


1. Introduction
The epigenome is a multitude of chemical compounds that instruct the genome how to function. These functions include DNA methylation, chromatin modifications, nucleosome positioning and alterations in the noncoding RNA (ncRNA) profile (Verma, Reference Verma2015), which may affect gene expression and its regulation without altering the DNA sequence. With the rapid development of bio-technologies, many unknown epigenetic phenomena have been revealed, which has led to an increasing interest in epigenetic mechanisms of individual development and of diseases.
Epigenetic modulation exerts either a positive or negative feedback pathway, leading to silencing of one of the two X chromosomes in female cells in early development (Gendrel & Heard, Reference Gendrel and Heard2014), genomic imprinting (Gupta et al., Reference Gupta, Chakraborty, Taggar, Kumar, Sharma and Singh2014a) and paramutation (Brink, Reference Brink1973). The epigenetic pattern and changed phenotype appear after mitosis or meiosis (Huang et al., Reference Huang, Jiang and Zhang2014). Although various epigenetic programs have been identified, only three main categories are widely accepted, which are as follows: DNA methylation, histone modification and ncRNAs. DNA methylation is the most studied; this includes the methylation of the fifth carbon of cytosine. The DNA methylation pattern is dynamically regulated by DNA methyltransferases (DNMTs) during development. DNA methylation pattern includes endogenous transposable elements repression, chromosome alignment and segregation, second X chromosome control via inactivation in females and modulation of imprinted gene expression. Histone modifications are characterized by histones that can be covalently modified at their flexible N- or C-terminal tails, as well as globular domains. This phenomenon is associated with DNA methylation. It is also regarded as one of the key components of chromatin packaging (Henikoff & Shilatifard, Reference Henikoff and Shilatifard2011). Histones function both positively and negatively in gene expression regulation; histones are also mainly governed by post-translational histone modifications (PTMs) and specific histone variants (Kimura, Reference Kimura2013). PTMs regulate transcription and other DNA-templated functions, which are dynamically mediated by specific modifying enzymes (Fan et al., Reference Fan, Krautkramer, Feldman and Denu2015). PTMs can be classified into several categories, including lysine acetylation, lysine and arginine methylation, arginine citrullination, lysine ubiquitination, phosphorylation, fatty acylation and ADP-ribosylation, which affect DNA function individually or collectively. NcRNAs are comprised of short and long ncRNAs. These molecules have been highlighted in biological processes with the development of deep sequencing and transcriptome analyses; however, these molecules have been previously regarded as junk RNAs.
Therefore, epigenetic programs are essential to basic biologic events that are associated with physiological and pathological processes, including skeletal genesis, bone remodelling and bone metabolic disorders (Fan et al., Reference Fan, Krautkramer, Feldman and Denu2015). This review will discuss the current knowledge of epigenetics in the skeletal system and will strive to shed new light on the understanding of epigenetic roles in bone and cartilage tissues.
2. DNA methylation
DNA methylation is the methylation of the fifth carbon of cytosine, which is known as one of the most important epigenetic modifications. It plays a considerable role in genome stability, gene expression and individual development in both prokaryotes and eukaryotes.
DNA methylation, which occurs on gene promoters, is linked to transcriptional repression (Chae et al., Reference Chae, Park, Lee, Nephew and Sun2013). Although it happens mostly in CpG sites, non-CpG sites have also been observed to be methylated. However, the role of non-CpG methylation remains unclear. Conversely, gene enhancers that have undergone DNA methylation are correlated with active gene expression (Moore et al., Reference Moore, Le and Fan2013). Except for DNA methylation of promoters and enhancers, DNA methylation emerges on different genomic regions with different functions. DNA methylation, which occurs on intergenic regions, represses the expression of potentially harmful genetic elements when the methylation of the CpG islands impairs the binding of transcription factors, the recruiting of repressive methyl-binding proteins and gene silencing. However, more studies are needed to determine how DNA methylation of the gene body contributes to gene regulation.
