



Introduction
PARP Enzymes are characterized by the presence of the characteristic PARP domain in the genes and proteins of the family (Ref. Reference Lüscher1). The ‘immediate’ family embodies 18 genes in humans (PARP1-4, PARP5a, PARP5b, PARP6-17) (Refs Reference Lüscher1, Reference Ame, Spenlehauer and de Murcia2). However, based on structural and functional homology the ‘extended’ family of the PARP enzymes is way wider (Ref. Reference Lüscher1). Classical PARP enzymes catalyse the cleavage of NAD + to nicotinamide and ADP-ribose units which are transferred to acceptor target proteins, thus inducing protein mono-ADP-ribosylation (MARylation) or poly-ADP-ribosylation (PARylation) that in turn modulate the biological properties of the acceptor proteins (Refs Reference Lüscher1, Reference Bonfiglio3). MARylation and PARylation are ancient reactions and are present in all domains of life (bacteria, plants, fungi and animals) (Ref. Reference Otto4). For a better understanding of the mechanisms involved in ADP-ribosylation we refer the readers to renowned reviews: (Refs Reference Lüscher1, Reference Slade5, Reference Schreiber6, Reference Ryu, Kim and Kraus7, Reference Bai8, Reference Ishiwata-Endo9).
PARP enzymes have widespread physiological and pathophysiological tasks (Ref. Reference Bai8). The bulk of the cellular PARylation is attributed to PARP1 and PARP2 (Refs Reference Schreiber10, Reference Szanto11), and there is a strong structural and functional homology between PARP1 and PARP2 (Refs Reference Szanto12, Reference Yelamos, Schreiber and Dantzer13). Recent studies have shed light on separate functions of PARP1 and PARP2 (e.g. (Ref. Reference Farres14)) and hereby we will describe the biological roles of PARP2 and decipher which ones are PARP2-specific and which are shared with other PARP enzymes.
PARP2 expression pattern and its regulation
The gene of PARP2 is located on chromosome 14 in humans. Translation of the PARP2 mRNA yields a protein product of 66.206 kDa molecular weight. The gene of PARP2 shares a bidirectional promoter with RNase P that is a rare lineup of genetic elements (Ref. Reference Ame15). A functional TATA box and DSE/Oct-1 elements were identified in the promoter (Ref. Reference Ame15). N-MYC was shown to induce the transcription of PARP2 (Ref. Reference Zhang16). In addition, the expression of PARP2 is modulated by microRNAs (Refs Reference Liu17-Reference Zhou24) (Table 1), and in turn, PARP2 was demonstrated to regulate the expression of miRNAs (Refs Reference Chacon-Cabrera25, Reference Mateu-Jimenez26). The expression of PARP2 can be induced by genotoxic agents or by the absence of PARP1 or PARP3 in multiple models including plants, as Arabidopsis (Ref. Reference Boltz27), non-vertebrates as sea urchin (Ref. Reference Reinardy and Bodnar28) or Caenorhabditis (Ref. Reference Neumann29) and vertebrates (Refs Reference Ame, Spenlehauer and de Murcia2, Reference Otto4).
Table 1. MicroRNAs modulating the expression of PARP2

We assessed the gene expression pattern of PARP2 using the Gtexportal database (Fig. 1) that we compare to the literature data (Refs Reference Schreiber10, Reference Dantzer30, Reference Quenet31, Reference Johansson32). In line with the literature, the mRNA expression of PARP2 was the highest in the central nerve system (Refs Reference Schreiber10, Reference Johansson32), in particular, in the neocortex (Ref. Reference Schreiber10), while Gtexportal identified the cerebellum with the highest expression of PARP2 mRNA. Besides, notable expression was detected in spinal ganglia, stratum granulosum of the dentate gyrus and the stratum pyramidale of the hippocampus and the olfactory bulb (Ref. Reference Schreiber10).
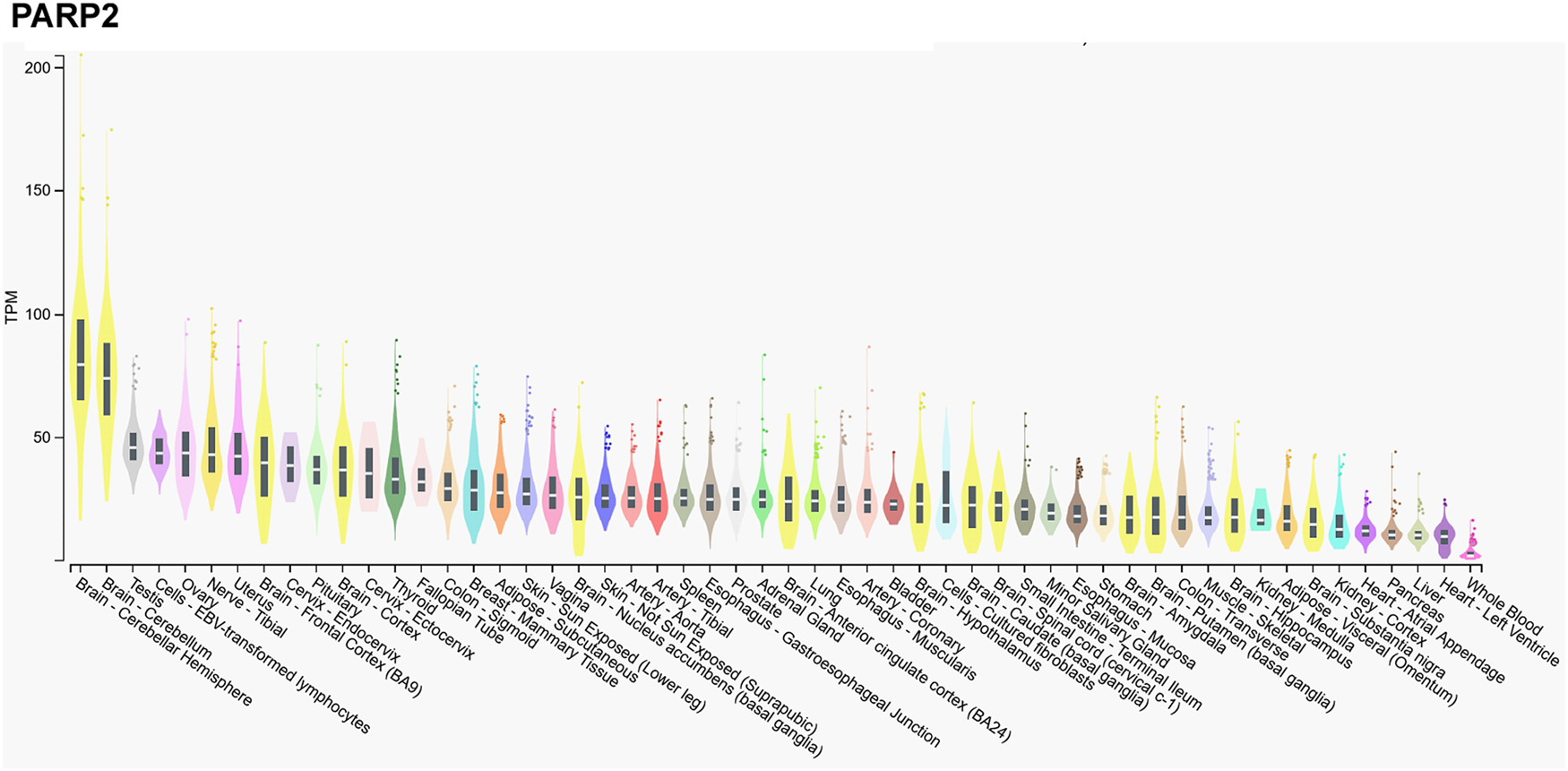
Figure 1. mRNA expression pattern of PARP2. Data was retrieved from the Gtexportal (https://gtexportal.org/home/gene/PARP2). The database was accessed 2023. 09. 20.
