



Introduction
Endoplasmic reticulum (ER) is one of the largest and dynamic organelles serving the myriads of functions inside the cell, such as protein synthesis and folding, lipid biogenesis and calcium metabolism (Ref. Reference Schönthal1). The protein synthesis and secretion function in the ER are accomplished by the ribosomes on rough ER. In contrast, smooth ER is central to the metabolism of lipid and carbohydrate, maintenance of calcium homeostasis, synthesis of fatty acids and detoxification (Ref. Reference Schönthal1). Proteins, which are co-translationally translocated as unfolded polypeptide chain inside the ER, are assisted by molecular chaperones, which mediate protein folding. In the ER, chaperone lectins like calnexin, calreticulin and chaperones of heat shock family (like BiP (also known as GRP78), GRP94), play a significant role in ensuring proper folding of proteins. Calnexin, the transmembrane protein; and its soluble analogue calreticulin recognise the N-linked glycoprotein in the ER, preventing the aggregation of glycoprotein and assuring proper folding before translocating from ER. However, calnexin and calreticulin re-bind the misfolded proteins and retain them in the ER till they are properly or folded or degraded. On the contrary, BiP recognises the hydrophobic region exposed in the misfolded proteins in the ER lumen and assists in the proper folding of proteins (Ref. Reference Halperin, Jung and Michalak2). BiP releases the protein after proper folding for further transport, but the unfolded or improperly folded proteins remain constrained by BiP and lead to stimulation of unfolded protein response (UPR) pathways, or by ER-associated protein degradation (ERAD) process (Ref. Reference Chakrabarti, Chen and Varner3). There is continuous on-going crosstalk between UPR pathways and ERAD, the two protein quality control processes work coherently to re-establish imperturbability in ER (Ref. Reference Hwang and Qi4). If these adaptive and survival pathways fail to pacify the ER stress and restore ER homeostasis, the cell is pushed towards apoptosis (Ref. Reference Hetz5).
UPR acts as a cytoprotective signalling cascade activated downstream in response to the disruption of ER cell homeostasis leading to the state of ER stress. Homeostasis in respect to ER is a physiological condition, wherein equilibrium is maintained between the demand of protein folding and the cellular ability to fold the protein accurately (Ref. Reference Ozcan and Tabas6). ER stress can arise under various situations, including internal factors such as genetic mutations that impair the protein folding ability of ER. There are other external causative agents of ER stress: altered metabolic conditions (nutrient/glucose deprivation), bacterial or viral infections, pharmacological interventions (e.g. tunicamycin, thapsigargin) which lead to over-burden on ER due to vigorous protein synthesis, imbalance in calcium homeostasis and post-translational modifications (Ref. Reference Zhang and Wang7). UPR relieves the stress by protecting the cell from the cytotoxic effect of malformed protein by attenuating the translation process, up-regulation of chaperones, which assist in protein folding and activating ERAD pathways which leads to proteasomal degradation and clearance of malformed proteins. Apart from the ERAD pathways, the terminally misfolded proteins can also be packaged into autophagosome and engulfed without releasing anything into the cytoplasm or affecting neighbouring cells (Ref. Reference Xu, Bailly-Maitre and Reed8). However, the persistent ER stress often culminates into the cell death process known as apoptosis, as a defence mechanism (Ref. Reference Metcalf9).
Unfolded protein response
Before the discovery of UPR, in the late 1980s, scientists observed an upregulation of a set of genes translating the proteins known as heat shock proteins (HSPs) like BiP/GRP78 during cellular stress conditions. They bind the denatured, unstable and misfolded proteins and assist in stabilising and proper folding of proteins, relieving the stress. Later, in the 1990s, scientists started their attempt at better understanding the UPR using Saccharomyces cerevisiae as a model organism and identified one of the UPR proximal sensors: IRE1. IRE1 would oligomerise and autophosphorylate itself, thereby activating the endoribonuclease domain that splices the HAC1 encoding mRNA translating into Hac1 protein that translocates to the nucleus and acts as a transcription factor (Refs Reference Hollien10, Reference Cox, Shamu and Walter11). The discovery of another novel kinase, protein kinase R-like endoplasmic reticulum kinase (PERK) involved in ER stress facilitated further elucidation of UPR. PERK protein was found to be embedded in the ER membrane and under stress, phosphorylates Ser-51 residue of eIF2α which has the downstream effect of downregulation of protein translation, relieving some stress from ER. Finally, the third branch of UPR triad was discovered in the late 1990s and known as activating transcription factor (ATF6). ATF6 is a 90 kD transmembrane glycoprotein embedded in the ER, which during stress conditions cleaves into 50 kD nuclear protein. After the characterisation of different branches on the UPR, in the early 2000s, further study on IRE1 pathway unveiled a novel mechanism of IRE1-mediated splicing of XBP1 mRNA and activation of genes involved in UPR from the spliced product acting as a transcription factor (Ref. Reference Back12). The gradual unfolding of the intricate mechanism of UPR over the years has highlighted the importance of protein folding in the cell, failure of which becomes the source of various human diseases. Before describing the disorders and the role of UPR in them and how to use UPR as a therapeutic, the mechanistic and structural understanding of the UPR is indispensable.
UPR signalling cascade is initiated by the three transmembrane ER stress sensors which activate the transcription by specific transducers: IRE1 (transmembrane protein kinase and endoribonuclease with spliced XBP1 as a transducer), PERK (transmembrane ER kinase with ATF4 as a transducer) and ATF6 (activating transcription factor 6 with cleaved ATF6 as a transducer) (Fig. 1). These sensors are maintained in an inactive state by a molecular chaperon protein BiP/GRP78, which upon disruption of ER homeostasis dissociates from the ER stress sensors, thereby converting them to the active state.
