


1. Introduction
Transposable elements are dispersed repetitive sequences that are, or were at some time in the past, capable of moving from one chromosomal location to another. DNA-based elements comprise one major class of transposable elements. Elements of this class are transposed directly from DNA to DNA in a ‘cut and paste’ manner, while retrotransposable elements move via RNA intermediates by ‘copy and paste’ mechanisms. The transposition reaction of a DNA-based element is catalysed by an enzyme called transposase, and the gene encoding the enzyme is usually located inside the element itself. In the ‘cut’ process of the transposition reaction, although the transposase enzyme excises the entire element from the chromosome, it recognizes only the terminal regions of the element. Because of this property, a copy of the element in which the transposase gene is defective, or even missing, can be transposed as long as the enzyme is provided by a complete, transposase-gene-carrying copy coexisting in the same host cell. Copies of complete and defective forms are thus called autonomous and non-autonomous copies, respectively.
The hAT family is one of the major families of DNA-based elements, and was named after the hobo, Activator and Tam3 elements of Drosophila, maize and snapdragon, respectively (Calvi et al., Reference Calvi, Hong, Findleys and Gelbert1991; Atkinson et al., Reference Atkinson, Warren and O'Brochta1993). Elements of this family have been found in a wide range of organisms, including animals, plants and fungi (Kempken & Windhofer, Reference Kempken and Windhofer2001; Rubin et al., Reference Rubin, Lithwick and Levy2001). In elements of this family, deletion of internal regions is the most common cause of transition from an autonomous copy to a non-autonomous copy (Fedoroff et al., Reference Fedoroff, Wessler and Shure1983; Streck et al., Reference Streck, MacGaffey and Beckendorf1986; Warren et al., Reference Warren, Atkinson and O'Brochta1994), and the underlying mechanism of such deletions is considered to be premature interruption of gap repair after excision of the element (Rubin & Levy, Reference Rubin and Levy1997). With one exception, all elements of the hAT family known to date comprise both autonomous and non-autonomous copies, or only non-autonomous copies, the latter case constituting the great majority. The exception is the Tol2 element of medaka Oryzias latipes, in which no copy suffering a deletion was found among more than 200 copies carried by medaka originating from a local population (Koga & Hori, Reference Koga and Hori1999), or among more than 200 copies in medaka collected at another 12 geographical locations (Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000). We proposed, as the reason for this exceptional situation regarding an hAT family element, that Tol2 is a recent invader of the medaka genome. The almost complete lack of nucleotide sequence variation among Tol2 copies supports this view (Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000).
The scale of our previous surveys appears to have been sufficient for proposing the above hypothesis that Tol2 is a young element in its host species. It was, however, possible that structural variation is present at such a low frequency that it could not be detected in our previous surveys. If a low-frequency variant were found and had features that turned out to lead to future proliferation in natural populations, the current status of the variant would serve as useful information for comparison with the results of future studies. One big difference in the research infrastructure between the time of our previous surveys and now is the availability of genome sequence databases of medaka. In the present study, we first examined the databases for structural variation of Tol2, and obtained information suggesting the presence of a copy suffering an internal deletion. After confirming that the deletion exists in the fish genome and is not an artefact that occurred at the stage of library preparation for the databases, we examined by PCR the distribution of this deletion in natural medaka populations. We surveyed samples from 58 locations (about five times the number of locations sampled in our previous surveys), and found that eight of these samples carried at least one copy of the deletion variant. Possible causes of the occurrence of this internal deletion in limited, but wide, areas are discussed.
2. Materials and methods
(i) Database search
Searches for variation in the nucleotide sequence of the Tol2 element were conducted using the BLAST program of Ensembl (http://www.ensembl.org) and the medaka genome browser of the National Institute of Genetics (http://dolphin.lab.nig.ac.jp/medaka/). The query sequence for initial searches was the entire sequence of the Tol2 element (EMBL D84375).
(ii) Fish samples
We used fish samples from 58 mass-mating stocks originally collected at various geographical locations (Fig. 1). We first conducted surveys using the 12 samples that had been materials for our previous surveys (Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000), and the 32 samples that had been used in another study to examine the distribution of the Tol1 element (Koga et al., Reference Koga, Wakamatsu, Sakaizumi, Hamaguchi and Shimada2009). Having found the short version of Tol2 in some samples originating from local populations in northern Japan, we added six samples collected in or around these regions, and eight more samples from locations in areas where the sampling density had previously been relatively low. Two commonly used laboratory strains, HNI and AA2, were also included in our analysis.