Tissues of the reproductive organs, as the ovary or testis, have relatively high PARP2 expression (Ref. Reference Johansson32), suggesting an involvement in spermatogenesis (see in a later chapter, (Refs Reference Dantzer30, Reference Quenet31)) that is verified by the Gtexportal data (Fig. 1). Similar, immune-related tissues, as the thymus and the white pulp of the spleen and Peyer patches in mice have notable PARP2 mRNA expression (Refs Reference Schreiber10, Reference Yelamos33). PARP2 expression decreases towards the centre of the thymus as lymphocytes differentiate and maturate (Refs Reference Schreiber10, Reference Yelamos33). Interestingly, metabolic tissues had a relatively low PARP2 expression in mice and in humans, despite the role of PARP2 in metabolic regulation (Refs Reference Bai and Canto34, Reference Vida35). Notable PARP2 mRNA expression was detected in the cortex of the kidneys, adrenal glands, stomach and intestinal epithelium (Refs Reference Schreiber10, Reference Johansson32).
In contrast, mRNA expression of PARP1 is not high in the cerebellum, or the testicular or ovarian tissues (Fig. 2), suggesting functional differences between PARP1 and PARP2.
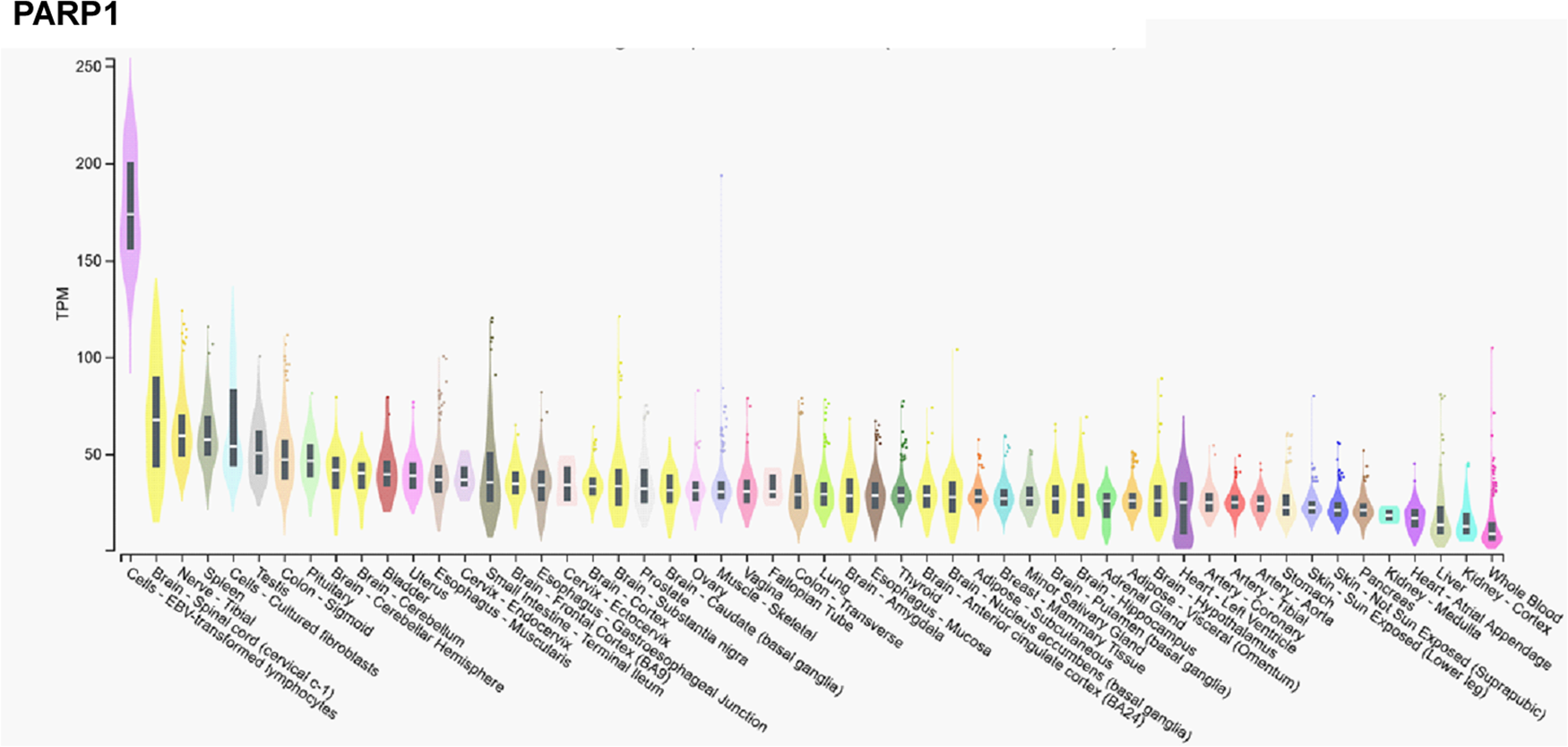
Figure 2. mRNA expression pattern of PARP1. Data was retrieved from the Gtexportal (https://gtexportal.org/home/gene/PARP2). The database was accessed 2023. 09. 20.
PARP2 structure and enzymatic activity
The molecular structure of PARP2
PARP2 is a multidomain protein (Fig. 3). The N-terminus of PARP2 is intrinsically disordered (Refs Reference Obaji, Haikarainen and Lehtio36, Reference Riccio, Cingolani and Pascal37); although originally it was considered to be organized to a SAP domain (Refs Reference Leger38, Reference Altmeyer39). Within the N-terminus, a functional nuclear localization signal (Refs Reference Haenni40, Reference Chen41), a nucleolar localization signal (Ref. Reference Meder42) and a caspase-3 cleavage site were identified (Ref. Reference Menissier-de Murcia43). Importin alpha is responsible for the nuclear import of PARP2 and importin alpha activity depends on the acetylation of lysine 36 in PARP2 (Refs Reference Haenni40, Reference Chen41).

Figure 3. The domain structure of PARP2. ART, ADP-ribosyl transferase domain; casp3, caspase 3 cleavage site; casp8, caspase 8 cleavage site; NLS, nuclear localization signal; NoLS, nucleolar localization signal.
The subsequent WGR domain is the primary site for DNA binding of PARP2 (Refs Reference Obaji, Haikarainen and Lehtio36, Reference Riccio, Cingolani and Pascal37, Reference Obaji, Haikarainen and Lehtio44). Nevertheless, it appears that all domains of PARP2 partake in nucleic acid binding (Ref. Reference Riccio, Cingolani and Pascal37), although the N-terminus seems disposable for DNA binding (Ref. Reference Langelier, Riccio and Pascal45). The WGR domain holds DNA ends in the proximity of each other (Ref. Reference Obaji46). The nucleic acid structures that can relieve the autoinhibition of PARP2 (Ref. Reference Obaji46) and activate PARP2 are discussed in the next chapter. The N-terminus and the C-terminal catalytic domain are separated by a caspase-8 cleavage site (Refs Reference Benchoua47, Reference Soni48). The catalytic domain of PARP2 shows high sequence and structural homology to the catalytic domain of PARP1 (Ref. Reference Oliver49) (Fig. 4).