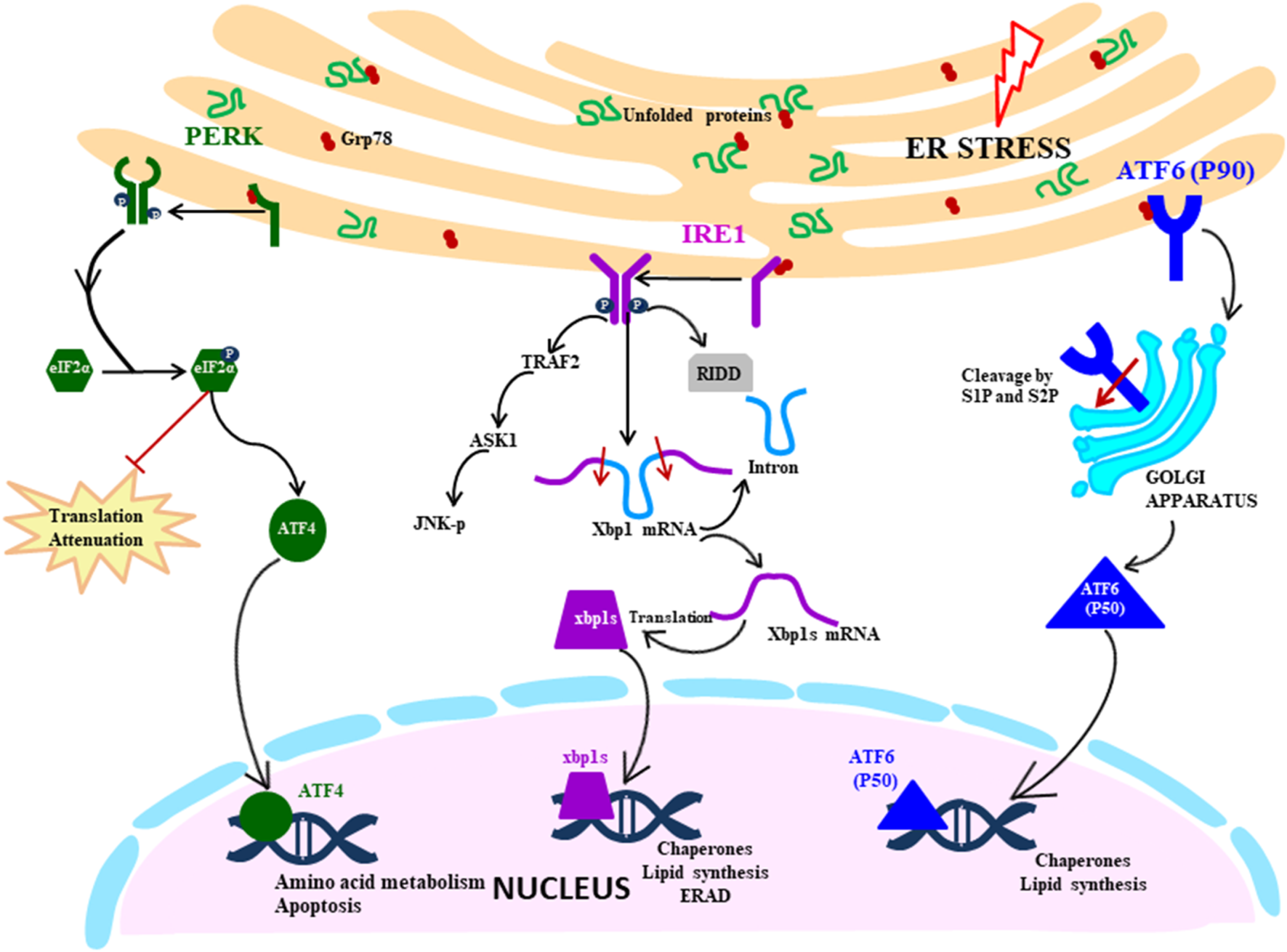
Fig. 1. UPR signalling cascade: Proximal sensors of UPR (PERK, IRE1 and ATF6) are released from GRP78 in response to the ER stress due to accumulation of unfolded proteins. PERK pathway is activated by oligomerisation and autophosphorylation of PERK, which facilitates the phosphorylation of eIF2α. It leads to the translation attenuation, but ATF4 bypass this blockage and gets activated and controls the genes in the nucleus. IRE1 pathway is activated by a similar mechanism of dimerisation and autophosphorylation, but downstream it splices the XBP1 mRNA, which then acts as a transcription factor in the nucleus. It also degrades mRNA by RIDD (regulated IRE1 dependent decay) and activates the JNK pathway (c-Jun N-terminal kinases) that initiates the apoptosis. ATF6 is activated by translocating to Golgi where it is cleaved by S1 and S2 proteases. The N-terminal segment moves to the nucleus and acts as a transcription factor.
IRE1 pathway
Among the three signalling pathways of the UPR, IRE1 arm of UPR response is the most ancient and evolutionarily conserved in yeast, mammalian species, fish and frogs (Ref. Reference Back12). IRE1 in mammals exist in two isoforms: IRE1α being ubiquitously expressed, whereas IRE1β expression is restricted to gastrointestinal tract (GI) and respiratory tract. IRE1 is ~110 kD type I transmembrane protein functioning as a bifunctional enzyme with protein serine/threonine-protein kinase domain and endoribonuclease domain residing in the cytoplasmic region. After sensing the ER stress, IRE1 sensor dissociates from BiP and instigates the UPR response by homodimerisation and trans-autophosphorylation and activates the kinase domain. After that, the kinase domain undergoes conformational changes induced by phosphorylation of the activation loop, thereby triggering the endoribonuclease domain's activity (Ref. Reference Ron and Hubbard13). The IRE1 (inositol requiring enzyme 1) pathway can also be directly activated by the unfolded proteins in a BiP-independent manner, as proposed by Gardner and Walter (2011) in the yeast model (Ref. Reference Gardner and Walter14). Activated IRE1 initiates the cytoprotective UPR response averting the increasing demand for protein folding to restore ER homeostasis. However, if the ER stress is not mitigated, IRE1 triggers the death response by performing regulated IRE1 dependent degradation (RIDD) of mRNA that either culminates into preserving the homeostasis or inducing cell death. RIDD demonstrates constitutive activity where it is increased at the time of ER stress. However, the basal level of RIDD activity is constitutively present and necessary to maintain the ER's homeostatic condition (Ref. Reference Maurel15). During UPR, the endoribonuclease domain of IRE1 is associated with splicing of 26 nucleotide intron from XBP1 mRNA, causing a frameshift thereby rendering it competent for undergoing translation into an active transcription factor which translocates to the nucleus (Ref. Reference Back12). Within the nucleus, the transcription factor interacts with promoters of genes encoding for chaperone proteins and those involved in retrograde transport of misfolded protein from ER to cytosol and in ERAD and initiate their transduction process to mitigate ER stress (Refs Reference Xu, Bailly-Maitre and Reed8, Reference Lai, Teodoro and Volchuk16). During persistent ER stress, IRE1 plays a role in switching pro-survival signalling to pro-apoptotic signalling with the induction of a reactive response like apoptosis (cell suicide) as a last resort to eliminate excessively stressed cells (Ref. Reference Szegezdi17). Apoptosis is a form of ‘programmed cell death’ pathway wherein it acts as a homeostatic and defence mechanism engulfing damaged and dysfunctional cells thereby eliminating them. During ER stress, activated IRE1 (phosphorylated) is also associated with a cytosolic receptor TRAF2 (tumour necrosis factor (TNF) receptor-associated factor) which recruits apoptosis signal-regulating kinase (ASK1). ASK1 activates c-Jun N-terminal kinase (JNK) which is a mitogen-activated protein kinase, involved in inducing apoptosis (Refs Reference Szegezdi17, Reference Fung, Liao and Liu18).