Fig. 1. Original collection sites of the 58 fish samples used in the present study. The diamond-shaped symbols indicate samples in which the deletion D1 was found, and the circles indicate samples in which D1 was not found. The collection sites were follows: (1) Higashidori, (2) Honjo, (3) Yokote, (4) Kajikawa, (5) Niigata-Tayuhama, (6) Niitsu, (7) Shirone, (8) Teradomari, (9) Ojiya, (10) Hamochi, (11) Nanao, (12) Kaga, (13) Maizuru, (14) Ine, (15) Kumihama-Nagae, (16) Toyooka, (17) Kasumi, (18) Hamasaka, (19) Hanamaki, (20) Sendai, (21) Kawachi, (22) Mito, (23) Odawara, (24) Fuji, (25) Nagoya, (26) Saori, (27) Hikone, (28) Kumano, (29) Kobe, (30) Saigo, (31) Miyoshi, (32) Hohoku, (33) Iwakuni, (34) Kamiura, (35) Aki, (36) Tosanakamura, (37) Saiki, (38) Saito, (39) Ashibe, (40) Kashima, (41) Sato, (42) Ginoza, (43) Sokcho, (44) Toseong, (45) Sachon, (46) Guoje, (47) Kwangi, (48) Jindo, (49) Sinpyong, (50) Samsan, (51) Guhang, (52) Buyong, (53) Simcheon, (54) Daebu, (55) Paltan, (56) Shanghai, (57) Ilan and (58) Kunming. The broken lines show rough boundaries of the four local populations. The two collection sites with asterisks are those belonging to a population different from that indicated in the map: (17*) Southern Japan and (54*) East Korea.
Medaka shows geographical variation. According to data on isozyme frequencies (Sakaizumi, Reference Sakaizumi1986) and nucleotide sequences (Takehana et al., Reference Takehana, Naruse and Sakaizumi2005), there are four geographical populations: Northern Japan, Southern Japan, East Korea, and China and West Korea. This population structure is also illustrated in Fig. 1.
Philippine medaka Oryzias luzonensis is a species closely related to Oryzias latipes (Naruse Reference Naruse1996; Takehana et al., Reference Takehana, Naruse and Sakaizumi2005). This species does not harbour the Tol2 element in its genome (Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000). We used genomic DNA of this species in a test for the detection ability of our PCR analysis.
(iii) Preparation of genomic DNA
We extracted genomic DNA from whole bodies of adult fish by the method described in Koga et al. (Reference Koga, Inagaki, Bessho and Hori1995) . The DNA concentration in the solvent (10 mm Tris (pH 8·0)) was adjusted to 20 ng/μl. Restriction enzyme digestion was conducted by treating 1 μg of genomic DNA with 20 units of enzyme. After 1 h of incubation for digestion, DNA was precipitated with ethanol and dissolved in 50 μl of the solvent, 20 ng/μl being the expected concentration on the assumption of no loss during treatments.
(iv) PCR analysis
We used ExTaq polymerase (Takara Bio Inc., Otsu, Japan) and reagents supplied with the enzyme for our PCR analysis. The reaction mixture was 25 μl in total volume, containing 0·4 μm of each primer, 2·0 mm MgCl2, 1×buffer, 0·1 μl of the polymerase, 100 ng (5 μl) of genomic DNA and extra DNA if necessary. The primers used were as follows: 1L (nt 1757–1784 of D84375), 1R (nt 2315–2288), 2L (nt 1–28) and 2R (nt 4682–4655). The PCR conditions were, unless otherwise noted, as follows: (120 s at 94°C), 32 cycles of (10 s at 98°C, 15 s at 64°C, 15 s at 72°C) and (30 s at 72°C). After the completion of PCR, 5 μl of the reaction solution was electrophoresed on a 90-mm-long agarose gel.