Figure 4. The sequence and structure of the catalytic domain of PARP2 is similar to the catalytic domain of PARP1. A. The amino acid sequence of PARP1 and PARP2 were aligned using the Uniprot align algorithm. The blue lines represent identical amino acids. B. The amino acid sequence of PARP1 and PARP2 were aligned using the Blast algorithm at NCBI. The middle sequence represents the consensus amino acids between the PARP1 and PARP2. C. The structure of the catalytic domain of PARP2 (3KJD (Ref. Reference Karlberg50)) and PARP1 (3L3L (Ref. Reference Gandhi51)) is presented using the RCSB PDB viewer.
Léger et al. (Ref. Reference Leger38) suggested that PARP2 can bind to RNA and get activated, however, Nakamoto and co-workers (Ref. Reference Nakamoto52) provided evidence that non-specific RNA binding does not lead to enzymatic activation.
The regulation of the catalytic activity of PARP2
PARP2 is an ADP-ribosyl transferase enzyme (Ref. Reference Lüscher1) that catalyses the transfer of ADP-ribose units onto target proteins yielding branched PAR chains (Ref. Reference Chen53). Furthermore, PARP2 can continue the elongation of a formerly attached ADP-ribose unit(s) (mono or oligo-ADP-ribose units) to form PAR (Ref. Reference Kosova54). PARP2 targets glutamate, serine (Refs Reference Bartlett55, Reference Bilokapic56, Reference Gaullier57, Reference Kurgina58) and its own lysine residues (Ref. Reference Haenni59). PARP2 auto-PARylation takes place in the WGR and the helical domain and on lysine 36 and 37 in the N-terminus (Ref. Reference Haenni59). PARP2 is usually responsible for 5–15% of cellular PARP activity (Refs Reference Schreiber10, Reference Szanto11, Reference Chen53, Reference Ame60, Reference Shieh61). Nevertheless, PARP1 and PARP2 target distinct acceptor proteins (Ref. Reference Gibson62), suggesting that these enzymes have discrete biological roles. DNA binding by PARP2, in contrast to PARP1, is not modulated by auto-PARylation (Ref. Reference Kutuzov63). PAR is an important interaction surface for DNA repair factors and other proteins; branching and the physical buildup of PAR plays key role in selecting PAR interacting partners (Refs Reference Reber and Mangerich64, Reference Löffler65). It is important to note that PARP2 plays key role in generating branched PAR chains, while not influencing the number of ADP-ribose moieties in the chain (Ref. Reference Chen53), hence PARP2 may impact on selecting PAR interactors. The KM value of PARP2 for NAD + falls within the range of cellular and nuclear NAD + , apparently, normal cellular NAD + levels are not limiting for PARP2 (Ref. Reference Houtkooper66). When comparing the affinity of PARP2 for NAD + to the NAD + -affinity of other relevant enzymes it is visible that the KM value of PARP2 is similar to that of SIRT1, lower than PARP1 or CD38, while much higher than tankyrase-1 (Table 2). Furthermore, while the kcat/KM value of PARP2 is lower than that of PARP1, it is clearly higher than that of SIRT1, CD38 or tankyrase-1 (Table 2). Taken together, PARP2 can limit NAD + for slower NAD + -dependent enzymes or enzymes with lower affinity for NAD + (e.g. tankyrase 1) (Refs Reference Mohamed18, Reference Bai67) and can likely reduce the available cellular NAD + pool when activated, however, not to the same extent as the activation of PARP1. In other words, as a function of the level of PARP2 activation, PARP2 can represent a burden on cellular NAD + levels.
Table 2. K M and the k cat/K M values of a set of NAD + -dependent enzymes

The structure of the catalytic domain of PARP2 shows high sequence and structural homology to PARP1 (Refs Reference Oliver49, Reference Karlberg50, Reference Ame60). There is an additional three amino acid insertion in the loop connecting the β-strands k and l in PARP1 (Refs Reference Oliver49, Reference Karlberg50, Reference Ame60). PARP2 has a narrower catalytic cleft that likely explains the lower substrate affinity and turnover rate of PARP2 as compared to PARP1 (Refs Reference Szanto11, Reference Ame60, Reference Shieh61).
PARP2 can homodimerize with itself or heterodimerize with PARP1 at the DNA damage sites (Refs Reference Sukhanova71, Reference Huber72). In addition, HPF1 is also an important interaction partner of PARP2. HPF1 was originally described as a factor shifting the amino acid-specificity of PARPs to serine residues (Refs Reference Bonfiglio3, Reference Gibbs-Seymour73). HPF1 forms complexes with PARP1 and PARP2 to complete the PARylation enzymatic site of PARP2 and PARP1 (Ref. Reference Suskiewicz74), importantly however, HPF1 is important but it is not necessary to enable PARP activity. The complex works most efficiently at sub-stoichiometric ratios of HPF1 relative to PARP1 and PARP2 (Ref. Reference Langelier75). HPF1 restricts elongation, stimulates initiation and induces the DNA-dependent, as well as, the DNA-independent, auto-PARylation of PARP2 and the trans-PARylation of nucleosomal histones (Refs Reference Kurgina58, Reference Kurgina76). The PARP2-HPF1 complex has profound roles in regulating chromatin structure as it binds to two nucleosomes and exposes the broken DNA section to facilitate ligation (Refs Reference Bilokapic56, Reference Bacic77). In line with that, PARP2, unlike PARP1, can bridge DNA ends very strongly as shown in single molecule atomic force microscopy experiments (Ref. Reference Bell and Molloy78). Furthermore, the PARP2/HPF1 complex can PARylate and activate a chromatin remodeller, ALC1 (also known as CHD1L; Chromodomain-helicase-DNA-binding protein 1-like) (Ref. Reference Bacic77).
In line with the DNA binding of PARP2, pharmacological PARP inhibitors trap PARP2 on the chromatin in complex with ALC1 (Refs Reference Gaullier57, Reference Murai79, Reference Blessing80, Reference Langelier81, Reference Lin82). PARP2 trapping to DNA is dependent on the R140 amino acid in the WGR domain (i.e. trapping depends on DNA binding) and the H415 amino acid in the catalytic domain (i.e. trapping depends on the suppression of the PARP1- and PAR-dependent rapid exchange of PARP2) (Ref. Reference Lin82).
PARP2 also possesses 5′-deoxyribose phosphate (5′-dRP) lyase activity, that is comparable to the 5′-dRP lyase activity of PARP1, but much weaker than that of polymerase β (Ref. Reference Kutuzov63).
A large set of natural and synthetic lipids were shown to modulate the activity and expression of PARPs (Ref. Reference Szanto83), among them, the expression of PARP2 (Refs Reference Zhang84, Reference Marton85, Reference Sun, Gatie and Kelly86) (Table 3). In addition to their effects on PARP2 expression, serum lipids can render PARP2 to the insoluble fraction of the cells (Ref. Reference Sun, Gatie and Kelly86). Lipid-dependency is a more widespread phenomenon among PARPs (Ref. Reference Szanto83).
Table 3. Lipid species modulating the expression of PARP2

Posttranslational modifications
PARP2 undergoes multiple posttranslational modifications, including acetylation (Ref. Reference Haenni59). Acetyl groups are introduced by the histone acetyl-transferase P/CAF (Ref. Reference Haenni59). PARylation and acetylation of PARP2 can take place on the same amino acids (Lysine 36, Lysine 37) pointing towards a competition between these two posttranslational modifications. Lysine 36 partakes in alpha-importin binding and, hence, regulates the nuclear translocation of PARP2 (Ref. Reference Haenni40). Furthermore, PARP2 expression affects the acetylation pattern in cells (Ref. Reference Chacon-Cabrera25). Competition between PARylation and acetylation was already suggested and appears to be a broader phenomenon, not restricted to PARP2 (Refs Reference Bartlett55, Reference Cantó, Sauve and Bai87). Further PARylation sites in PARP2 were mapped to the WGR and the HD domains. Sun et al. have suggested that the ubiquitin-proteasome system is involved in PARP2 turnover (Ref. Reference Sun, Gatie and Kelly86).