PERK pathway
During ER stress, the immediate response of the cell is the attenuation of translation globally, which is primarily mediated by the PERK pathway of the UPR (Ref. Reference Lai, Teodoro and Volchuk16). Like other ER stress sensors, monomeric PERK is associated with BiP in resting cells, which gets dissociated during stressed conditions leading to PERK mediated UPR signalling cascade. The luminal domain of PERK pathway is like IRE1, it oligomerises on the ER membrane and trans-autophosphorylates itself. PERK (PKR-like ER kinase) is a transmembrane protein Ser/Thr kinase that phosphorylates Ser-51 residue on eukaryotic initiation factor 2 (eIf2α) preventing the formation of pre-initiation complex and inhibiting the translation machinery as an attempt to reduce the load on ER (Refs Reference Xu, Bailly-Maitre and Reed8, Reference Lai, Teodoro and Volchuk16, Reference Schröder19). Although eIF2α-P leads to inhibition of translation, it selectively facilitates the translation of specific mRNA with short reading frame upstream at 5′UTR, translation of ATF4, and a bZIP transcription factor is upregulated in this way (Refs Reference Hollien10, Reference Schröder19). ATF4 regulates the expression of genes involved in protein synthesis, response to antioxidant stress and amino acid synthesis. ATF4 also regulates the expression of genes that encode proteins which determine the cell fate: the prosurvival (chaperone protein) or pro-apoptotic proteins (CHOP) which induces apoptosis in various ways. C/EBP-homologous protein (CHOP) is a transcription factor acting as a positive regulator of pro-apoptotic proteins (Bim) and negative regulator of anti-apoptotic proteins (Bcl2) (Ref. Reference Iurlaro and Muñoz-Pinedo20). ATF4/CHOP also targets GADD34 which acts as a negative regulator of the PERK pathway by dephosphorylating eIF2α, re-establishing global translation machinery and increasing the protein load on ER, indirectly directing the cells towards cell death (Ref. Reference Diehl and McQuiston21).
ATF6 pathway
ATF6 belonging to the family of bZIP transcription factors has a novel mechanism of initiating the pathway which is very distinct from the other two UPR sensors, the common link being held inactivated by BiP which dissociates in response to ER stress. ATF6 is a transmembrane protein usually existing in two isoforms: ATF6α and ATF6β which are ubiquitously present in tissues. The ER luminal domain sense the disturbance in ER homeostasis and the cytosolic domain acts as a transcription factor. During ER stress, BiP dissociates ATF6, which translocate to the Golgi apparatus where the cytoplasmic domain is cleaved by S1P (serine protease) and S2P (metalloproteases). The N-terminal cytosolic segment acts as a transcription factor which eventually migrates to the nucleus and regulates expression of various genes involved in ER stress response encoding chaperones, including CHOP, involved in pro-apoptotic pathway and protein involved in the ERAD pathway. ATF6 also cross-regulates IRE1 pathways by inducing the expression of XBP1 mRNA, thereby assisting in relieving the ER stress. ATF6 mediated upregulation of pro-apoptotic CHOP transcription factor plays a role in inducing apoptosis during prolonged stress condition by the regulation of members of the Bcl-2 family of proteins (Ref. Reference Logue22).
Role of UPR in human diseases
After understanding the different pathways of the UPR, it is imperative to gain insight into the role of UPR in disease prognosis as the accumulation of unfolded/misfolded protein contributes to the aetiology of diseases in humans (Table 1).
Table 1. Role of UPR in disease progression and control
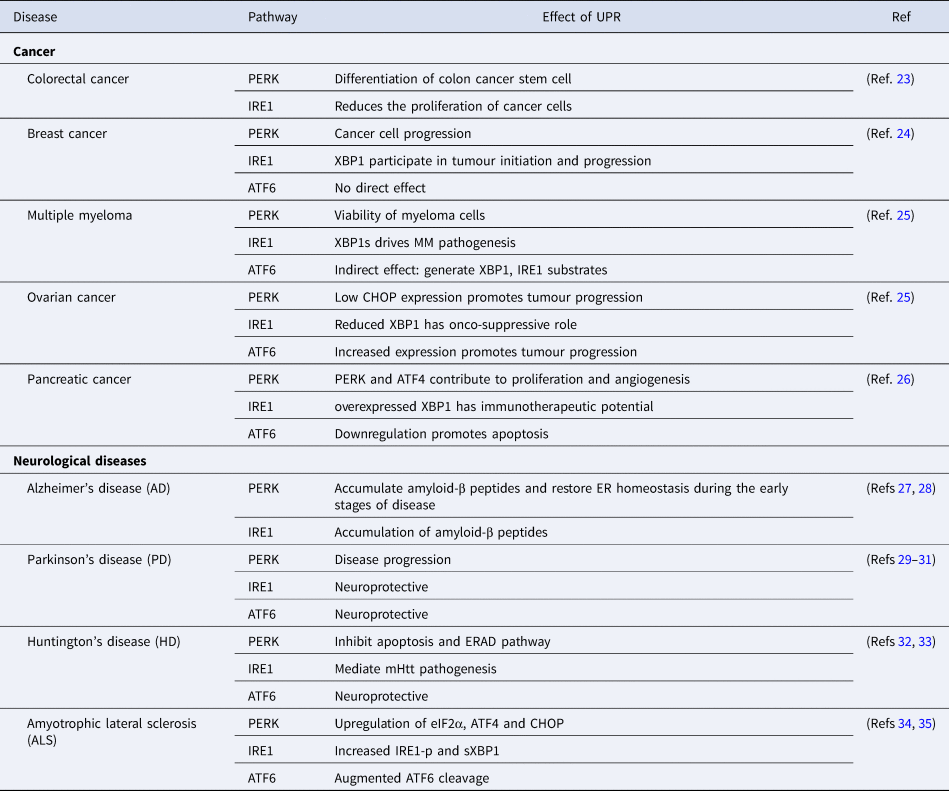
ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; CHOP, C/EBP-homologous protein; eIF2α, eukaryotic initiation factor 2-α; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; IRE1, inositol requiring enzyme 1; mHtt, mutant Huntington gene; MM, multiple myeloma; PERK, PKR-like ER kinase; XBP1, X-box binding protein-1.