3. Results
(i) Identification and confirmation of an internally deleted Tol2 copy
It is a general feature of genome sequence databases that repetitive sequences, especially those of 1 kb or more in length, tend to be underrepresented. This is because assembly of shotgun sequence reads for contigs is harder with repetitive than with non-repetitive sequences. In fact, a BLAST search against the sequence files of the medaka Hd-rR strain, using the entire sequence of the Tol2 element (4682 bp) as a query, detected no contigs including the entire sequence, and there were only two hits showing alignment of more than 100 bp (data not shown) despite the presence of 13 ‘full-length’ Tol2 copies per haploid genome in this strain (Koga et al., Reference Koga, Iida, Hori, Shimada and Shima2006). For this reason, we could not expect clear evidence for the existence, if present, of structural variation of Tol2. We then analysed individual shotgun reads for differences from the Tol2 sequence. This analysis suggested that a Tol2 copy lacking a 119-nt block (nt 1793–1911) is present in the genome of the HNI strain. The ‘full-length’ Tol2 element carries a pair of internal inverted repeats (‘internal’ is prefixed here to distinguish from the terminal inverted repeats; designated as IIRs) of 302 bp (nt 1434–1735) and 303 bp (nt 1786–2088) and the 119-nt block is included in the right repeat unit (Figs 2 and 3). The two units are separated by 50 bp (nt 1736–1785), possibly forming a hairpin structure (Izsvak et al. Reference Izsvak, Ivics, Shimoda, Mohn, Okamoto and Hackett1999).
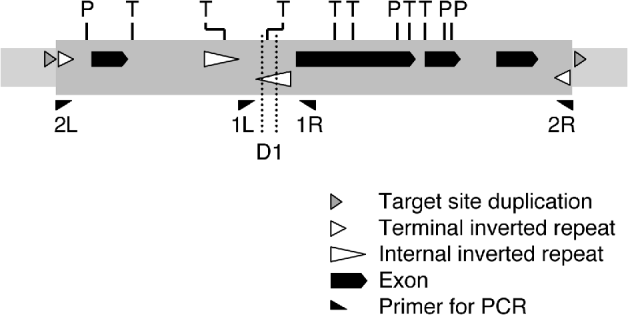
Fig. 2. Structure of Tol2. The structure of Tol2-F is shown, together with the position of the internal deletion D1 found in Tol2-D1. P and T indicate recognition sites for PvuII and TaqI, respectively. The elements are not exactly to scale.

Fig. 3. Nucleotide sequence of part of Tol2. The region containing the IIRs is shown here (nt 1321–2400 of D84375). The regions of the left (L) and right (R) repeat units are indicated by vertical lines and short arrows. The segment with lower-case letters is the 119-bp deletion D1. The long arrows indicate the PCR primers.
Because inverted repeat structures are known to be fragile during bacterial artificial chromosome (BAC) amplification in host bacteria (Doherty et al., Reference Doherty, Lindeman, Trent, Graham and Woodcock1993), there was a possibility that the deletion was an artefact that had been generated during the process of the genome library preparation. To determine if this had happened, we performed PCR to amplify the region of the right repeat unit from genomic DNA, using primers 1L and 1R. The expected length of a PCR product was 532 bp if it had originated from the ‘full-length’ Tol2 copy, and 413 bp if from a Tol2 copy that had suffered the 119-bp deletion. PCR amplification from genomic DNA of the HNI strain resulted in two bands with mobilities corresponding to these sizes (Fig. 4, lane 2). We cloned and sequenced the product fragments (0·53 kb and 0·41 kb), and thereby confirmed that the deletion was exactly at the 119-nt block. Thus, the deletion was not an artefact that occurred during library preparation, but a structural change that occurred in one or more Tol2 copies present in the HNI strain. We designate this 119-bp-long deletion as D1, and a Tol2-copy suffering this deletion as Tol2-D1. When we need to specify a ‘full-length’ Tol2 copy, we use the designation Tol2-F.