Interaction partners of PARP2
PARP2 interacts with a large number of protein partners (Refs Reference Schreiber10, Reference Isabelle88, Reference Dahl89, Reference Alemasova90, Reference Jain91, Reference Butepage92, Reference Vasil'eva93). These proteins cover a wide array of functions such as cell cycle, cell death, DNA repair, DNA replication, transcription, metabolism, energy homoeostasis and RNA metabolism.
PARP2 has multiple PARylation targets including proteins involved in transcription, translation, mitochondrial organization, redox balance, DNA repair, PARylation machinery (e.g. HPF1) (Refs Reference Chen53, Reference Bilokapic56, Reference Troiani94, Reference Jankó95, Reference Vivelo96, Reference Jungmichel97, Reference Gagne98, Reference Fontana99). Nucleic acid structures may also be sites of ADP-ribosylation (Ref. Reference Groslambert, Prokhorova and Ahel100), nevertheless, the role of PARP2 in that context is unknown and unproven. Of note, protein-bound PAR, built upon PARP activation, is an important binding surface for DNA repair factors (Ref. Reference Tartier101).
PARP2 in protecting genomic integrity and regulating chromatin structure
PARP2 was first identified as a DNA repair protein (Ref. Reference Ame60), similar to PARP1 and PARP3 (Refs Reference Schreiber6, Reference Rouleau102, Reference Boehler103). The involvement of PARP2 in DNA repair has been demonstrated not only in animal models (Refs Reference Szanto12, Reference Yelamos, Schreiber and Dantzer13), but in plants too (Refs Reference Song104, Reference Gu105, Reference Taipakova106, Reference Zhang107). PARP2 expression correlates with exposure to genotoxic noxae in humans (Ref. Reference Niazi108). In line with the involvement of PARP2 in DNA repair the loss of PARP2 leads cell cycle arrest in G1 upon genotoxic insult (Ref. Reference Menissier-de Murcia43).
PARP2 accumulates at DNA damage sites (Refs Reference Mahadevan109, Reference Chalmers110) with slower pace than PARP1 (Ref. Reference Mortusewicz111). PARP2 binds to both double and single-strand breaks (Ref. Reference Sukhanova112); to single-strand breaks as a monomer (Ref. Reference Obaji, Haikarainen and Lehtio36), while to double-strand breaks as homodimer or as a heterodimer with PARP1 (Refs Reference Schreiber10, Reference Obaji, Haikarainen and Lehtio36). Interestingly, the caspase-cleaved N-terminal fragment of PARP1 can inhibit the DNA-induced catalytic activity of PARP2 (Ref. Reference Deeksha and Rajakumara113).
Damaged DNA structures that activate PARP2 contain single nucleotide gaps (Ref. Reference Ukraintsev114) and 5′ phosphate (Refs Reference Obaji, Haikarainen and Lehtio44, Reference Langelier, Riccio and Pascal45, Reference Taipakova106, Reference Talhaoui115), suggesting that these sites are ligation-competent ends. PARP2 had better affinity for nucleotide gaps as compared to AP sites (Ref. Reference Ukraintsev114). Such sites include 8-oxoguanine or the apurinic/apirimidinic sites (Ref. Reference Lebedeva116) that can be inflicted by low dose ionizing radiation (Refs Reference Menissier-de Murcia43, Reference Chalmers110, Reference Boudra117, Reference Farres118), laser irradiation (Refs Reference Mortusewicz111, Reference Koczor119) or DNaseI treatment (Ref. Reference Ame60). PARP2 bind to the intact AP site via Schiff base formation (Ref. Reference Kutuzov63). PARP2 bridges DNA ends very strongly (Ref. Reference Bell and Molloy78). This is a more restricted repertoire of DNA damage sites as compared to PARP1 (Refs Reference Zandarashvili120, Reference Dawicki-McKenna121). PARP2, similar to PARP1, can PARylate and MARylate double-strand break termini in oligonucleotide models of double and single-strand breaks (Ref. Reference Zarkovic122).
PARP2 was first shown to be involved in single-strand break repair, as in the absence of PARP2 base excision repair (BER) slows down (Ref. Reference Schreiber10). As discussed above, PARP2 can interact with nucleotide intermediates of BER, although, PARP2 has lower affinity to the early intermediates of BER than PARP1 (Ref. Reference Sukhanova71). Furthermore, PARP2 can interact with most BER proteins (Ref. Reference Vasil'eva93) including X-ray repair cross-complementing protein 1 (XRCC1), PARP1, DNA polymerase β and DNA ligase III, Flap endonuclease 1 (FEN1) or Y-box-binding protein 1 (Refs Reference Schreiber10, Reference Alemasova90, Reference Kutuzov123, Reference Kutuzov124, Reference Hanzlikova125). PARP2 is necessary for the recruitment of DNA polymerase β and XRCC1, however, the absence of PARP2 does not inhibit DNA polymerase β and XRCC1 recruitment totally; this is achieved only in the simultaneous absence of PARP1 and PARP2 (Refs Reference Schreiber10, Reference Koczor119). Importantly, the NAD + hydrolase activity of PARP2 can also play role in XRCC1 and DNA polymerase β recruitment, as NAD + availability is also vital for the recruitment of these factors (Ref. Reference Koczor119). Interestingly, PARP2 can inhibit DNA polymerase β activity and, hence, slow down the resealing of the site of a missing DNA base, while inducing the activity of ligase IIIα (Refs Reference Kutuzov123, Reference Kutuzov124).
Successful retrotransposition requires the activation of DNA repair machinery (Ref. Reference Yamaguchi, Kajikawa and Okada126). Single strand breaks, associated with LINE-1 retrotransposon integration, recruit PARP2 to the integration site (Ref. Reference Miyoshi, Makino and Moran127). PARP2 is activated at the integration site and supports the recruitment of the members of the integration machinery and, hence, successful LINE-1 integration (Ref. Reference Miyoshi, Makino and Moran127).
Similar to PARP1 (Ref. Reference Bryant128), PARP2 is also involved in the resolution of double-strand breaks. In the absence of PARP2 double-strand breaks accumulate (Ref. Reference Nicolas129). PARP2 was shown to be involved in non-homologous end joining (NHEJ) (Refs Reference Jia130, Reference Robert, Dantzer and Reina-San-Martin131), as well as, in homologous recombination (HR; i.e. interaction with the Ku proteins) in animals (Refs Reference Yelamos, Schreiber and Dantzer13, Reference Jia130, Reference Robert, Dantzer and Reina-San-Martin131) and in plants (Ref. Reference Chen132). In line with its involvement in double-strand break repair, the absence of PARP2 hampers the maturation of thymocytes (Ref. Reference Nicolas129) and B cells (Ref. Reference Robert, Dantzer and Reina-San-Martin131), or the resolution of blocked replication forks (Refs Reference Bryant128, Reference Malanga and Althaus133, Reference Richards134, Reference Ronson135). Importantly, PARP2 restricts the accumulation of the resection barrier factor 53BP1 at DNA damage sites and tunes DNA repair towards the resection-dependent CtIP-dependent DNA end-resection (Ref. Reference Fouquin136). The PARP2-dependent PARylation of polymerase δ3 at replication forks play key role in stabilizing the stalled replication forks (Ref. Reference Richards134). Interestingly, mitomycin C-induced double-stranded DNA damage induces the expression of PARP2 and other double-strand break repair proteins (Ref. Reference Kang137).