Diabetes
Diabetes is a chronic metabolic condition characterised by elevated blood glucose levels leading to a condition called hyperglycaemia, due to inappropriate secretion of insulin or dysfunctioning of β-cells. The primary function of pancreatic β-cells is maintaining the blood glucose levels by synthesising and releasing insulin that mediates the uptake of glucose from the blood into the liver, where it acts as an energy reservoir and into muscle and fat cells where they can be converted to fat for long-term usage (Ref. Reference Eizirk, Cardozo and Cnop36). Diabetes mellitus is categorised into type 1 and type 2 diabetes. Type I diabetes is associated with impaired secretion of insulin due to the destruction of pancreatic β-cells. Whereas during type 2 diabetes individuals develop insulin resistance, deregulating the movement of blood sugar into the cells, leading to chronic hyperglycemic state (Refs Reference Ozcan and Tabas6, Reference Eizirk, Cardozo and Cnop36). Excess blood glucose leads to dysfunctional β-cells with impaired ability to secrete insulin, disturbing the balance in ER and disrupting the pro-insulin secretion, folding and processing. Early activation of UPR plays a protective role in relieving insulin resistance, regulating insulin production in β-cells and in modulating the capacity of chaperone proteins according to the increased load of protein (Ref. Reference Eizirk, Cardozo and Cnop36). UPR induced during obesity in mouse models also leads to the repression of insulin receptor signalling resulting in the development of systemic insulin resistance eventually developing into type 2 diabetes as demonstrated by Ozcan et al. (2004) (Ref. Reference Ozcan37). However, extended induction of UPR pathways possibly interfere with β-cells function and can lead to cell death via apoptosis. Apoptosis may cause deprivation of insulin, as a consequence of the destruction of pancreatic β-cells (Ref. Reference Demirtas38).
β-cells have highly developed ER with high expression of IRE1α and PERK participating in the secretion of insulin. IRE1 pathway is critical for biogenesis of insulin by regulating the translation of proinsulin mRNA, assisting in cleavage of the pre-proinsulin signal peptide, recruitment of ribosomes to the ER and in protecting the β-cells from oxidative and inflammatory stress (Ref. Reference Hassler39). IRE1-dependent activation of JNK eventually results in the development of insulin resistance in a mouse model with deleted XBP1 (Ref. Reference Ozcan37). Contrary to the above-mentioned results, a recent paper by Brown et al. (2020) (Ref. Reference Brown40) demonstrates the development of insulin resistance in ER stressed cells, independent of downstream UPR signalling pathways including the JNK signalling cascade. Recent studies have also shown some evidence of the involvement of these pro-apoptotic downstream targets of the PERK pathway, CHOP in exacerbating the collapse of β-cells during type 2 diabetes (Refs Reference Wang and Kaufman41, Reference Song42). The ATF6 branch of UPR, have been shown to interfere with gluconeogenesis, a metabolic process that results in the formation of glucose, thereby lowering the blood glucose level. The overall accumulating evidence suggests that the UPR plays a vital role in regulating blood glucose levels where it has both protective roles in relieving insulin resistance and contributing to restoring blood glucose homeostasis and the opposing role of contributing to insulin resistance or collapse of β-cells (Ref. Reference Eizirk, Cardozo and Cnop36).
The importance of UPR can be verified in Wolcott-Rallison syndrome, a rare autosomal recessive syndrome wherein mutation in PERK leads to dysfunctional β-cells, leading to infant diabetes linked with skeletal dysplasia and growth retardation (Ref. Reference Hassler39).
Cancer
Cancerous cells are characterised by high proliferation rates and metastasis under stressful microenvironment like hypoxic, oxidative stress, low glucose or compromised protein folding conditions (Ref. Reference Wang and Kaufman43). Several studies have revealed the crucial role of ER stress and UPR in the development and progression of various types of cancerous cells like multiple myeloma, glioblastoma, carcinoma of breast, liver, lung, oesophagus and stomach (Refs Reference Oakes and Papa44–Reference Fernandez46). During the initial stages of cancer progression, the transformation of a tumour cell is initiated by the loss of tumour suppressor genes or activation of oncogenes, followed by uncontrolled cell division. These processes overwhelm the ER with the increased load of protein folding, thereby stimulating UPR. UPR plays a pro-tumorigenic role by contributing to the hyper-proliferation induced by oncogene (Ref. Reference Madden47). However, the UPR signalling cascade has also been reportedly associated with anti-tumorigenic processes like senescence as reported by Denoyelle et al. (2006) (Ref. Reference Denoyelle48). The above-mentioned studies highlight the possibility of the opposing role of UPR in maintaining the equilibrium between tumour cell progression and senescence depending on the stage of the tumorigenesis process. Hence, the transformation step acts as a checkpoint wherein the cells with controlled activation of the UPR, progresses to the next stage or are eliminated by cell death mechanism (Ref. Reference Madden47).