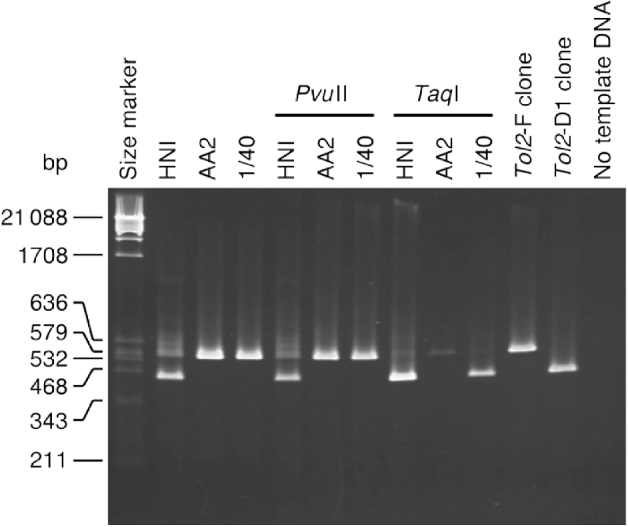
Fig. 4. Results of PCR to test for the detection ability. The size marker is λ phage DNA digested with PvuII. The sizes of the marker fragments are shown in the left margin. Template DNA was used without restriction enzyme digestion, or was digested with the enzymes shown above the lanes before PCR.
(ii) Preparation of virtual medaka genome containing a single Tol2-D1 copy
Tol2 constitutes multicopy sequences dispersed in the medaka genome. We wanted to attempt to detect Tol2-D1 copies by PCR, but we thought that Tol2-F copies coexisting in the genomic DNA would act as competitors for primers. An ideal survey of fish samples for the presence/absence of Tol2-D1 would be one having the ability to detect a single Tol2-D1 copy coexisting with many Tol2-F copies. As material for a test of the detection ability, we prepared a virtual medaka genome containing a single Tol2-D1 copy and 39 Tol2-F copies.
The reason for setting 40 as the total number of Tol2 copies was as follows. We had already examined the copy numbers per diploid genome by genomic Southern blotting (Koga & Hori, Reference Koga and Hori1999; Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000). We analysed a total of 24 fish samples, obtaining an average copy number of 17, the highest copy number 30 and the lowest copy number 10. Thus, the great majority of medaka samples were considered to contain 40 or fewer Tol2 copies.
We first amplified the entire Tol2 region from genomic DNA of an HNI fish by PCR using primers 2L and 2R, which represented the terminal regions of Tol2. In this PCR, we set the time period of the extension step at 6 min. Two bands 4·7 and 4·6 kb in length were observed, as expected, on an electrophoresis gel (data not shown). We cloned these bands into a plasmid (pT7Blue-2; 3·1 kb in length) by the TA-cloning method.
The haploid genome size of medaka has been estimated to be 0·68–0·85 Gb (Tanaka, Reference Tanaka1995). Here we use the upper limit of the estimate, 0·85 Gb. If we assume 39 copies of Tol2-F (4·7 kb) in a diploid genome, the Tol2 regions correspond to a fraction of 1·1×10−4 [(39×4·7 kb)/(2×0·85 Gb)] of the genome. Similarly, one copy of Tol2-D1 (4·6 kb) corresponds to a fraction of 2·7×10−6 [(4·6 kb)/(2×0·85 Gb)]. We thus added, to 10 μg of genomic DNA of O. luzonensis, 1·8 ng [(1·1×10−4)×(7·8/4·7)×10 μg] of DNA of the clone of Tol2-F, and 45 pg [(2·7×10−6)×(7·7/4·6)×10 μg] of DNA of the clone of Tol2-D1. We designate the DNA sample prepared in this way ‘1/40’.
(iii) Test for detection ability
We conducted PCR to amplify the region between primers 1L and 1R from DNAs of the HNI strain, the AA2 strain, and the ‘1/40’ sample (Fig. 4). Before PCR amplification, genomic DNA was treated in three ways: no restriction enzyme digestion, digestion with PvuII and digestion with TaqI.
PvuII and TaqI have several recognition sites in the Tol2 sequence (Fig. 2). In the region of D1, there is one site for TaqI. It can thus be expected that digestion of DNA with TaqI would prevent amplification of fragments from Tol2-F copies and lead to a higher efficiency of amplification from Tol2-D1 copies. PvuII does not have a recognition site in the region between 1L and 1R.
PCR amplification from undigested DNA of the HNI fish (Fig. 4, lane 2) yielded two bands (0·53 and 0·41 kb), indicating that the HNI fish contains both types of Tol2 copies. Only the 0·53-kb band was observed in the lane for the 1/40 sample. It was thus likely that the PCR was not sufficiently powerful to detect a single Tol2-D1 copy coexisting with 39 Tol2-F copies. The AA2 fish also exhibited only the 0·53-kb band, but this does not necessarily mean that all copies in this fish were Tol2-F copies.
