an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
3. Results
(i) Introduction of plasmids into embryos and their recovery
We performed two experiments: A and B. In experiment A, the indicator plasmid (pInd263GFP) and the functional helper plasmid (pHel851aa) were injected into 2- or 4-cell-stage X. laevis embryos. Experiment B was a negative control experiment on transposase function in which the defective helper plasmid (pHel316aa) was used in place of the functional helper plasmid. In experiments A and B, 154 and 168 embryos, respectively, were injected with the DNAs, and 112 and 136 of these reached the tailbud stage. There was no apparent difference in the survival rate between A and B (χ2=3·07, d.f.=1, P>0·1).
It is a prerequisite for a successful detection of excision that the injected DNA molecules are incorporated into the nucleus, because the transposase enzyme is supplied only when the transposase gene in the helper plasmid is transcribed. We monitored the nucleus incorporation of DNA by examining the expression of the GFP gene that had been embedded in the indicator plasmid. The frequencies of GFP-expressing embryos were 57% (64/112) and 65% (88/136) in experiments A and B, respectively, with no apparent difference between these two frequencies (χ2=1·48, d.f.=1, P>0·4). No apparent difference was observed, either, in the spatial patterns of the GFP expression by microscopy of the embryos. We selected 12 embryos expressing the GFP gene most strongly (A1–A12) from embryos of experiment A, and similarly 12 embryos (B1–B12) from experiment B. We then recovered plasmid DNA separately from these 24 embryos.
(ii) PCR analysis of recovered plasmids
Using the recovered DNAs as templates, we conducted two kinds of PCR analyses with a pair of primers (P1L and P1R) that encompass the Tol1 element on pInd263GFP.
The first analysis was to confirm the recovery of DNA molecules of the indicator plasmid. The distance between the two primers on pInd263GFP was 2·4 kb, and fragments of this size were observed with all the samples (Fig. 2, upper panel). The amount of PCR products varied among individual samples, but there was no apparent difference in the degree of variation between the two groups A and B. The recovery of the indicator plasmid was thus confirmed. Precise excision of the Tol1 portion from pInd263GFP was expected to result in a PCR product of 535 bp in length. A band of this size was not seen with any of the samples, indicating that plasmid DNA molecules that had undergone excision, if present, were much fewer than intact plasmid DNA molecules.

Fig. 2. Detection of excision products in X. laevis embryos by PCR. A1–12 and B1–12 are embryos of experiments A and B, respectively, used for the PCR analyses. The upper panel is a photograph of gel electrophoresis after PCR to amplify the entire Tol1 region. The PCR conditions were: [94°C, 120 s], 33× [94°C, 20 s; 64°C, 20 s; 68°C, 150 s] and [68°C, 60 s]. Five microlitres of the 20 μl PCR reaction mixture was electrophoresed on a 1·0% agarose gel. Bands of 2·4 kb were observed with all the samples of both A and B. The lower panel shows the result of PCR to effectively amplify excision products. The extension time was shortened to 40 s, and the other conditions were the same as those for the first PCR. The PCR reaction mixtures were electrophoresed on a 2·0% agarose gel. Bands of about 535 bp were observed with the A1–A12 samples, and not with the B1–B12 samples.
The second PCR analysis was conducted with a shorter extension time (40 s instead of 150 s) which was expected to be insufficient to amplify the entire Tol1 region (Fig. 2, lower panel). In this case, all embryos of experiment A (A1–A12) produced DNA fragments of about 535 bp and, in contrast, no PCR product was observed with the embryos of experiment B. These results suggested occurrence of deletion of the Tol1 portion from pInd263GFP in embryos of experiment A and absence of such events in embryos of experiment B.
(iii) Sequencing analysis of PCR products
To determine the breakpoints of the rearrangements of pInd263GFP, we then performed a sequencing analysis of the PCR products obtained from the 12 embryos from experiment A. We reamplified the PCR products with nested PCR, using primers P2L and P2R, and then cloned them into a plasmid vector. A single clone was chosen at random from colonies derived from each embryo. Therefore, these samples represented products of independent deletion events. Fig. 3 shows the alignment of the nucleotide sequences of the 12 PCR products. All or most of the Tol1 sequence was found to have been deleted in all the samples, proving the occurrence of the Tol1 excision while the indicator plasmid was present in frog cells. In 11 of the 12 samples (A1–A6, and A8–A12) the entire Tol1 region had been deleted, and parts of the TSD sequences of 1–7 nucleotides had been left behind at their breakpoints. In the case of A7, 39 nucleotides at the Tol1 right end had been left and 77 nucleotides in the left Tol1 flanking region, including one unit of the TSD, had been taken away. Another feature was substitution or addition of G residues that was observed with 7 of the 12 samples.