After successful transformation, cancer cell advances to the cell proliferation stage wherein the IRE1 branch of UPR plays a crucial role as concluded in the studies of colon cancer, breast cancer etc. (Refs Reference Li49, Reference Logue50). The blocking of IRE1 expression in cancer cells reportedly results in a decrease in cell proliferation in vitro and in vivo, which is associated with a cell cycle arrest in the G1 phase. The activation of IRE1 and PERK branches of UPR also facilitates the sustenance of cancer cell in hypoxic and nutrient deprivation environments. During the hypoxic condition, XBP1 possibly interacts with HIF (hypoxia-inducible factor) transcription factors (Ref. Reference Chen51) and the PERK pathway increases the antioxidant responses and redirects the cells towards autophagy, contributing to cell survival during hypoxic conditions (Refs Reference Marciniak52, Reference Rouschop53). Under the conditions of nutrient deprivation, the UPR pathways facilitate the reprogramming of metabolic pathways by elevating the hexosamine biosynthetic pathway that mediates the survival of cancer cells (Ref. Reference Wang54). UPR also participates in the cancer cell progression by alleviating the anti-tumorigenic response by modulating the immune response via ER stress-mediated factors and by repressing the NK cells (natural killer) and T cell-mediated cytotoxicity (CD8 + ) (Refs Reference Fang55, Reference Mahadevan56). The tumour cells undergo angiogenesis wherein the new vasculature network is formed for the supply of nutrients and oxygen to the cancer cell, thereby assisting the growth of cells. IRE1 pathway has a contrasting role during angiogenesis stage of cancer development wherein IRE1–XBP1s signalling have pro-angiogenic and XBP1–RIDD signalling having anti-angiogenic consequence (Ref. Reference Lhomond57). PERK pathway also has a pro-angiogenic role during angiogenesis mediated by direct interaction of a downstream target ATF4 with VEGF-A (vascular endothelial growth factor) promoter encoding for a pro-angiogenic soluble factor (Ref. Reference Wang58).
Angiogenesis provides a medium for cancer cells to metastasize and spread to other organs. The PERK and IRE1 branches of UPR mediate the migration and invasion of cancer cells, however similar to angiogenesis, the activation of XBP1s and RIDD activity has a contrasting effect in the case of glioblastoma (Refs Reference Lhomond57, Reference Tanjore59). UPR also plays a role in conferring resistance to cancerous cells against chemotherapeutic agents with tumour cells activating different branches of the UPR to overcome the effect of chemotherapy and sustaining their survival (Ref. Reference Kim60). Lhomond et al. (2018) has recently demonstrated the upregulation of IRE1 RNase activity in the presence of paclitaxel (chemotherapeutic drug) leading to induction of expansion of tumorigenic cells in triple-negative breast cancer cells (TNBC) (Ref. Reference Lhomond57). The PERK pathway activates the Nrf2 transcription factor that mediates the expression of MRP1, thereby conferring MRP1-mediated chemo-resistance (Ref. Reference Salaroglio61).
In conclusion, cancer cells efficiently integrate UPR for their progression and dispersal with different branches having various roles at different stages of cancer cell development. However, further research is needed to understand the factors controlling the balance between IRE–XBP1s (pro-tumorigenic) and IRE1–RIDD (anti-tumorigenic) signalling, which plays an indispensable role in cancer cell development.
Neurodegenerative disease
Neurodegenerative diseases are a set of debilitating conditions resulting in progressive degeneration of neurons in different parts of the nervous system characterised by loss-of-function of the central nervous system (CNS) or peripheral nervous system (PNS) (Ref. Reference Hetz and Saxena62). Various neurological diseases share a common attribute: unusual aggregation of misfolded protein in the brain triggering UPR to restore protein homeostasis.
Alzheimer's disease (AD) is a neurological disorder which causes gradual degeneration of brain cells (neurons) leading to dementia and characterised by severe memory loss. The pathophysiology of the disease is marked by increased production and accumulation of amyloid-β plaques and deposits of neurofibrillary tangles with hyperphosphorylated τ protein (Ref. Reference Hetz and Saxena62). The accumulation of amyloid-β peptides and τ protein induces the ER stress response by disturbing the Ca+2 homeostasis in the cytosol and interfering with ER proteostasis (ERAD), respectively (Refs Reference Casas-Tinto27, Reference Abisambra63).
UPR is associated with early stages of AD marked by an expression of BiP and activation of PERK and IRE1 pathways correlating with the aggregation of abnormal τ protein. PERK pathway has a contradictory role in the progression of AD: Aids in disease progression by the accumulation of amyloid-β-peptides and assists in restoring ER proteostasis having neuroprotective effect during early stages of disease progression (Ref. Reference Lee64). A recent study has provided evidence of the direct correlation of IRE1 activation with exacerbation of AD-associated pathology, which was further corroborated by the reports of a decrease in expression of amyloid precursor protein during IRE1 genetic ablation. The activation of IRE1 signalling leads to activation of genes involved in amyloid precursor protein metabolism, thereby accelerating the amyloid signalling cascade leading to accumulation of amyloid-β peptides, a hallmark of Alzheimer's disease (Ref. Reference Duran-Aniotz28). However, the XBP1s has a neuroprotective effect in Drosophila and in vitro cell culture, wherein it prevents the accumulation of Ca+2 averting the amyloid-β peptide toxicity (Ref. Reference Casas-Tinto27). Contrary to the above-mentioned studies, Hashimoto et al. (2018) (Ref. Reference Hashimoto65) reported that there is no induction of ER stress during overexpression of amyloid-β peptide or amyloid precursor proteins or τ protein. Based on these results, it can be concluded that the role of UPR in Alzheimer disease progression needs to be further elucidated.
Parkinson's disease (PD) is a neurological disorder characterised by degeneration of dopaminergic neurons in substantia nigra pars compacta in the midbrain and aggregation of misfolded α-synuclein fibrils in Lewy bodies leading to dysfunctional motor and cognitive ability of patients (Ref. Reference Colla29). The aggregation of α-synuclein impair the ATF6 branch of the UPR by inhibiting the transition of COP-II vesicles from the ER-Golgi, resulting in the decreased ERAD function and increased chances of cells adopting the apoptotic route (Ref. Reference Credle66). The accumulated α-synuclein also disrupts the calcium homeostasis by activating the calcium pump in the ER, SERCA (sarco–endoplasmic reticulum Ca-ATPase), possibly facilitating the stimulation of ER stress (Ref. Reference Betzer67).
The ER stress is believed to play a dual role in PD progression with UPR relieving the ER stress during the initial stages and triggering neurodegeneration in the case of persistent ER stress. A recent study validated the activation of a PERK/ATF4 pathway in mouse models as an early marker of PD. Inhibition of the PERK pathway was found to have neuroprotective outcome by augmenting the survival of dopaminergic neurons restoring the levels of the synaptic proteins, attenuating neurodegeneration process (Ref. Reference Mercado30). XBP1 transcription factor has been shown to have a neuroprotective effect on the survival of dopaminergic neurons as shown by the loss of protein homeostasis in the dopaminergic neuron of animals deficient in XBP1 (Ref. Reference Hetz and Saxena62). Recent studies demonstrated the protection against dopaminergic neuron degeneration induced by α-synuclein during XBP1 overexpression in C. elegans and mouse models (Refs Reference Cui31, Reference Ray68). Overall, it can be concluded that during the early stages of PD, different branches of UPR are stimulated rending neuroprotection attenuating degeneration of dopaminergic neurons.