PCR using PvuII-digested DNA (lanes 5–7) resulted in band patterns with no detectable difference from those of undigested DNA (lanes 2–4). We can infer from this result that restriction enzyme digestion and subsequent manipulations (such as ethanol precipitation that might cause loss of short restriction fragments) had no positive or negative effects on the detection ability of the PCR analysis.
Digestion with TaqI resulted in clear differences in band patterns. The HNI fish (lane 8) exhibited a 0·41-kb band and no 0·53-kb band, and the 0·41-kb band was broader than that in lane 2. The 1/40 sample (lane 10) exhibited a 0·41-kb band, in contrast to the absence of a band of this size in lane 4. Although this band was narrower than the corresponding band of the HNI fish (lane 8), it was distinguishable as a clear band. The AA2 fish exhibited a faint 0·53-kb band and no 0·41-kb band. A plausible explanation for this result is that all Tol2 copies in this fish are Tol2-F copies. The faint 0·53-kb band can be thought to be a product from a small amount of DNA fragments left uncut after the TaqI treatment, for reasons such as DNA methylation at the TaqI-recognition site or stochastic non-association of enzyme and substrate.
These results indicate that digestion of genomic DNA with TaqI has a positive effect on the ability to detect a D1-suffering copy in subsequent PCR analysis, and that our method is sufficiently powerful to detect a single Tol2-D1 copy coexisting with 39 Tol2-F copies.
(iv) Distribution of deletion
We examined 58 fish samples for the presence of Tol2-D1 in their genomes. Fig. 5 a shows the results of PCR amplification in which genomic DNA was used without restriction enzyme digestion. The primary purpose of this analysis was to confirm that the quality of genomic DNA was high enough for PCR and that the PCR worked. All samples exhibited a 0·53-kb band, and a 0·41-kb band was observed as an additional band in five samples (5, 16, 19, 21 and 23). Fig. 5 b shows the results of PCR amplification from TaqI-digested genomic DNA. The 0·41-kb band was observed in all five samples that exhibited this band in Fig. 5 a, and in three additional samples (6, 10 and 14). Thus, these eight samples carry in their genomes at least 1 Tol2-D1 copy together with Tol2-F copies, and the other 50 samples can be thought to have only Tol2-F copies. Five of the eight samples exhibiting the 0·41-kb band originated from the Northern Japan population, and the other three samples from the Southern Japan population.

Fig. 5. Survey of fish samples for D1. The numbers above the lanes indicate the sample collection sites shown in Fig. 1. M stands for the size marker. (a) PCR with genomic DNA not digested with a restriction enzyme. (b) PCR with TaqI-digested genomic DNA.
(v) Sequencing analysis of PCR products
We cloned and sequenced the 0·41-kb bands from the eight samples, and confirmed that their deletion breakpoints were identical to that of the 0·41-kb band of the HNI strain (Fig. 2).
4. Discussion
(i) Origin of D1
In the present study, we first searched genome sequence databases of medaka for variation in the sequence of Tol2, and obtained information suggesting the presence of an internally deleted Tol2 copy. We confirmed, by cloning and sequencing, the occurrence of the deletion, which we named D1. We then established an effective method to detect D1, and surveyed fish samples originally collected at 58 locations. Eight of these samples were found to carry Tol2-D1 copies.
Medaka inhabits East Asia, including Japan, Korea and China. Although the fish samples we used in this study may not represent well the four geographical populations of medaka, we can see a clear feature of the distribution of D1: the occurrence of D1 is restricted to the Northern Japan population and the eastern part of the Southern Japan population. This is, however, not a small area, considering that medaka is a freshwater fish species, mountain ranges divide the two populations, and medaka eggs and embryos are not tolerant to drying. The maximum distance between collection sites in which D1 was found is 600 km (between 14 and 16), and this distance appears to be too far for fish to migrate in a short time. The age of the deletion is likely to be fairly old if we assume that the Tol2-D1 copies we found originated from a single mutational event. Even if this is true, however, the age of D1 is not likely to be as old as that of the species. This is because Tol2 is highly homogeneous at the nucleotide sequence level (Koga et al. Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000): the Tol2 sequences (4682 bp, including exons of a total of 2319 bp) were virtually identical between samples from the Northern Japan and Southern Japan populations, while 49 nucleotide substitutions have accumulated in a 2051-bp-long region for the tyrosinase gene (exons, 1045 bp; introns, 1006 bp).