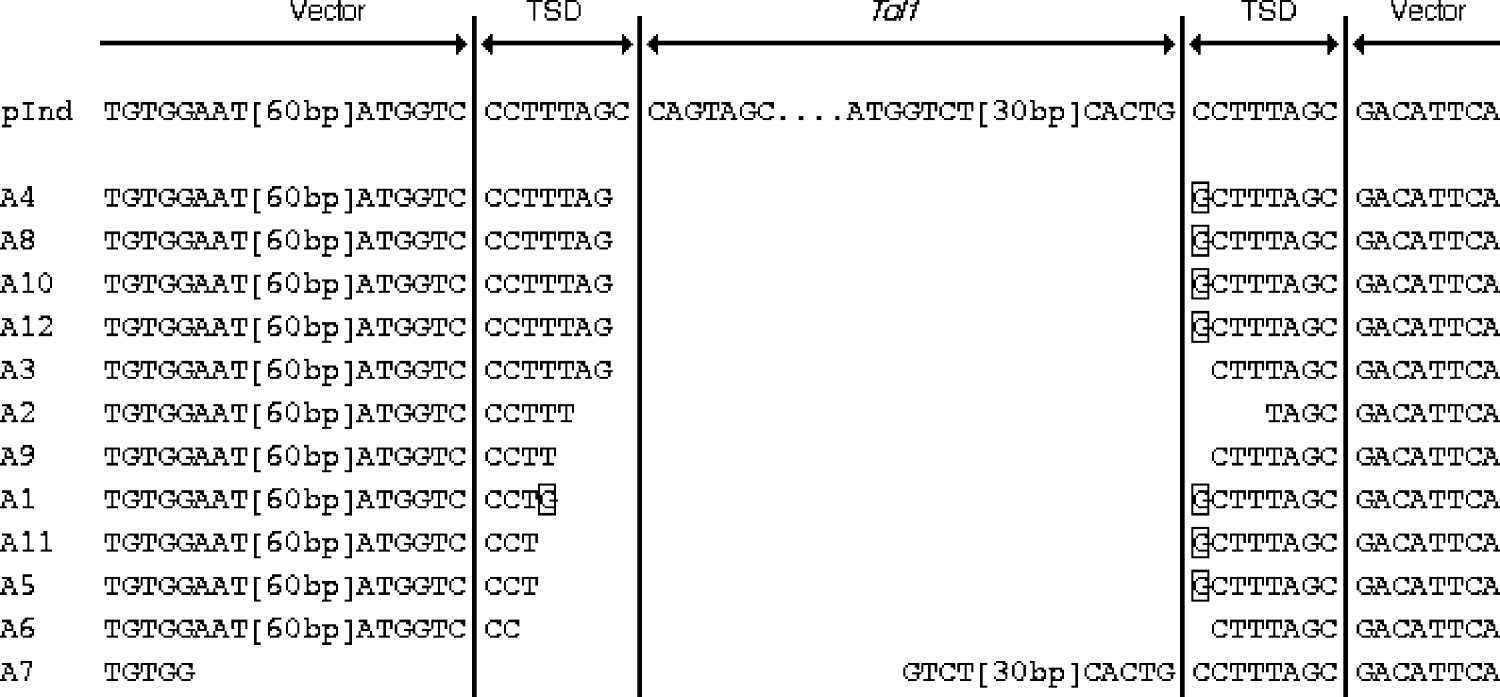
Fig. 3. Alignment of nucleotide sequences of excision breakpoints and their vicinities. pInd at the top is the sequence of pInd263GFP, shown for reference. TSD stands for target site duplication, which was present in pInd263GFP. The framed nucleotides indicate residues that are not seen in pInd263GFP. Two segments in the vector and Tol1 portions are indicated by their lengths in brackets. Their sequences are: [60 bp], nt 504–445 of L09137 (pUC19); [30 bp], nt 1821–1850 of D84375 (Tol-tyr).