DNA methylation is accomplished by DNMTs, including these five enzymes: DNMT1, DNMT3A, DNMT3B, DNMT3L and DNMT2. DNMTs have been reported to be involved in the maintenance and de novo of DNA methylation (Uysal et al., Reference Uysal, Akkoyunlu and Ozturk2015). For example, DNMT1 mainly maintains DNA methylation patterns (Elliott et al., Reference Elliott, Sheaffer, Schug, Stappenbeck and Kaestner2015). On the other hand, DNMT3A and DNMT3B are essential for de novo methylation in early development (Okano et al., Reference Okano, Bell, Haber and Li1999). DNMT3L cooperates with both DNMT3A and DNMT3B and enhances the initiation of DNA methylation (Cheng & Blumenthal, Reference Cheng and Blumenthal2008). The fifth DNA methyltransferase, DNMT2, targets RNA methylation in mammals, rather than participating in genome methylation. This phenomenon modifies the 38th cytosine residue in the anticodon loop of certain tRNAs and enhances the stability of tRNAs (Ashapkin et al., Reference Ashapkin, Kutueva and Vanyushin2016). For example, DNMT2 affects polypeptide synthesis during haematopoiesis by modulating the stability and fragmentation of tRNAs (Tuorto et al., Reference Tuorto, Herbst, Alerasool, Bender, Popp, Federico, Reitter, Liebers, Stoecklin, Gröne, Dittmar, Glimm and Lyko2015).
DNA methylation changes the nucleic acid structure and the gene phenotype when it occurs at a certain gene region, which makes DNA methylation a potential new biomarker in biological research (Lee et al., Reference Lee, Yoshida, Abe, Nakabayashi, Wakeda, Hata, Marquette, Blum, Sode and Ikebukuro2016). The earliest approach that detected DNA methylation was through the quantification of total methylated cytosines in a chunk of DNA (Umer & Herceg, Reference Umer and Herceg2013). With development in technology, genome-wide analyses, such as next-generation sequencing (NGS) technologies, have been widely used in genome-wide and locus-specific DNA methylation analyses. Particularly, novel approaches emerge with pertinence, sensitivity and speed. For example, the global estimation of 5-methylcytosine content can be detected by high-performance capillary electrophoresis with UV-V detection, liquid chromatography with electrospray ionization mass spectrometric detection and LUminometric Methylation Assay (Berdasco et al., Reference Berdasco, Fraga and Esteller2009). These methods provide information about the disease process and progress and can be useful in various clinical settings and in drug screening (Umer & Herceg, Reference Umer and Herceg2013). Locus-specific DNA methylation analysis provides insights into early epigenetic reprogramming events and identifies rare cells with unique epigenetic signatures (Cheow et al., Reference Cheow, Quake, Burkholder and Messerschmidt2015). Many methods are available, including methylation-specific PCR, MethyLight, combined bisulfite conversion restriction analysis, bisulfite (Sanger) sequencing, bisulfite pyrosequencing and methylation-sensitive high-resolution melting (Umer & Herceg, Reference Umer and Herceg2013). Although genome-wide and locus-specific analyses provide a comprehensive understanding of DNA methylation, higher coverage and accuracy are required to detect DNA methylation. Firstly, restriction landmark genomic scanning, methylation-specific arbitrarily primed PCR, methylation-sensitive representational differential analysis and amplification of intermethylated sites are widely used, with some inevitable limitations for samples with large quantities of DNA. These approaches are labour-intensive and include complex procedures. Furthermore, microarray-based (DMH, CHARM, HELP assays, MeDIP-Chip and Illumina Infinium) and sequencing-based approaches (bisulfite treatments [BS-Seq, MethylC-Seq, reduced representational bisulfite sequencing (RRBS) and methyl CpG-binding domain (MBD)-isolated genome sequencing (MIGS)]) enhance the sensitivity, simplify the experimental setup and reduce the sequence redundancy in genome-scale DNA methylation analysis (Umer & Herceg, Reference Umer and Herceg2013). In addition to these widely used DNA methylation detection methods, some novel approaches have subsequently emerged, which include an electrochemical detection system and a methyl-sensitive fluorescence polarization assay. The former technique targets MBD and a glucose dehydrogenase-fused zinc finger protein (Lee et al., Reference Lee, Yoshida, Abe, Nakabayashi, Wakeda, Hata, Marquette, Blum, Sode and Ikebukuro2016). The latter technique recognizes the palindromic target sequence CCGG through restriction endonucleases, namely, MspI and HpaII (Shiratori et al., Reference Shiratori, Feinweber, Knothe, Lötsch, Thomas, Geisslinger, Parnham and Resch2016). The developments in DNA sequencing technologies, as well as methods to identify and map 5-hydroxymethylcytosine are expected to augment our current understanding of epigenomics.
3. Histone modifications
Eukaryotic DNA is tightly packaged within the 2–10 µm nucleus, which requires several metres of DNA to be compact (Rothbart & Strahl, Reference Rothbart and Strahl2014). These packages are mainly comprised of core nucleosome particles, which are formed of 147 DNA base pairs wrapped around an octamer of histones (two copies each of H2A, H2B, H3 and H4). The central histones modulate the function and dynamics of the chromatin by the presence of specific histone variants and PTMs (Biterge & Schneider, Reference Biterge and Schneider2014).