An explanation alternative to single origin is that D1 was generated multiple times. However, the results we have obtained so far appear to support the possibility of single origin. First, the breakpoint of D1 was identical among the eight locations. Second, D1 was not found in samples in western parts of the Southern Japan population (14 collection points) or Korea (13 collection points). If there is a nucleotide substitution specifically found in Tol2-D1 copies, the hypothesis of a single origin could be tested in detail. There is, however, little expectation of such a situation at present because Tol2 is highly homogeneous in nucleotide sequence (Koga & Hori, Reference Koga and Hori1999; Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000).
(ii) Autonomy of Tol2-D1
The transposase gene of Tol2 contains four exons, and all three introns have nucleotide blocks that fit the consensus sequences of splicing donor sites (/GTRAGT; Alberts et al., Reference Alberts, Johnson, Lewis, Raff, Roberts and Walter2007) and splicing acceptor sites (Y n NYAG/; Alberts et al., Reference Alberts, Johnson, Lewis, Raff, Roberts and Walter2007), just with one exceptional nucleotide in the third intron (Koga et al., Reference Koga, Suzuki, Maruyama, Tsutsumi and Hori1999). D1 is located inside the first intron (Fig. 2). In the Tol2-D1 sequence, there are no nucleotide blocks that appear to fit the above consensus sequences at or around the breakpoint. For this reason, it is not likely that D1 affects the structures of mature mRNAs for the Tol2 transposase. It is, however, not known if D1 creates a novel enhancer or abolishes an existing enhancer. Gene expression experiments with cloned Tol2-D1 fragment will be required to determine whether Tol2-D1 is autonomous.
(iii) Possible mechanisms for spread of D1
On the assumption that Tol2-D1 copies had a single origin with respect to D1, we discuss below possible mechanisms for the spread of D1 in natural medaka populations.
It is a commonly accepted idea that natural selection acts against the transposition activity of transposable elements, and selection pressure is relatively weak against non-autonomous copies (Hartl et al., Reference Hartl, Lohe and Lozovskaya1997). If Tol2-D1 is non-autonomous, weakened natural selection may be a factor contributing to the spread of D1, in addition to stochastic changes in copy number.
An effect of D1 on the secondary structure of DNA might be another factor. Tol2-F has IIRs in the first intron of the transposase gene (Fig. 2). The repeats are considered to have originated from insertion of a pair of MITEs (miniature inverted-repeat transposable elements) called Angel, and energy calculations have suggested that they form a stable hairpin structure (Izsvak et al., Reference Izsvak, Ivics, Shimoda, Mohn, Okamoto and Hackett1999). In fact, one often encounters unusual events while handling clones of Tol2 in molecular biology experiments. For example, faint extra bands often appear in photos of gel electrophoresis, extension of complementary strands by DNA polymerase is often halted in the IIR region in reactions for DNA sequencing, and plasmid clones carrying IIRs are frequently rearranged unless one uses host bacterial strains that carry the uvrC and umuC mutations. If the IIR regions also form hairpin structures in host cells, they may give rise to a high rate of scission of chromosomal DNA at their positions, and a decrease in the efficiency of DNA duplication. D1 is located inside the right repeat unit of the IIRs. One conceivable factor that might have facilitated the spread of D1 is reduced instability of DNA sequences of this region due to a decreased size of the hairpin structure. The scale of possible hairpin structures differs between Tol2-F and Tol2-D1. The stem regions of Tol2-F1 are about 300 bp in length, with a nucleotide identity of 96·1%. The loop region is 50 bp in length. In the case of Tol2-D1, the stem regions are about 180 bp, the nucleotide identity is 95·6%, and the loop region is 177 bp. Thus, in Tol2-D1, the stem regions are shorter, and the hairpin structure is expected to be less rigid. These changes may lead to a decrease in the instability of chromosome DNA at the IIR regions of inhabitant Tol2 elements. Thus, with Tol2-D1, the survival rate of host cells per Tol2 copy may be higher, or the energy cost for chromosome duplication may be lower. To test this speculation, experiments examining DNA structures using Tol2 clones will be needed, and continuous future surveys of natural medaka populations for the distribution of D1 will also be required. The current status of D1 reported here is expected to provide information with which results of future surveys can be compared for determination of factors controlling population dynamics of this DNA-based element.