4. Discussion
We introduced, by microinjection, the indicator plasmid (pInd263GFP) into 2- or 4-cell-stage embryos together with either the functional helper plasmid (pHel851aa) or the defective helper plasmid (pHel316aa). Between these two experiments, no apparent difference in the frequency or spatial patterns of the GFP expression was observed at the tailbud stage of embryos, indicating that the efficiency of DNA intake into the nucleus was not significantly different. We further confirmed the recovery of the indicator plasmid in similar amounts from the embryos in experiments A and B. Therefore, the cause of the clear difference in the amount of PCR products between the two experiments was the difference in the nucleotide sequences of the two helper plasmids. Such a difference is present only in a 6-nucleotide segment: two codons for the 317th and 318th amino acids in pHel851aa and two stop codons in pHel316aa. It can thus be concluded that the deletion of the Tol1 region from the indicator plasmid was mediated by an enzyme produced from pHel851aa and was not mediated by a protein from pHel316aa.
The significance of the above conclusion is that the Tol1 element can undergo excision in cells of X. laevis. This contention is supported by the fact that deletion of the Tol1 region is associated with various footprint sequences, as commonly observed with many DNA-based transposable elements, including hobo of Drosophila (Atkinson et al., Reference Atkinson, Warren and O'Brochta1993), Activator of maize (Wessler et al., Reference Wessler, Baran, Varagona and Dellaporta1986), Tam3 of snapdragon A. majus (Coen et al., Reference Coen, Carpenter and Martin1986), mariner of Drosophila (Bryan et al., Reference Bryan, Garza and Hartl1990) and Tc1 of the nematode C. elegans (Plasterk, Reference Plasterk1991).
The substitution or addition of G (C in the other strand) observed with 7 samples is a phenomenon of interest. This is unlikely to be an artefact of DNA sequencing because we have examined the clones for sequences of both strands. The occurrence of this change in 7 independent excision events might reflect some DNA repair mechanism that is specific to this frog species or amphibians in general. We have so far sequenced more than 20 similar PCR products from the medaka fish and more than 20 from mammalian culture cells, but such an event has not been observed in these other species (data not shown).
Excision is part of the transposition reaction of DNA-based transposable elements. Taking into account the fact that elements of the hAT family have been proposed to transpose in a non-replicative manner (Kunze, Reference Kunze, Saedler and Gierl1996), it is likely that the entire transposition reaction can proceed in X. laevis cells. We are now attempting detection of the rest of the transposition reaction: reintegration of Tol1 into the chromosomes. However, this detection requires a suitable marker gene and a complex detection system, and about 1 year to raise and cross frogs to observe their progeny.
Of the 12 independent excision products examined, not one was a complete recovery of the original nucleotide sequence. However, this does not imply that reintegration of the excised elements into the chromosomes is expected to be similarly imperfect. It is a common phenomenon among DNA-based transposable elements that the entire element is cut out precisely at the element ends and inserted into a new position but the resultant vacant donor site is accompanied by addition or deletion of nucleotides. This is considered to be due to interruption of the repair of the double-strand break using the sister chromatid or homologous chromosome as a template (Plasterk, Reference Plasterk1991) and/or non-homologous end joining (Weinert et al., Reference Weinert, Min and Rio2005). We have recently cloned two Tol1 elements which were introduced into mouse chromosomes and their flanking regions, and we found that the entire 1855 bp nucleotide block had been successfully transposed, along with the 8 bp target site duplication which originated from the recipient chromosome (Koga et al., in press).
Several DNA-based transposable elements, such as Sleeping Beauty and Tol2, are currently used as genetic manipulation tools for the frog (Sinzelle et al., Reference Sinzelle, Vallin, Coen, Chesneau, Du Pasquier, Pollet, Demeneix and Mazabraud2006; Hamlet et al., Reference Hamlet, Yergeau, Kuliyev, Takeda, Taira, Kawakami and Mead2006). However, Tol1 has an advantage in its ability to carry large DNA fragments (Koga et al., in press). Therefore, Tol1 is not merely a new addition to the biotechnology arsenal for the frog but a useful tool that extends our biotechnology repertoire.
We are grateful to Hiroshi Hori, Akira Kawahara and Samuel S. Chong for helpful discussions. This work was supported by grant no. 19570003 to A. K. from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant to A. K. from the Yamada Science Foundation.