On the other hand, histone variants are the key players in the shape of chromatin structure and the regulation of fundamental cellular processes, such as chromosome segregation and gene expression (Vardabasso et al., Reference Vardabasso, Hasson, Ratnakumar, Chung, Duarte and Bernstein2014). Histone variants replace the canonical histones to change DNA expression timings (DNA replication independent) and mRNA characteristics (Biterge & Schneider, Reference Biterge and Schneider2014). This phenomenon alters the stability, dynamics and accessibility of DNA (Weber & Henikoff, Reference Weber and Henikoff2014), More importantly, histone variants play important roles in disease progression in certain cases. For example, MacroH2A is a histone variant that is overexpressed in patients with Huntington's disease and steatosis-associated hepatocellular carcinoma (Biterge & Schneider, Reference Biterge and Schneider2014). H3.3 is the variant of H3 that replaces H3K27me3 in paediatric glioma; it is associated with reduced survival in patients with paediatric glioma (Chan et al., Reference Chan, Fang, Gan, Hashizume, Yu, Schroeder, Gupta, Mueller, James, Jenkins, Sarkaria and Zhang2013). Furthermore, subsequent studies have reported that other core histone variants, namely H2A.X, H2A.Z, CENP-A and linker histone H1 variants, are linked to biological development of diseases (González-Romero et al., Reference González-Romero, Riveracasas, Frehlick, Méndez, Ausió and Eirínlópez2012).
For PTMs, covalent modification occurs in histones at their flexible N- or C-terminal tails, as well as globular domains. PTMs also play important roles in many biological processes, including in DNA stability and expression. With the advancement of approaches to biochemical systems, PTMs can be classified into the following categories: lysine acetylation, lysine and arginine methylation, arginine citrullination, lysine ubiquitination, phosphorylation, fatty acylation and ADP ribosylation. These processes are precisely modulated by enzymes that transfer specific chemical groups to implement different modification. Histone acetylation is catalysed by diverse enzymes, in which two converse enzymes, namely, histone acetyltransferases and deacetylases, modulate the acetylation status of histones (Bannister & Kouzarides, Reference Bannister and Kouzarides2011). Additionally, a large number of histone methyltransferases catalyse the methylation on the ε-amino group of lysine residues and form the most prevalent histone methylation (Fan et al., Reference Fan, Krautkramer, Feldman and Denu2015). PTMs have diverse functions with enzyme involvement. For example, histone acetylation, which is the so-called opening up of chromatin, makes DNA more accessible to other protein factors (Fan et al., Reference Fan, Krautkramer, Feldman and Denu2015). Histone methylation level is associated with activating transcription, resulting in different chromatin states, influencing aging and aging phenotypes (McCauley & Dang, Reference McCauley and Dang2014; Berr et al., Reference Berr, Zhang and Shen2016). Furthermore, cross-talk between different histone modifications, which may constitute a histone code, have been observed, which may help fine tune overall control (Bannister & Kouzarides, Reference Bannister and Kouzarides2011).
Thousands of experiments have provided enormous data repositories in terms of the genome-wide binding pattern of modified histones by ChIP-seq (Rivera & Ren, Reference Rivera and Ren2013). Although ChIP-seq is the gold standard for mapping PTMs, limited resolution, dependence on antibodies and the need for large amounts of starting material have limited its application. To break the limitations, researchers have proposed a ChIP-exo technique to provide a single bp accuracy, in which an exonuclease precisely trims the ChIP DNA of the cross-linking site into a small fragment (Rhee & Pugh, Reference Rhee and Pugh2011). Furthermore, a nano-ChIP-seq protocol, which is combined with a high-sensitivity small-scale ChIP assay and a tailored procedure, generates high-throughput sequencing libraries from scarce amounts of ChIP DNA (Adli & Bernstein, Reference Adli and Bernstein2011). This nano-ChIP-seq does not only decrease the need for starting materials, but it also makes the entire procedure faster. Additionally, many novel approaches have emerged with comprehensiveness, high-resolution and short setup features, including ChIP-bisulfite-sequencing (ChIP-BS-seq) (Brinkman et al., Reference Brinkman, Gu, Bartels, Zhang, Matarese, Simmer, Marks, Bock, Gnirke, Meissner and Stunnenberg2012), bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) (Challen et al., Reference Challen, Sun, Mayle, Jeong, Luo, Rodriguez, Mallaney, Celik, Yang, Xia, Cullen, Berg, Zheng, Darlington, Li and Goodell2014) and other high-resolution mass spectrometry assays (Lin & Garcia, Reference Lin and Garcia2012).