PARP2 also partakes in chromatin, telomere and centromere maintenance. The deletion of PARP2 induces spontaneous chromosome and chromatid breaks and an increase in the number of DNA ends lacking detectable telomere repeats, which is explained by the loss of interaction between PARP2 and TRF-2 (Ref. Reference Dantzer138). Interestingly, the contribution of PARP2 to telomere stability appears to be limited (Ref. Reference Boltz27). PARP2 localizes to centromeres in a cell-cycle dependent manner and plays role in accurate chromosome segregation (Ref. Reference Dantzer30) through interacting with kinetochore proteins, centromere protein A (CENPA), centromere protein B (CENPB) and mitotic spindle checkpoint protein BUB3 in prometaphase and metaphase (Ref. Reference Saxena139). Furthermore, PARP2 interacts with topoisomerase I and topoisomerase IIb and through that interaction regulates chromatin condensation (Refs Reference Malanga and Althaus133, Reference Meyer-Ficca140). In line with that, female lethality due to X chromosome instability was reported in PARP1±PARP2−/− mice (Ref. Reference Menissier-de Murcia43). Furthermore, it is tempting to speculate that early embryonic lethality in PARP1/PARP2 double knockout mice (Ref. Reference Menissier-de Murcia43) or in ATM/PARP2 double knockout mice (Ref. Reference Huber72) may be due to genomic instability.
PARP2 in transcriptional regulation
Regulation of chromatin structure: epigenetic marks, chromatin dynamics
In contrast to the well-characterized PARP1 (Ref. Reference Ryu, Kim and Kraus7), little is known about the role of PARP2 in transcription. PARP2 may impact on transcription through multifaceted processes, in which the regulation of chromatin dynamics plays pivotal role.
PARP2 can bind to nucleosomes (Ref. Reference Maluchenko141). In addition, PARP2 can modulate chromatin dynamics and DNA/promoter accessibility through modulating histone acetylation status by regulating the recruitment of histone deacetylase 5 and 7 and histone methyltransferase G9a to promoters independently of its PARP activity inducing the repression of cell cycle genes (Ref. Reference Liang142). PARP2 also interacts with histone acetyl-transferase P/CAF (Ref. Reference Haenni59) and transcriptional intermediary factor-1β and heterochromatin protein-1α (Ref. Reference Quenet143). In addition to regulating histone acetylation, PARP2, in complex with HPF1, can PARylate nucleosomal histones (Refs Reference Kurgina58, Reference Kurgina76). ADP-ribosylation or PARylation is considered a histone mark on its own right (Ref. Reference Karch144). Furthermore, serine ADP-ribosylation likely interplays with histone marks (Ref. Reference Bartlett55) highlighting a complex involvement of histone ADP-ribosylation in the modulation of chromatin structure.
PARP2 cooperates with chromatin modelling factors, among these, topoisomerase I and IIβb (Refs Reference Malanga and Althaus133, Reference Meyer-Ficca140). Topoisomerase activation generates DNA strand breaks (Ref. Reference Ju145), and PARP2 likely assists in the resolution of the DNA nicks, similar to PARP1 (Ref. Reference Ju145) and through that PARP2 supports the resolution of torsional stress upon the relaxation or compaction of chromatin and when DNA unwinds for transcription. As mentioned earlier, the PARP2-HPF1 complex can bridge two nucleosomes and display the ligation-prone broken DNA ends for ligation (Refs Reference Bilokapic56, Reference Bacic77, Reference Bell and Molloy78). Furthermore, the PARP2-HPF1 complex PARylates and activates ALC1 (Ref. Reference Bacic77), and cooperates with Cockayne syndrome group B protein at actively transcribed DNA regions (Ref. Reference Bilkis146).
Interaction of PARP2 with factors of RNA polymerase I and II
The DNA repair machinery, wherein PARP2 plays major role, has strong interconnections with transcription (Refs Reference Páhi147, Reference Pankotai148). In agreement with that, PARP2 is involved in RNA polymerase I and II-mediated transcription events.
PARP2 interacts with nucleophosmin/B23 (Ref. Reference Meder42) which is involved in rRNA transcription (Ref. Reference Derenzini149). Interestingly, the deletion of PARP2 does not modify rRNA expression, while the inhibition of RNA polymerase I removes PARP2 from the nucleolus (Ref. Reference Meder42), hence, the involvement of PARP2 in rRNA transcription is not deciphered.
PARP2 mediates the expression of multiple RNA polymerase II-mediated genes (Refs Reference Farres14, Reference Chacon-Cabrera25, Reference Marton85, Reference Antal150, Reference Szanto151) through the modulation of multiple transcription factors and cofactors (Table 4). PARP2 can impact on transcription through acting as a cofactor of transcription complexes (Refs Reference Bai152, Reference Gui153), by PARylating transcription factors (Refs Reference Dahl89, Reference Jankó95), by regulating the expression of transcription factors (Refs Reference Szanto11, Reference Bai67, Reference Marton85, Reference Szanto151, Reference Maeda154) or by limiting NAD + for the enzymatic activity of sirtuins and, hence, impact on the deacetylase activity of sirtuins (Refs Reference Szanto11, Reference Mohamed18, Reference Chacon-Cabrera25, Reference Bai67, Reference Zhang84, Reference Motyl155). DNA binding and the enzymatic activity of PARP2 is apparently required for its involvement in transcriptional regulation. These phenomena may appear in a concerted, joint fashion. Genes whose expression changes in the absence of PARP2 are involved in intermediary metabolism (mitochondrial, carbohydrate, fatty acid and cholesterol/steroid metabolism) (Refs Reference Szanto11, Reference Mohamed18, Reference Chacon-Cabrera25, Reference Bai67, Reference Zhang84, Reference Marton85, Reference Szanto151, Reference Bai152, Reference Motyl155), redox balance (Ref. Reference Jankó95) or inflammation (Ref. Reference Dahl89).
Table 4. Known PARP2-transcription factor/cofactor interactions

AR, androgen receptor; ER, oestrogen receptor; RXR, retinoid X receptor; PPAR, peroxisome proliferator activated receptor; TTF1, Thyroid transcription factor-1; SREBP1/2, Sterol regulatory element-binding protein 1/2; NRF2, nuclear factor erythroid 2–related factor 2.
The PARP2-SIRT1 interaction requires a deeper discussion due to its complex and contradictory nature. SIRT1 is an NAD + -dependent protein deacetylase (Refs Reference Smith157, Reference Imai158), therefore, PARP2 activity may limit NAD + for SIRT1 (the catalytic properties of PARP2 and SIRT1 were compared earlier in the enzymology chapter), while in the absence of PARP, NAD + levels increase, thereby increasing the activity of SIRT1. Indeed, multiple studies have found SIRT1 induction upon the silencing or deletion of PARP2 (Refs Reference Szanto11, Reference Mohamed18, Reference Chacon-Cabrera25, Reference Bai67). However, increase in NAD + levels were not always confirmed. In our hands, we have not found increases in NAD + levels in all models in which PARP2 was silenced. Of note, in relation to PARP1, Ryu and co-workers (Ref. Reference Ryu159) showed that changes to compartment-specific NAD + metabolism plays pivotal role in regulating PARP1-dependent biological processes, as adipocyte differentiation; that appears as a charming scenario with regards to PARP2. An alternative pathway to explain the higher SIRT1 activity in the absence of PARP2 leans on the observation that SIRT1 expression is induced in models in which PARP2 was silenced (Refs Reference Szanto11, Reference Bai67). PARP2 can act as a repressor of the SIRT1 promoter binding in the close proximity of the transcription start site (−1– −91 region) (Ref. Reference Bai67) that aligns well with the gene repression capacity of PARP2 through rearranging chromatin structure (Ref. Reference Liang142). The deletion of PARP2 does not interfere with SIRT2 or SIRT3 activation (Ref. Reference Bai67).