(iv) Possibility of participation of a vector
All the hypotheses we have discussed so far are based on the assumption that Tol2-D1 copies, whether they are of a single origin or multiple origins, have spread only by migration of fish and inheritance by fish. Now below we will discuss another scenario by challenging this assumption.
We now assume participation of a vector that carries the transposable element from fish to fish or from an external source to fish. We also assume that the vector is capable of moving over a long distance in a short time. Viruses and parasitic organisms are examples of such vectors. With these assumptions, we can explain all of the following results: medaka exhibits a high level of nucleotide sequence variation (Sakaizumi et al., Reference Sakaizumi1986; Takehana et al., Reference Takehana, Naruse and Sakaizumi2005; Kasahara et al., Reference Kasahara, Naruse, Sasaki, Nakatani, Qu, Ahsan, Yamada, Nagayasu, Doi, Kasai, Jindo, Kobayashi, Shimada, Toyoda, Kuroki, Fujiyama, Sasaki, Shimizu, Asakawa, Shimizu, Hashimoto, Yang, Lee, Matsushima, Sugano, Sakaizumi, Narita, Ohishi, Haga, Ohta, Nomoto, Nogata, Morishita, Endo, Shin, Takeda, Morishita and Kohara2007), Tol2 is homogeneous in nucleotide sequence among different medaka populations and even among different species (Koga & Hori, Reference Koga and Hori1999; Koga et al., Reference Koga, Shimada, Shima, Sakaizumi, Tachida and Hori2000), and Tol2-D1 is found in different medaka populations (this study). An example of a scenario along this line is that variation accumulated in the medaka genome while medaka inhabited East Asia for a long time, Tol2 was introduced recently into medaka through wide-range invasion by a Tol2-carrying vector, and the variant Tol2-D1 was already present at the time of the invasion.
Recent bioinformatic studies have revealed that horizontal transfer of DNA-based transposable elements has occurred frequently in vertebrates (Ray et al., Reference Ray, Feschotte, Pagan, Smith, Pritham, Arensburger, Atkinson and Craig2008; Pace et al., Reference Pace, Gilbert, Clark and Feschotte2008; Novick et al., Reference Novick, Smith, Ray and Boissinot2010). For many of them, there is evidence for invasion of multiple species. The most striking example with respect to the width of the host range would be the Space Invaders element (Pace et al., Reference Pace, Gilbert, Clark and Feschotte2008), which is found in the genomes of several mammals (such as bat and opossum), and in lizard and frog. These authors considered that DNA viruses, especially poxviruses, are good candidates for a vector. There are also several reports suggesting horizontal transfer of transposable elements mediated by DNA viruses (Friesen & Nissen, Reference Friesen and Nissen1990; Jehle et al., Reference Jehle, Nickel, Vlak and Backhaus1998; Piskurek & Okada, Reference Piskurek and Okada2007).
As for medaka and the Tol2 element, there is no information about an exogenous vector at present. Nevertheless, the scenario invoking such a vector is, in our opinion, worth considering because Tol2 is highly active in a wide range of organisms, once artificially introduced into genomes (Balciunas et al., Reference Balciunas, Wangensteen, Wilber, Bell, Geurts, Sivasubbu, Wang, Hackett, Largaespada, McIvor and Ekker2006; Hamlet et al., Reference Hamlet, Yergeau, Kuliyev, Takeda, Taira, Kawakami and Mead2006; Keng et al., Reference Keng, Ryan, Wangensteen, Balciunas, Schmedt, Ekker and Largaespada2009).
We are grateful to Drs Elizabeth Nakajima, Lira Yu, Hiroshi Hori and Shinichi Morishita for helpful discussions. The fish samples were obtained from the medaka division of the National BioResource Project of Japan. This work was supported by grant no. 19570003 from the MEXT of Japan to A. K., and by Global COE program A06 of Kyoto University.