4. NcRNAs
Previously, mRNA function had been mainly considered to provide protein-coding information only (Kumari & Sampath, Reference Kumari and Sampath2015). With the development of deep sequencing and transcriptome analysis, only a tiny portion of the biological genome corresponds to protein-coding sequences; in addition, most genomic loci produce large transcripts rather than proteins, which are defined as ncRNAs (Perez et al., Reference Perez, Jang and Alevizos2013). Although these ncRNAs had been regarded as junk RNAs for a long time, they have now been identified to have a role in many physiological processes, including the maintenance of self-renewal, direction of cell lineage (Guan et al., Reference Guan, Zhang, Liu and Belmonte2013) and expression of hundreds of genes (Varela et al., Reference Varela, Roberts and Wood2013).
NcRNAs are classified into the following two categories based on their length: small and long ncRNAs (lncRNAs) (Hirose et al., Reference Hirose, Mishima and Tomari2014). Small ncRNAs include microRNAs (miRNAs), small interfering RNAs (siRNAs) and Piwi-interacting RNAs (piRNAs). MiRNAs are one of the most widely studied small ncRNAs. A total of 52% of miRNAs are located in human intergenic regions, 40% lie within the intrinsic regions of genes and the final 8% are exonic (Hsu et al., Reference Hsu, Huang, Hsu, Lin, Tsou, Tseng, Stadler, Washietl and Hofacker2006). Mature miRNAs have similar physiological roles to other RNAs, such as transcriptional activation and inhibition, epigenetic repression and controlled degradation rates (Mott & Mohr, Reference Mott and Mohr2015). Furthermore, by targeting the complementary sequence of miRNAs, they modulate over 60% of the translation of protein-coding genes (Mott & Mohr, Reference Mott and Mohr2015). Not only in the modulation of gene translation, miRNA also present intricate functions in different tissues (Guo et al., Reference Guo, Ingolia, Weissman and Bartel2010). For example, miR-125b up-regulates and displays oncogenic potential in colon cancer and haematopoietic tumours. Conversely, miR-125b down-regulation contributes to malignant transformation in mammary tumours and hepatocellular carcinoma (Banzhaf-Strathmann & Edbauer, Reference Banzhaf-Strathmann and Edbauer2014). siRNAs are usually paired with mRNAs with perfect complementarity and are directed in their endonucleolytic cleavage and destruction (Hirose et al., Reference Hirose, Mishima and Tomari2014). On the other hand, piRNAs play a pivotal role in germline genome protection from transposons (Hirose et al., Reference Hirose, Mishima and Tomari2014). lncRNAs are the second class of ncRNAs, which regulate the RNA and protein content of a cell on the transcriptional and post-transcriptional level (Melissari & Grote, Reference Melissari and Grote2016). These molecular mechanisms include dosage compensation, chromatin regulation, genomic imprinting and nuclear organization (Yan et al., Reference Yan, Arfat, Li, Zhao, Chen, Yin, Sun, Hu, Yang and Qian2016). In addition, a small number of lncRNAs modulate the recruitment of RNA polymerase II and induce chromatin remodelling in global or local gene expression in trans or cis (Shi et al., Reference Shi, Sun, Liu, Yao and Song2013). Therefore, lncRNAs are present in pathological conditions such as cancer and cardiovascular disease, which makes lncRNAs a novel potential biomarker for clinical usage (Schmitz et al., Reference Schmitz, Phillip and Herrmann2016).
Moreover, a small part of ncRNAs codes for proteins including early nodulin 40 and MtHAP2-1 in plants. At the same time, some mRNAs may modulate post-transcriptional level gene expression, which is independent of their encoded proteins. These two kinds of RNAs are called cncRNAs (Kumari & Sampath, Reference Kumari and Sampath2015), which have shed new light on the understanding of ncRNAs. A total of 300 alternatively spliced bifunctional RNAs may be observed in the human genome (Ulveling et al., Reference Ulveling, Francastel and Hubé2011). The exploration of protein-coding and noncoding functions for cncRNA loci may be highlighted in future studies (Kumari & Sampath, Reference Kumari and Sampath2015).
NcRNA realization has been expanded with the advent of NGS (Ilott & Ponting, Reference Ilott and Ponting2013). In particular, lncRNAs have been identified by RNA-seq technology (Li et al., Reference Li, Zhang and Zhou2014). Moreover, RNA-seq data integration detects active transcription to further understand the reported lncRNAs (Chen et al., Reference Chen, Chen, Lai, Lin, Wu, Tung, Liu, Shih, Chen, Lin, Ma, Huang, Wu, Hong, Chu, Wu, Li and Chen2016 a). These findings may help to discover novel lncRNAs and may improve the characterization of identified lncRNAs.