PARP2 in oxidative stress
Oxidative stress or high frequency irradiation damages DNA (Refs Reference Pacher, Beckman and Liaudet160, Reference Pacher and Szabo161, Reference Hegedűs162, Reference Hegedűs163) induce the DNA damage-dependent PARP enzymes (PARP1-3) (Ref. Reference Lüscher1). The activation of PARP1 in pathologies associated with oxidative stress declutches biochemical pathways that ultimately lead to mitochondrial dysfunction, cell death, inflammation and organ failure (Refs Reference Virag and Szabo164, Reference Bai165, Reference Berger166, Reference Curtin and Szabo167). In this chapter we will assess pathologies where PARP2 plays role in oxidative stress-induced organ damage (Table 5).
Table 5. Oxidative stress-mediated pathologies with the involvement of PARP2
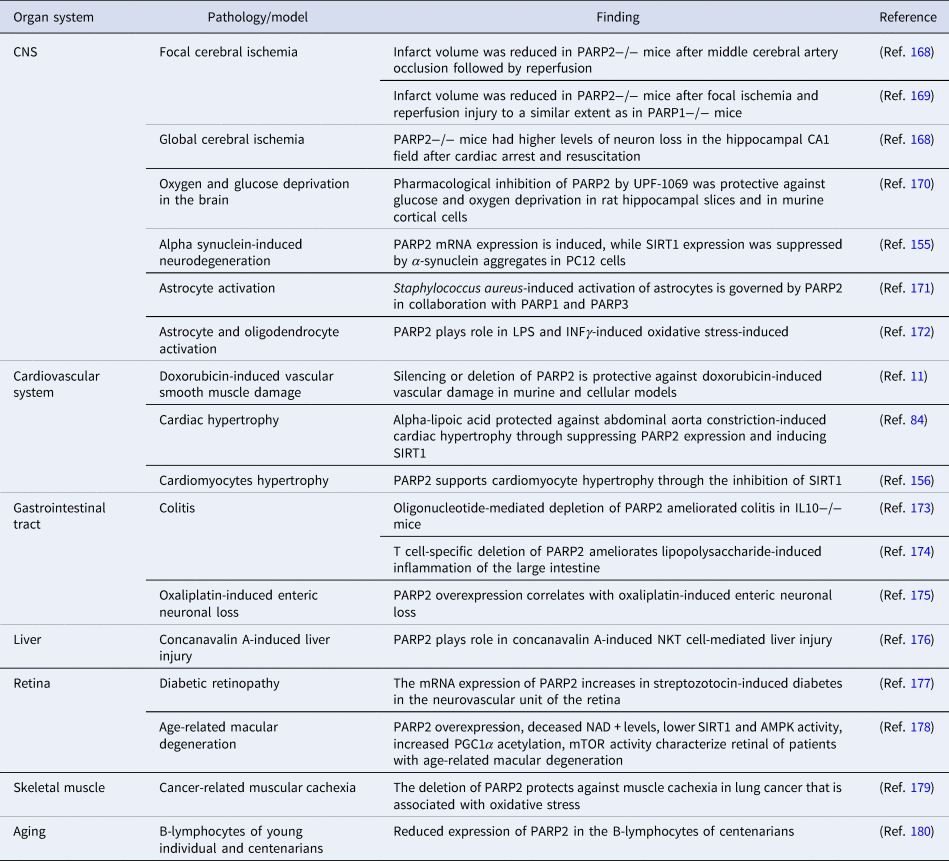
AMPK, AMP-activated protein kinase; CNS, central nerve system; INFγ, interferon γ; IL, interleukin; LPS, lipopolysaccharide; mTOR, mechanistic/mammalian target of rapamycin; NKT, natural killer T cell; PGC1α, peroxisome proliferator activated receptor cofactor 1α.
Interestingly, increased oxidative stress was detected when PARP2 was silenced in cultured cells (Refs Reference Mateu-Jimenez26, Reference Jankó181). One of the studies reported unchanged nitrosative stress alongside increases in oxidative stress (Ref. Reference Jankó181). Reactive oxygen species (ROS) stemmed from multiple sources, such as the mitochondria (Ref. Reference Jankó181) and the impaired capacity for scavenging ROS (Ref. Reference Jankó95). Impaired ROS scavenging is, at least in part, due to the PAR-mediated cellular relocalization of nuclear factor erythroid 2 (NFE2)-related factor 2 (NFE2L2, or NRF2). NRF2 is a transcription factor that plays key role in inducing the expression of genes coding for enzymes with antioxidant activity (Ref. Reference Smolková182). NRF2 can be PARylated by PARP2 (Ref. Reference Jankó95). PARylated NRF2 is anchored to the nucleus (Ref. Reference Jankó95). In accordance with that, while the silencing of PARP2 increased the cytosolic fraction of NRF2 and led to the rearrangement of the expression of numerous genes associated with redox homoeostasis (Ref. Reference Jankó95). Silencing, inhibition or deletion of PARP1 was also reported to induce oxidative stress (Ref. Reference Mateu-Jimenez26) and to regulate NRF2 activity, however, with an entirely different mechanism (Ref. Reference Wu183).
Silencing, genetic deletion or pharmacological inhibition of PARP2 proved to be beneficial in oxidative stress-associated pathologies of the central nervous system, cardiovascular system, the gastrointestinal tract, the liver, the retina and skeletal muscle (Table 5). Of note, multiple studies reported PARP2-related gene expression changes in response to UPF-1069 treatment. UPF-1069 is a commercially available PARP2-selective pharmacological inhibitor, however, UPF-1069 has limited selectivity towards PARP2 over PARP1 (Ref. Reference Moroni170). Importantly, the pathways stemming from oxidative stress-induced PARP2 activation were identified in plants (Refs Reference Vanderauwera184, Reference De Block185) and also in nematodes (Ref. Reference Neumann29) suggesting strong evolutionary conservation.
The PARP2-dependent pathways that contribute to tissue damage are multi-pronged and involve NAD + depletion (Ref. Reference Mohamed18), NAD + /SIRT1-mediated mitochondrial damage (Ref. Reference Szanto11), the induction of inflammation (Ref. Reference Popoff173) and the regulation of cell death (Refs Reference Cohausz and Althaus186, Reference Cohausz187). These pathways are discussed in-depth elsewhere in the review. These pathways overlap with the PARP1-dependent tissue damage pathways. Nevertheless, there are clear-cut differences between PARP2 and PARP1. For example, in a secretagogue-induced murine pancreatitis model the deletion of PARP1, but not the deletion of PARP2 suppressed the inflammatory response (Ref. Reference Mota188). While PARP1 activation leads to nuclear translocation of AIF from the mitochondria (Ref. Reference Yu189), the deletion of PARP2 does not (Ref. Reference Li169). Moreover, the deletion of PARP1 provided protection in murine models of contact hypersensitivity and irritative dermatitis, while the deletion of PARP2 was without effect (Refs Reference Brunyanszki190, Reference Bai191). Furthermore, apparently, PARP2 has model specific roles, as a function of focal or global ischemia-reperfusion the deletion of PARP2 can deteriorate or ameliorate neuron loss (Ref. Reference Kofler168) and such difference is unlikely to be explained by the global or focal nature of ischaemia. This observation may have importance when interpreting conflicting results in difference models and calls for verifying observations in other models or in human tissue samples.