Huntington disease (HD) is a genetic neurological disorder resulting in the gradual degeneration of brain cells deteriorating a person's physical skills and cognitive ability, caused by a mutation in the Huntingtin gene (htt). The huntingtin gene is located in the cytosol and not within the ER lumen; hence there are concerns over it directly leading to UPR. However, there are some plausible explanations addressing these concerns: mhTT (mutation in huntington gene) impair the ERAD process or perturbs the vesicular trafficking leading to an increase in protein load on ER and disturbance of calcium homeostasis (Ref. Reference Vidal69).
The mutation in the huntingtin gene also leads to an increase in the number of polyglutamine repeats (polyQ). The polyQ toxicity has been responsible for perturbation of ER homeostasis triggering UPR in various model systems during HD. The aggregation of glutamine repeats in HD patients triggers the PERK-eIF2α pathway to induce autophagy as a cellular defence mechanism evading ER-mediated cell death (Ref. Reference Ganz70). However, HD did not alter the expression of the downstream targets of the pathway: ATF4 or CHOP. IRE1 also contributes to HD pathogenesis in vivo by inhibiting the autophagy via IRE1–TRAF2 signalling cascade leading to a degeneration of brain cells in diseased individuals (Ref. Reference Lee32). Knockdown of XBP1 gene induces autophagy leading to progressive degradation of mHtt and polyglutamine repeat peptide (Ref. Reference Vidal33). ATF6 has been reported to delay memory loss as a consequence of downstream regulatory element antagonist modulator (DREAM) inhibition during HD in mouse model having a neuro-protective function. DREAM is a calcium neuronal sensor regulating Ca+2 and protein levels (Ref. Reference López-Hurtado71).
Amyotrophic lateral sclerosis (ALS) is a rare debilitating neurological disorder caused by progressive deterioration of motor neurons exhibiting disruption in voluntary movement. It is a fatal disease characterised by muscle weakness, paralysis, atrophy and onset of respiratory deterioration. Protein misfolding and accumulation in the affected tissue are the pathological signatures of initiation of disease pathogenesis caused by gene mutations implicating ER stress as an upstream event in ALS (Ref. Reference Walker and Atkin72). Gene mutation in the SOD1 gene (superoxide dismutase), which encodes for metalloenzyme that catalyses the conversion of superoxide radicals into dioxygen and hydrogen peroxides, caused SOD1-linked ALS activating the UPR sensors (Refs Reference Walker and Atkin72, Reference Valentine, Doucette and Zittin Potter73). The overexpression of the mutant SOD1 protein resulted in increased expression of BiP (chaperone protein) in the Cos7 cell line and transgenic mouse models, highlighting the role of mSOD1 in the initiating ER stress (Refs Reference Kanekura34, Reference Tobisawa74). SOD1 is primarily a cytosolic protein; therefore mSOD1 is hypothesised to activate ER stress from the cytosol possibly through the interaction with BiP (Ref. Reference Tobisawa74), impairing ER-Golgi transport (Ref. Reference Atkin75) or by interacting with Derlin-1 (a component of ERAD process) (Ref. Reference Nishitoh76). Apart from SOD1, recent studies have uncovered a direct link between several other mutations like TDP-43, FUS, VAPB with ALS as these mutations also lead to accumulation of clusters of disease-causing proteins (Ref. Reference Kanekura34). VAPB (vesicle-associated membrane protein-associated protein B) is also predicted to be involved in activation of the UPR pathway via IRE1/XBP1 as demonstrated in yeast cells, wherein Scs2 (the yeast homolog of VAPB) reportedly regulate inositol metabolism and ablation of the gene, increasing the vulnerability of the cell to the ER stress. These studies demonstrated that the VAPB as a positive inducer of the UPR, but it negatively modulates ATF6 (Ref. Reference Gkogkas35).
Modulation of UPR by viruses
Various studies have highlighted the close connection between UPR and virus infection where the host cell activates UPR to curb virus infection and on the other hand, the virus hijacks and manipulates UPR in the host cell for its advantage. The viral infection can often lead to the accumulation of misfolded proteins, perturbation of ER membrane; interfere with glycosylation of host protein, disturbance of calcium concentration or misusing the ER membrane during the viral particle release. Viruses manipulate the different branches of UPR very precisely to use the host translational machinery to synthesise viral protein and impair the synthesis of host proteins involved in the generation of the immune response (Ref. Reference Jheng, Ho and Horng77). Viruses also exploit the ERAD process forcing the degradation of host immune molecules or envelope proteins of viruses to evade the host cells' immune response (Ref. Reference Chan78). Viruses also hijack certain ERAD factors and facilitate the formation of double-membrane vesicles that are essential for viral replication process (Ref. Reference Noack, Bernasconi and Molinari79). Emerging shreds of evidence have indicated that different viruses can elicit different branches of UPR during the infection process (Table 2).
Table 2. Modulation of the UPR pathways by viruses

ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; CHOP, C/EBP-homologous protein; EDEM, ERAD-enhancing α-mannosidase; eIF2α, eukaryotic initiation factor 2-α; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; GADD, growth arrest and DNA damage-inducible gene; GRPs, glucose-regulated proteins; HPV, Human Papillomavirus; IRE1, inositol requiring enzyme 1; JNK signalling, c-Jun N-terminal kinase signalling; nsp, non-structural protein; NS4B, non-structural protein 4B; PERK, PKR-like ER kinase; PDI, protein disulphide isomerase; scAAV, self-complementary adeno-associated virus; UPR, unfolded protein response; UPRE, unfolded protein response element; XBP1, X-box binding protein-1.
Among the members of the alphavirus genus, Semliki Forest virus (SFV) activates the PERK branch of UPR (Ref. Reference Barry82), whereas Chikungunya virus (CHIKV) and Sindbis (SINV) virus differentially modulate the different branches. CHIKV infection triggers the IRE1 and ATF6 branches of UPR and suppresses the PERK branch via interaction of viral nsp4 protein with eIF2α, whereas SINV activates the PERK and IRE1 branches of the UPR pathway (Ref. Reference Rathore, Ng and Vasudevan84).