5. Bone and cartilage diseases associated with epigenetics
Mesenchymal stromal cells (MSCs) are typical adult progenitor cells, which possess multidifferentiation capacity in vitro and in vivo (Takahashi et al., Reference Takahashi, de Andrés, Hashimoto, Itoi and Oreffo2015). MSCs are one of the promising candidate cells for skeletal regeneration due to their convenient isolation and immune-modulatory capability. The epigenetic changes of MSCs are essential in the differentiation of MSCs into bone and cartilage. These processes include DNA methylation, histone modifications and miRNAs.
(i) Osteogenic and chondrocyte differentiation of MSCs by epigenetics
A coordinated cascade of transcription factors and epigenetic modifications drive gene transcription, and cause specific cell fate, and this is indispensable for terminal differentiation of multipotent stem cells (Meyer et al., Reference Meyer, Benkusky, Sen, Rubin and Pike2016). For example, the osteogenic and adipogenic differentiation of MSCs are partly regulated by transcription factors, including peroxisome proliferator-activated receptor-γ (PPAR-γ) and Runt-related transcription factor 2 (Runx2) (James, Reference James2013), and various epigenetic alterations (Glemžaitė & Navakauskienė, Reference Glemžaitė and Navakauskienė2016). These processes are accompanied by a loss of the Brachyury gene. Brachyury inactivation is associated with the methylation of its promoter, which represses stem cell-associated genes (Dansranjavin et al., Reference Dansranjavin, Krehl, Mueller, Mueller, Schmoll and Dammann2009). In addition to the Brachyury gene, the cytosine methylation accumulation at the endogenous thyroid hormone receptor interactor 10 (Trip10) promoter reduces Trip10 expression, which accelerates osteogenic differentiation (Hsiao et al., Reference Hsiao, Lee, Hsu, Tseng, Jin, Sun, Hung, Yeh, Yan, Lai, Sun, Lu, Chang, Tsai, Huang and Leu2010). Osteocalcin (OC) is a non-collagenous bone matrix protein, which is partly regulated by DNA methylation and histone modifications. In MSC osteogenic differentiation in vitro, OC expression is usually increased with the decrease in DNA methylation. Additionally, OC is activated by the accumulation of H3 and H4 acetylation. Conversely, OC is inhibited by the decrease in H3 and H4 acetylation in the OC promoter and coding regions. This event occurs in the proliferative period of osteogenic differentiation (Takahashi et al., Reference Takahashi, de Andrés, Hashimoto, Itoi and Oreffo2015). For the H3 sub-family, the acetylation levels of H3K9Ac and H3K9me2 are associated with the activation and silencing genes, respectively, which are modulated by vitamin D receptors at specific gene promoters (Tan et al., Reference Tan, Lu, Huang, Dong, Kong, Li, Gao, Guo and Huang2009). For the epigenetic modulation of miRNAs, the function of miRNA has been reported in the inhibition of mRNA translation and degradation (Wei et al., Reference Wei, Shi, Zheng, Zhou, Inose, Wang, Guo, Grosschedl and Karsenty2012; Takahashi et al., Reference Takahashi, de Andrés, Hashimoto, Itoi and Oreffo2015). For example, miR-206 overexpression inhibits the differentiation of osteoblasts by the target of connexin 43 (Inose et al., Reference Inose, Ochi, Kimura, Fujita, Xu, Sato, Iwasaki, Sunamura, Takeuchi, Fukumoto, Saito, Nakamura, Siomi, Ito, Arai, Shinomiya and Takeda2009). MiR-34 inhibits mouse osteoblast proliferation and differentiation by targeting SATB2, which is a nuclear matrix protein involved in osteoblast differentiation (Wei et al., Reference Wei, Shi, Zheng, Zhou, Inose, Wang, Guo, Grosschedl and Karsenty2012). MiR-27a and miR-489 down-regulate the osteoblast differentiation by targeting PEX7; on the other hand, miR-148b up-regulates the differentiation (Schoolmeesters et al., Reference Schoolmeesters, Eklund, Leake, Vermeulen, Smith, Force and Fedorov2009).