PARP2 in metabolic regulation
PARP enzymes have widespread metabolic roles (Refs Reference Bai and Canto34, Reference Vida35, Reference Marton192), with no exception for PARP2. In contrast to PARP1 (Refs Reference Asher193, Reference Bai194) no change to circadian entrainment was observed in PARP2 knockout mice (Ref. Reference Bai67) making it unlikely that central metabolic oscillatory circuits are affected by the deletion of PARP2.
As mentioned earlier, the deletion of PARP2 induces SIRT1 expression and SIRT1 activity. Through that pathway, the deletion of PARP2 induces mitochondrial biogenesis in metabolically relevant tissues, as the skeletal muscle and the liver (Refs Reference Mohamed18, Reference Bai67) and other tissues, as the cardiovascular system (Refs Reference Szanto11, Reference Zhang84, Reference Geng156), the retina (Ref. Reference Zhang178) and the central nervous system (Ref. Reference Motyl155). Interestingly, the deletion of PARP2 did not induce mitochondrial biogenesis in the brown adipose tissue (Ref. Reference Bai67), that is another key metabolic tissue important for maintaining energy balance. Therefore, the browning effect of PARP inhibitors are likely related to PARP1, but not to PARP2 (Refs Reference Bai194, Reference Nagy195). ROS production due to the cellular relocalization of NRF2 is another key pathway in inducing mitochondrial biogenesis in the absence of PARP2 (Refs Reference Jankó95, Reference Jankó181). Somewhat contradictious, despite the higher mitochondrial output, the mitochondrial network is fragmented in PARP2-silenced cells. In agreement with increases in mitochondrial oxidative capacity, chow-fed PARP2−/− mice showed higher oxygen consumption rates and were consequently leaner than their wild-type littermates (Ref. Reference Bai67). Similar to the aforementioned situation, PARP2 is involved in metabolic regulation in plants (Ref. Reference De Block185). Interestingly, in Arabidopsis PARP2 is present in the mitochondria (Ref. Reference Pham196) that is an interesting phenomenon, as in mammalian cells no PARylating activity was identified in the mitochondria (Refs Reference Bai165, Reference Köritzer197).
PARP2 impacts on lipid metabolism in the white adipose tissue (Ref. Reference Bai152), the skeletal muscle and the liver (Ref. Reference Bai67). In the white adipose tissue the deletion of PARP2 hampers the transcriptional activity of PPARγ (Ref. Reference Bai152) that leads to the downregulation of a set of PPARγ target genes as adipocytes protein 2 (aP2), CD36, lipoprotein lipase (LPL) and fatty acid synthase (FAS) that altogether lead to impaired fat accumulation and white adipose tissue function (Ref. Reference Bai152). In line with that, low level metaflammation was detected in the white adipose tissue of PARP2−/− mice (Ref. Reference Bai152). There was no sign of beiging/browning in the white adipose tissue of the PARP2−/− mice in line with the lack of mitochondrial biogenesis in the brown adipose tissue.
In the skeletal muscle and in the liver the deletion of PARP2 impacted on lipid metabolism through the induction of mitochondrial lipid oxidation (Ref. Reference Bai67). This process is primarily associated with increases in SIRT1 activation and the induction of the expression of uncoupling protein (UCP)-2, muscle isoform of carnitine O-palmitoyltransferase 1 (mCPT1b), acyl coenzyme A oxidase I (ACOX1), medium-chain specific acyl-CoA dehydrogenase (MCAD), malonyl-CoA decarboxylase (MCD), Ndufa2, cytochrome c (cyt c) and COX IV (Ref. Reference Bai67). Importantly, PARP2−/− mice have lower respiratory quotient as compared to their wild type counterparts during the active (dark) phase which points towards higher fatty acid oxidation. As PARP2−/− mice prefer fatty acid during their diurnal cycle (i.e. they are locked-in in fatty acid oxidation) that can be considered a form of metabolic inflexibility.
Another PARP2-mediated arch of lipid metabolism is cholesterol biosynthesis and cholesterol-utilizing downstream pathways as steroidogenesis. PARP2 acts as suppressor of sterol regulatory element-binding protein (SREBP)-1 and SREBP2 (Refs Reference Marton85, Reference Szanto151), hence the silencing or deletion of PARP2 induces SREBP1/2 mRNA and protein expression and the accumulation of the active, processed nuclear form of SREBP1 in the nucleus in the liver and the skeletal muscle (Refs Reference Marton85, Reference Szanto151). Subsequently, the expression of the SREBP-dependent genes was upregulated leading to higher hepatic and muscular cholesterol content (both esterified and non-esterified) (Refs Reference Marton85, Reference Szanto151). Higher cholesterol content coincided with stark changes to the composition of the membrane-constituent lipids (Refs Reference Marton85, Reference Szanto151) and slower lateral diffusion in the membranes (Ref. Reference Marton85).
In addition to changes in the tissue cholesterol homoeostasis, systemic cholesterol homoeostasis was also impacted upon deletion of PARP2. Serum HDL levels were lower in the PARP2−/− mice highlighting impaired cholesterol traffic both to the peripheral tissues and to the liver (Ref. Reference Szanto151) (of note, in mice HDL serves both directions of cholesterol transport). Furthermore, hepatic cholesterol output through ABCA1 was hampered in the absence of PARP2 (Ref. Reference Szanto151).
In skeletal muscle cholesterol was channelled towards sexual steroid biosynthesis through Star, HSD17b11 and Srd5a1 (Ref. Reference Marton85). The upregulation of these enzymes was due to the loss of the promoter suppression effects of PARP2 (Ref. Reference Marton85). These changes culminated in the overproduction of dihydrotestosterone, an anabolic steroid (Ref. Reference Marton85). Higher anabolic steroid levels coincided with improved muscular fibre strength (Ref. Reference Marton85). Sexual steroid biosynthesis was also upregulated in the skin of PARP2−/− mice marked by higher expression of Stard5, HSD17B3 and Cyp19A1, however, the intermediates were channelled towards oestrogen biosynthesis (Ref. Reference Antal150).
The deletion of PARP2 in spite of increased mitochondrial oxidative capacity led to higher serum glucose and lower serum insulin levels in the PARP2 knockout mice under both fed and fasted conditions (Ref. Reference Bai67). Glucose challenge in intraperitoneal glucose tolerance test verified these observations, high fat diet-fed PARP2−/− mice were unable to dispose of glucose, as glucose-induced insulin secretion was blunted in the PARP2−/− mice (Ref. Reference Bai67). Disrupted glucose-induced insulin secretion was due to the hypoplasia of the Langerhans islands and β cells that was linked to lower pdx-1 expression in beta cells (Ref. Reference Bai67).
PARP1 was associated with autophagy (Refs Reference Munoz-Gamez198, Reference Rodriguez-Vargas199, Reference Rodriguez-Vargas200, Reference Rodríguez-Vargas, Oliver-Pozo and Dantzer201, Reference Kataura202) and recent data connected PARP2 to autophagy as well (Refs Reference Chacon-Cabrera25, Reference Janko203, Reference Wyrsch204). The silencing of PARP2 increased the number of autophagic vesicles in an AMPK, mTORC1/2-dependent fashion (Ref. Reference Janko203). Autophagy can provide valuable nutrients (Ref. Reference Neufeld205) and even NAD + levels (Ref. Reference Kataura202) for cell growth therefore, enhanced autophagy in the absence of PARP2 may serve that role.