Among the Flaviviridae family Dengue virus (DENV) manipulates the host UPR pathway in a time-dependent manner with the PERK pathway being activated during the early phase of infection, then IRE1 and later ATF6. It exploits the UPR cascade to enhance its replication by suppressing host anti-viral innate immune response and apoptosis cascade and driving the cells towards autophagy (Ref. Reference Perera, Miller and Zitzmann107). Zika virus activates the three branches of UPR, it does not induce the expression of ATF4 but increases the expression of GADD34 attenuating protein translation (Ref. Reference Tan86). Hepatitis C virus (HCV) has been reported to cause chronic ER stress conditions, thereby activating all the three arms of UPR. The PERK pathway is carefully modulated by virus ensuring the optimal synthesis of protein as viral envelope proteins (E1 and E2) catalyses the phosphorylation of eIF2α and suppresses global translation machinery, whereas NS5A protein suppresses eIF2α phosphorylation (Ref. Reference Chan and Egan108). Viral non-structural proteins (NS4B) induce cleavage of ATF6 and XBP1 splicing activating ATF6 and IRE1 arm of the UPR pathway (Ref. Reference Li109). Japanese Encephalitis virus (JEV) takes advantage of the host metabolic pathway for its benefit by activating RIDD mRNA degradation pathway via induction of the IRE1–XBP1 pathway of UPR, positively regulating viral replication (Ref. Reference Bhattacharyya, Sen and Vrati110). JEV infection also induces the ATF6 and PERK pathways and induces expression of a chaperone protein CHOP resulting in apoptosis via p38-MAP kinase signalling cascade (Ref. Reference Sharma88). Another member of Flaviviridae family, West Nile virus (WNV) stimulates the UPR signalling by an upregulation in the expression of ER chaperones like BiP, calnexin, calreticulin, PDI etc. WNV infection leads to proteasomal degradation of ATF6 and activates IRE1 pathway as a consequence of an increase in the viral titre, but it has no role in viral replication. A study demonstrated the dual importance of the ATF6 pathway during WNV infection in modulating viral replication and host immune response (Ref. Reference Ambrose and Mackenzie111). Contrary to the wild type strain, WNV Kunjin strain also activates the PERK pathway by transient phosphorylation of eIF2α, ultimately leading to enhanced expression of pro-apoptotic gene CHOP, suggesting a possible role of CHOP in WNV-associated apoptosis. CHOP also controls the WNV replication as indicated by a study wherein deletion of CHOP leads to robust viral infection as compared to wild type (Ref. Reference Medigeshi89).
Influenza A virus (IAV) activates the ER stress by activating the IRE1 pathway which facilitates viral replication with little or no co-activation of ATF6 and PERK pathways (Ref. Reference Hassan90). A recent study provides evidence of identification of viral HA protein as non-self or misfolded by host UPR, thereby recruiting ERAD to degrade HA acting as an anti-viral response (Ref. Reference Frabutt91).
Respiratory syncytial virus (RSV), a member of Paramyxoviridae family also modulates host UPR pathways with activation of IRE1 and ATF6 pathways and suppression of the PERK pathway. The endoribonuclease domain of IRE1 has been shown to have an inhibitory effect on viral replication during the early stages of viral infection (Ref. Reference Hassan92).
Human immunodeficiency viruses (HIV), a member of Retroviridae family, modulates UPR signalling in a customised way for its benefit, with the induction of BiP being the first indicator of activation of UPR. HIV activates all the three branches of UPR as verified by the cleavage of ATF6 and phosphorylation of IRE1 and eIF2α, with IRE1 and PERK pathways showing a synergistic effect with anti-retroviral therapy (Ref. Reference Borsa99).
Pharmacological modulation of UPR
The role of UPR in the aetiology of various diseases, as mentioned above has attracted keen interest in probing the therapeutic implications of targeting these pathways. In-depth knowledge of the role of UPR in multiple conditions is crucial for translating this understanding in identifying novel therapeutic targets. Induction of UPR has a protective effect in various diseases like diabetes and neurological disorders, inhibition of UPR and increasing ER stress have a beneficial effect on various cancers and certain viral infections. There have been multiple studies concentrating on pharmacological interventions that modulate the UPR pathway to prevent the occurrence of disease or curb the severity of symptoms (Ref. Reference Li112). Various inhibitors target the different domains of the IRE1 pathway, which are in preclinical stages for the treatment of diseases like cancer, diabetes etc. The RNase domain inhibitors (e.g. MKC-3946, STF-083010) are composed of the aromatic aldehyde group in the core and adjacent hydroxyl group. The aldehyde group reacts with the amino group of lysine and forms imine derivatives, and the hydroxyl group forms hydrogen bonds with the tyrosine residue, impairing the catalytic cleavage of IRE1α (Ref. Reference Cross113). MKC-3946 inhibits the XBP1 splicing in the mouse model; leading to remarkable growth inhibition of multiple myeloma cells (Ref. Reference Mimura114). STF-083010 demonstrated the anti-myeloma activity in the multiple myeloma xenografts (Ref. Reference Papandreou115) and also protects against liver fibrosis in the mouse model (Ref. Reference Chen116). Another endoribonuclease inhibitor 3-ethoxy-5,6-dibromosalicyaldehyde has demonstrated the potential anti-viral effect against influenza virus (Ref. Reference Hassan90).
The inhibitors targeting the kinase domain (e.g. KIRA6) facilitate the maintenance of the monomeric inactive form of IRE1α as it disrupts the contact points at the back-to-back interface in the dimeric form (Ref. Reference Feldman117). KIRA6, the kinase domain inhibitor, has shown promising results in protecting the Akita diabetic mice from hyperglycaemia and safeguarding the pancreatic-β cells and increasing the insulin production (Ref. Reference Ghosh118). The competitive small molecule inhibitors of the PERK pathway (e.g. GSK2606414, GSK2656157) inhibit the interaction of PERK with ATP, thereby blocking the signalling cascade. These ATP-competitive inhibitors comprising the indoline core binds the kinase domain in the ATP-binding site, resulting in the conformational change that contributes to the kinase domain's inactivity (Ref. Reference Hetz, Axten and Patterson119). GSK2606414 inhibits the growth of pancreatic tumour cells (Ref. Reference Axten120), but facilitate the infection of reovirus in head and neck carcinoma cells and increase the production of viral protein (Ref. Reference McLaughlin121). The modified and pharmacokinetically more stable small molecule GSK2656157 exhibited anti-tumorigenic and anti-angiogenic properties in pancreatic cancer cell lines and mouse model (Ref. Reference Atkins122) and also increased the secretion of insulin-stimulated by glucose in hyperglycaemic mice at low concentration (Ref. Reference Kim123). The modulators of the PERK pathway (Salubrinal, Guanabenz and Sephin1) act by inhibiting the protein phosphatase complex thereby preventing the de-phosphorylation of eIF2α, a negative feedback loop to modulate protein translation during the ER stress (Ref. Reference Hetz, Axten and Patterson119).