The epigenetic mechanisms of chondrocyte differentiation of MSCs are scarce. In cartilage formation, DNA methylation levels of CpG-rich promoters of chondrocyte-specific genes are mostly at a low level (Ezura et al., Reference Ezura, Sekiya, Koga, Muneta and Noda2009). SOX9 is one of the master chondrogenic transcription factors, which is involved in chondrocyte differentiation and cartilage formation (Shi et al., Reference Shi, Wang, Acton, Eckert and Trippel2015). Age-dependent SOX9 expression is regulated by epigenetic mechanisms (Mak et al., Reference Mak, Singh, Turcotte and Ghert2015). Epigenetic studies revealed that DNA methylation levels increased at specific CpG islands of the Sox9 gene in mice articular chondrocytes (ACs) at 6 and 12 months old (Zhang et al., Reference Zhang, Lu, Miller, Barnthouse and Wang2016). Using the supplementation of 5-azacytidine, which is an inhibitor of DNA methylation, the DNA methylation level is reduced in the Sox9 promoter region, which elevates the level of Sox9 expression in ACs. This finding suggests that expression is associated with DNA methylation (Zhang et al., Reference Zhang, Lu, Miller, Barnthouse and Wang2016). Moreover, in DNA methylation, many kinds of histone modifications are involved in chondrocyte differentiation. For example, several transcription factors and coactivators, such as Scleraxis/E47 and p300, cooperatively modulate Sox9-dependent transcription through p300-mediated histone acetylation (Furumatsu & Asahara, Reference Furumatsu and Asahara2010). And a novel Runx2 enhancer is localized in the primary osteoblasts and is characterized by the presence of the histone variant H2A.Z, which contains sufficient elements to direct Runx2 expression to osteoblasts (Kawane et al., Reference Kawane, Komori, Liu, Moriishi, Miyazaki, Mori, Matsuo, Takada, Izumi, Jiang, Nishimura, Kawai and Komori2014). For the epigenetic modulation of ncRNAs, miR-101 and HOTTIP are up-regulated by targeting DNMT3B, which alters integrin-α1 methylation, which is a key protein of chondrocyte ossification (Kim et al., Reference Kim, Song, Han, Kim, Chun and Jin2013a).
(ii) Skeletal diseases associated with epigenetics
Epigenetic alterations are associated with the aetiology and pathology of bone and cartilage diseases. Osteoarthritis (OA) is a chronic multifactorial disease associated with specific genes. Specific transcription factors (TFs), cartilage-degrading enzymes, pro- and anti-inflammatory cytokines and extracellular matrix proteins contribute to OA development (Zhang & Wang, Reference Zhang and Wang2015). More importantly, advanced studies highlight the role of epigenetics in OA, including DNA methylation, histone modifications and miRNAs (Im & Choi, Reference Im and Choi2013) (Figure 1).
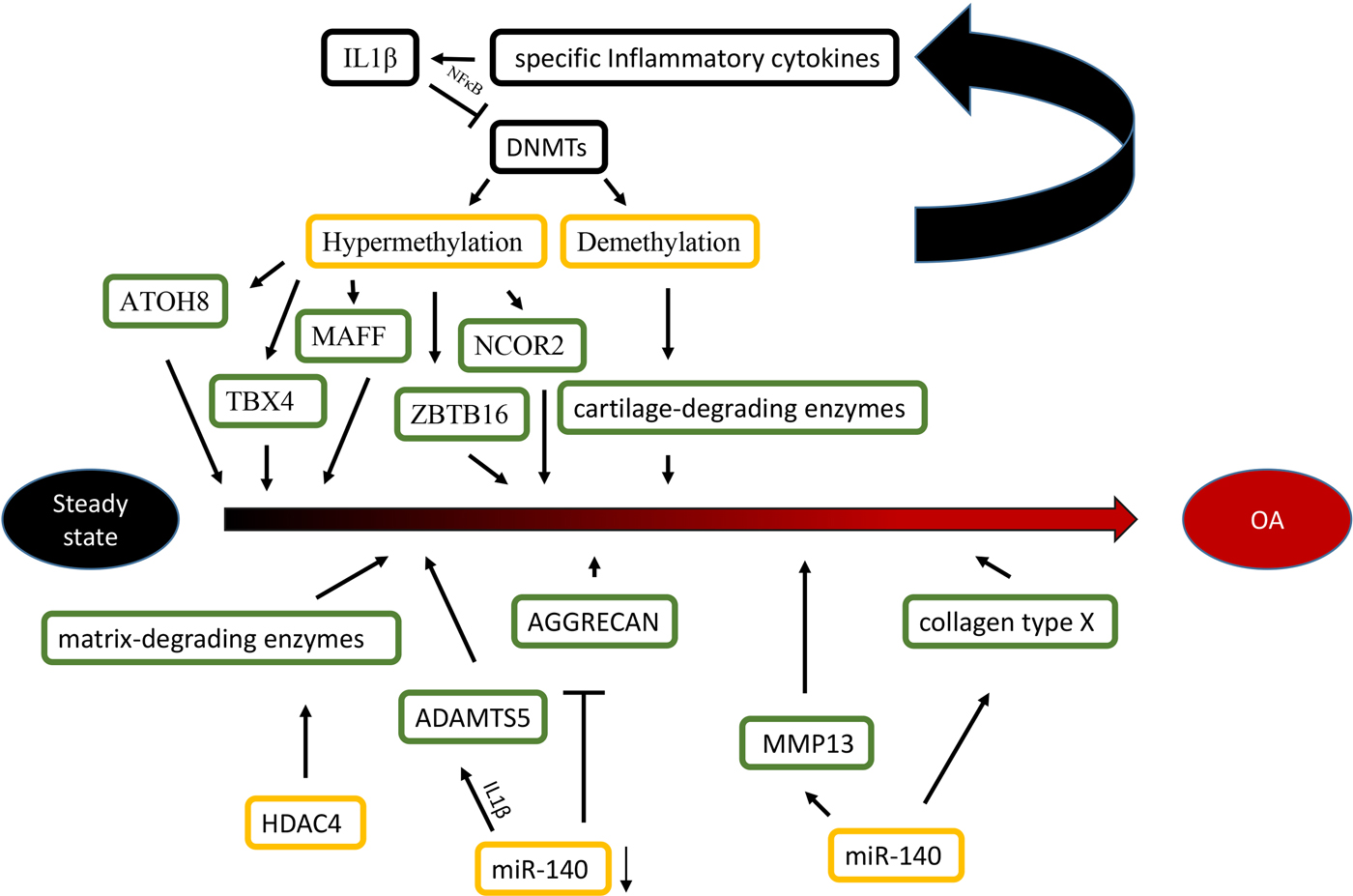
Fig. 1. Epigenetic mechanisms are associated with OA. Specific inflammatory cytokines were shown to accelerate the development of OA by directly targeting DNA methyltransferases, and the level of DNA methylation modulated the expression of specific inflammatory cytokines in chondrocytes reversely. For histone modifications, the overexpression of HDAC4 significantly led to a release of matrix-degrading enzymes, thus, contributing to bone loss associated with OA. For miRNAs, the reduction of miR-140 played an important role in OA progression by targeting ADAMTS5 and AGGRECAN. Conversely, The overexpression of miR-365 may accelerate the development of OA by targeting MMP13 and collagen type X (Col X).
Differences in the methylome between normal and OA knee articular cartilage have been identified, including 929 differentially methylated sites. Most of the methylated sites in OA (69%) are hypomethylated and enriched among gene enhancers in OA cartilage tissue (Jeffries et al., Reference Jeffries, Donica, Baker, Stevenson, Annan, Humphrey, James and Sawalha2014). For epigenetic changes of TFs, some OA related TFs (ATOH8, MAFF, NCOR2, TBX4, ZBTB16 and ZHX2) are significantly hypermethylated and down-regulated in OA cartilage. These results indicate that the DNA methylation level negatively affects the chondrocyte transcriptome and function in OA pathogenesis (Alvarez-Garcia et al., Reference Alvarez-Garcia, Fisch, Wineinger, Akagi, Saito, Sasho, Su and Lotz2016). For cartilage-degrading enzymes, the demethylation of MMP3, MMP9, MMP13 and ADAMTS4 promoters increase their gene expression in OA cartilage (Reynard, Reference Reynard2016). Additionally, inflammatory mediators modulate DNMTs, DNMT3B and DNMT3A, which target DNA methylation directly (Shen et al., Reference Shen, Abu-Amer, O'Keefe and McAlinden2017). IL-1β is a proinflammatory cytokine guiding the function of immune and proinflammatory cells and decreases the expression of DNMT3B and DNMT3A through NF-κB signalling (Shen et al., Reference Shen, Abu-Amer, O'Keefe and McAlinden2017). Another inflammatory chemokine, IL-8, shows increased demethylation in the promoter region in OA chondrocytes, which is correlated with enhanced IL-8 expression (Takahashi et al., Reference Takahashi, de Andrés, Hashimoto, Itoi and Oreffo2015). Furthermore, the DNA methylation level modulates the expression of specific inflammatory cytokines in chondrocytes reversely. Significant demethylation of CpG sites in the inflammatory chemokine IL-8 promoter increases the expression of IL-8 in the OA chondrocytes (Takahashi et al., Reference Takahashi, de Andrés, Hashimoto, Itoi and Oreffo2015).