Owing to different ongoing research instead of identifying the drug with therapeutic potential that targets specific branch of UPR, FDA has approved two protease inhibitors, bortezomib (Velcade) and carfilzomib (Kyprolis) for the treatment of multiple myeloma. Bortezomib has been termed as ‘blockbuster drug’ leading to the drastic improvement in multiple myeloma patients acting as an anticancer drug by killing cancerous cells (Refs Reference Goldberg124, Reference de Hungria125). Apart from this, various drugs are at different stages of clinical trials or still under experimental validation in pursuit of preventing disease progression (Table 3).
Table 3. Compounds modulating UPR pathways to prevent disease progression
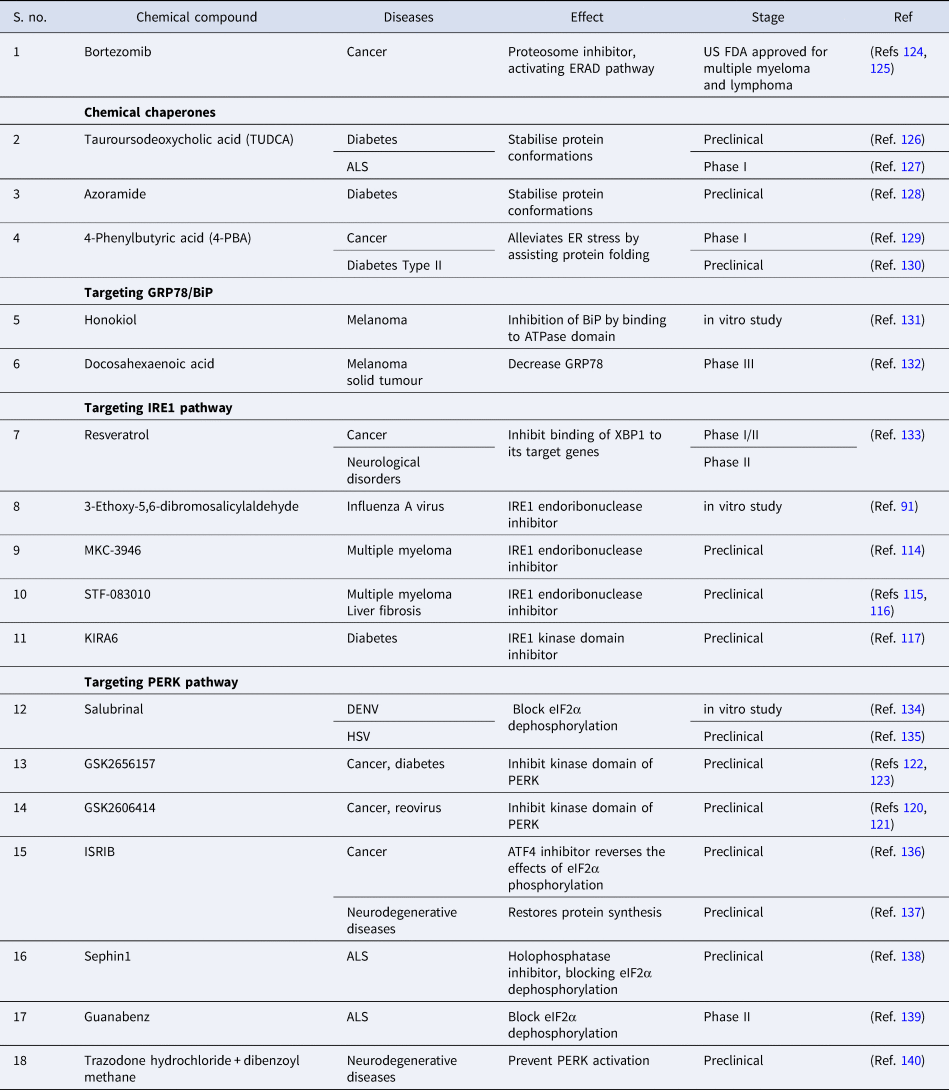
ALS, amyotrophic lateral sclerosis; ATF4, activating transcription factor 4; DENV, Dengue virus; ER, endoplasmic reticulum; eIF2α, eukaryotic initiation factor 2α; ERAD, endoplasmic reticulum-associated protein degradation; IRE1, inositol requiring enzyme-1; ISRIB, integrated stress response inhibitor; HSV, Herpes simplex virus; PERK, PKR-like ER kinase.
Conclusion
In recent years, modulation of host metabolic pathways like UPR has gained significant attention owing to their role in the pathogenesis of various diseases. There are numerous ongoing studies targeting different mediators of UPR to block disease progression, validated by the success story of Bortezomib; US FDA approved drug for the treatment of cancer (Ref. Reference de Hungria125). Various drugs like TUDCA, Guanabenz, Docosahexaenoic acid that re-establish proper protein folding or block different mediators of UPR like GRP78, eIF2α are currently in different phases of clinical trials for diseases like cancer, ALS (Refs Reference Elia127, Reference Bedikian132, Reference Bella139). Apart from them, many drugs are in the preclinical stages or the in vitro stages for treatment of diseases like diabetes, different types of cancers, neurodegenerative diseases and more. UPR also plays a crucial role in viral pathogenesis, wherein viruses hijack the UPR pathways and manoeuvre them for their benefit, opening another avenue that needs further research for designing novel antivirals. A recent study by Sureda et al. (2020) (Ref. Reference Sureda141) highlights the possibility that the recently identified SARS-CoV-2, the reason for the current pandemic situation, can perhaps manipulate UPR for its benefit, making it a potential target site for antiviral treatment. Despite the vast knowledge about UPR, it is imperative to understand the paradoxical nature of various arms of UPR in different diseases. There is also an urgent need to expedite the ongoing research on pharmaceutical drugs for the treatment of deadly diseases by targeting UPR, as an alternative mode of treatment apart from the conventional ways of treatment.
Acknowledgement
The authors did not receive any specific funding for this work.
Conflict of Interest
The authors declare there is no conflict of interests.





 Open access
Open access