With increasing data of genome-wide profiling of DNA methylation in OA, the alterations of DNA methylation affect more gene expression in OA. The demethylation of specific promoter and enhancer sites in GDF5, iNOS and SOST increase their gene expression in OA cartilage and isolated chondrocytes. Conversely, the increased methylation of SOX9, DIO2 and COL9 promoters caused reduced expression in OA cartilage (Iliopoulos et al., Reference Iliopoulos, Malizos and Tsezou2007; Verma and Dalal, Reference Verma and Dalal2011; Kim et al., Reference Kim, Park and Im2013b; Gupta et al., Reference Gupta, Niger, Buo, Eidelman, Chen and Stains2014b; Papathanasiou et al., Reference Papathanasiou, Kostopoulou, Malizos and Tsezou2015).
For histone modifications, HDAC4 is one of the key regulators in OA development (Lu et al., Reference Lu, Sun, Ge, Teng and Jiang2014). The HDAC4 expression level has a statistically negative correlation with OA severity (Lu et al., Reference Lu, Sun, Ge, Teng and Jiang2014). HDAC4 expression reduction significantly leads to a repression of the following matrix-degrading enzymes: MMP1, MMP3, MMP13, ADAMTS4 and ADAMTS5 (Lu et al., Reference Lu, Sun, Ge, Teng and Jiang2014). HDAC4 overexpression does not only decrease the expression of IL-1β, Cox2 and iNOS, but it also partially blocks IL-1β mediated effects in catabolic events in human OA chondrocytes (Thompson et al., Reference Thompson, Patel, Kelly, Wann, Hung, Chapple and Knight2015). However, HDAC4 inhibition has suppressed the expression of inhibited genes (Young et al., Reference Young, Lakey, Pennington, Jones, Kevorkian, Edwards, Cawston and Clark2005). For example, trichostatin A (TSA) and sodium butyrate HDAC inhibitors inhibit cartilage degradation by blocking key MMPs (MMP-1 and MMP-13) and aggrecan-degrading enzymes (ADAMTS4 and ADAMTS5). In addition, HDAC4 inhibitors, including vorinostat, TSA and sodium butyrate, are promising in the potential treatment of OA (Chen et al., Reference Chen, Bao, Hu, Feng and Wu2010; Makki & Haqqi, Reference Makki and Haqqi2016).
For miRNAs, while normal human articular cartilage expresses miR-140 in chondrocytes, its expression is significantly reduced in OA progression (Zhang et al., Reference Zhang, Liu, Xiao and Guo2012). The transfection of miR-140 in chondrocytes down-regulates IL-1β-induced ADAMTS5 expression and alleviates IL-1β-dependent repression of AGGRECAN gene expression (Miyaki et al., Reference Miyaki, Nakasa, Otsuki, Grogan, Higashiyama, Inoue, Kato, Sato, Lotz and Asahara2009). These findings indicate that decreased expression of miR-140 caused the abnormal gene expression profile seen in OA. MiR-365 overexpression in chondrocytes increases the expression of the matrix-degrading enzyme MMP13 and collagen type X, which may accelerate OA development (Yang et al., Reference Yang, Guan, Tian, Wang, Sun and Chen2016). Moreover, the interplay of histone modifications and miRNAs have been identified in the aetiology of OA. Subsequently, evidence shows that miR-365 directly targeted HDAC4, which led to HDAC4 expression down-regulation (Yang et al., Reference Yang, Guan, Tian, Wang, Sun and Chen2016), which accelerates the development of OA. Furthermore, a study of miR-381 has demonstrated that miR-381 overexpression promotes MMP13 and Runx2 expression by the inhibition of HDAC4, and miR-381 inhibitors increased HDAC4 expression and decreased Runx2 expression (Chen et al., Reference Chen, Sheng, Huang, Meng, Kang, Huang, Zhang, Liao and Zhang2016b). This finding provides us with a novel therapeutic method for the treatment of OA. This result may inspire researchers to study inhibitors that block the interaction between histone modifications and miRNAs in order to alleviate the severity of OA.
6. Future direction
This review summarizes the current advances in the study of epigenetics and discusses the epigenetic findings pertaining to the skeletal system. In addition to significant technological developments, emerging epigenetics research may provide new understanding of single genes, specific chromosome regions and the whole genome (Adli & Bernstein, Reference Adli and Bernstein2011). Moreover, modifications can be induced or inhibited by drugs to alleviate or cure disease; this finding needs further investigation. However, many challenges remain unsolved to fully use this epigenetic information. Thus; these challenges will require further research.
Acknowledgement
The authors thank the National Natural Science Foundation of China (No. 81472078、No. 31670951 and No. 31370992) and Shenzhen Basic Research Grant (No. JCYJ20170307100446585).