It should be noted that metabolic inflexibility, as it is the case upon PARP2 knockout, is an actionable feature of cancer cells (Ref. Reference Smith206). Furthermore, the physical presence and the activity of PARP2 is associated with a set of metabolic sensors (AMPK, mTORC1, mTORC2, SIRT1), that also serve in survival signalling. Both of these findings have important practical applications in terms of antineoplastic therapy, nevertheless, were not assessed in detail.
Selective role of PARP2 in biological processes related to high proliferative index
Characterization of mouse models with deficiency in PARP2 or in PARP1 has revealed selective functions of PARP2, over PARP1, in various biological processes associated with a high cell proliferation rate, including T cell development (Ref. Reference Yelamos33), spermatogenesis (Ref. Reference Dantzer30), erythropoiesis at steady-state (Ref. Reference Farres14) conditions, and hematopoiesis under stress conditions (Ref. Reference Farres118).
PARP2 is required for T cell development
T cell development takes place in the thymus from bone marrow derived thymic progenitors through highly regulated processes involving T cell receptor gene rearrangement, proliferation, selection, survival and differentiation (Ref. Reference Zúñiga-Pflücker207). These sequential steps are required for the differentiation of double-negative (DN; CD4- CD8-) thymocytes into double-positive thymocytes (DP; CD4+ CD8+ ). DP thymocytes undergo intensive negative and positive selection to differentiate into either CD4+ or CD8+ single positive thymocytes that migrate to the periphery as mature naïve T lymphocytes. Interestingly, PARP2-deficient, but not PARP1-deficient mice show a significant reduction in the number of DP thymocytes indicating that PARP2 plays a role in T cell development (Ref. Reference Yelamos33). This phenotype is associated with the function of PARP2 in preventing the accumulation of DNA double-strand breaks and the ensuing activation of a DNA damage response (DDR) during T cell receptor rearrangement in a p53-dependent manner (Refs Reference Yelamos33, Reference Nicolas129).
Selective role of PARP2 in spermatogenesis
Spermatogenesis is a complex process that takes place in the seminiferous epithelium in which diploid spermatogonial stem cells differentiate into male haploid germ cells (Ref. Reference Fayomi and Orwig208). Interestingly, PARP1 and PARP2 have a different distribution in the seminiferous epithelium, with PARP1 expression restricted to the peripheral cell layer, whereas PARP2 expression is homogeneously distributed throughout the seminiferous tubules (Refs Reference Schreiber10, Reference Tramontano, Di and Quesada209, Reference Tramontano, Malanga and Quesada210) suggesting a discrete role of PARP1 and PARP2 in spermatogenesis. Indeed, PARP2-deficient male mice show hypofertility connected with abnormalities in spermatogenesis including chromosome missegregation in metaphase I cells, impaired meiotic sex chromosome inactivation, large number of spermatocytes in meiotic prophase and metaphase I displaying features of apoptosis and high level of DNA damage in primary spermatocytes and haploid cells (Refs Reference Dantzer30, Reference Quenet31, Reference Tramontano, Malanga and Quesada210). PARP2 (similar to PARP1) partakes in genome remodelling during spermiogenesis (Refs Reference Quenet31, Reference Meyer-Ficca140)
Human studies have also verified these observations. Jha et al. (Ref. Reference Jha211), based on studies on the semen samples of 18 healthy and 12 infertile humans, showed that the expression of PARP2 correlated with genotoxic insults to semen. Furthermore, Sakugawa et al. (Ref. Reference Sakugawa212) have identified on a cohort of 18 Japanese men three single nucleotide polymorphisms (SNPs) in the region coding the PARP2 catalytic domain and two in the 3′ UTR associated with azoospermia.
PARP2 in hematopoiesis
Hematopoiesis is a highly regulated multistep process within the bone marrow in which multipotent hematopoietic stem cells self-renew and differentiate into a series of committed progenitors that give rise to all mature blood cell lineages, including lymphocytes, monocytes, granulocytes, platelets and erythrocytes (Ref. Reference Pucella, Upadhaya and Reizis213). To maintain hematopoietic homoeostasis through life, several pathways involved in cell cycle regulation, DNA repair and apoptosis must be tightly regulated in order to prevent genomic instability, haematological malignancies and bone marrow failure (Ref. Reference Delia and Mizutani214). Indeed, hematopoiesis is a process highly sensitive to genotoxic agents such as radiation or chemotherapy which causes myeloablation-associated site effects in patients (Refs Reference Delia and Mizutani214, Reference Renneville, Bernard and Micol215). Hence, understanding DDR pathways in hematopoietic stem and progenitor cells is critical to avoid off-target responses in patients under treatment with genotoxic agents.
Interestingly, PARP2-deficiency, but not the deficiency of PARP1, accelerated bone marrow failure and death of mice exposed to sublethal doses of irradiation. This phenotype is associated with an impaired DDR and increased p53-dependent and Puma-dependent apoptosis of hematopoietic stem-progenitor cells in the absence of PARP2 (Ref. Reference Farres118).
Furthermore, at steady-state conditions, PARP2, but not PARP1, deficiency leads to chronic anaemia in mice (Ref. Reference Farres14). The reduced circulating erythrocytes number in PARP2-deficient mice is associated with the accumulation of DNA damage in response to high replicative stress in erythroid progenitors, which limits their expansion through the activation of cell cycle checkpoints and apoptosis (Ref. Reference Farres14).
Immunomodulatory roles of PARP2
The aforementioned cellular roles of PARP2 may impact, in a specific manner, on the biology of immune cells of both the innate and the adaptive immune system and thus affect different immune-mediated functions. Interestingly, the involvement of PARP2 in the regulation of immune responses is evolutionarily conserved and can also be found among plants (Ref. Reference Feng216).
PARP2 in innate immune responses
The innate immune system consists of cells (neutrophils, macrophages, dendritic cells, mast cells, natural killer cells and other innate lymphoid cells) and soluble molecules that serve as the front line of host protection to infection, tissue injury and tumour progression. In addition, the innate immune system also stimulates adaptive immune responses. One of the major protective reactions of the innate immune system is inflammation, a process by which innate immune components are recruited to site of infection or damaged tissues. PARP2 has been shown to be involved in different aspects of the inflammatory response, which are specifically detailed below.
PARP2 in adaptive immune responses
Adaptive immune response is mediated by T and B lymphocytes. Despite the aforementioned role of PARP2 in T cell development at the thymus, PARP2 deficiency does not change the number of peripheral T cell subsets under basal conditions, although its effect on the functional activity of these cells is unknown. Noteworthy, PARP2-deficient T cells produce more interferon γ (IFNγ) than wild-type T cells by unknown mechanisms that would be interesting to explore given their possible functional implications (Ref. Reference Moreno-Lama217). Characterization of germline and B lymphocyte-specific PARP2−/− mice revealed that PARP2 deficiency has no impact on B lymphocyte development, in contrast to T lymphocyte development. Furthermore, PARP2 deficiency has no impact on antibody production in response to T-dependent and T-independent antigens (Ref. Reference Galindo-Campos218).
PARP2 in the regulation of inflammation
The role of PARPs in inflammation became evident in the late 1990s, when PARylation was implicated in monocyte/macrophage activation upon lipopolysaccharide (LPS) induction (Refs Reference Hauschildt219, Reference Heine220), and the genetic deletion or pharmacological inhibition of PAR synthesis was shown to exert anti-inflammatory effects in mice (Ref. Reference Szabo221). Since these initial findings, multiple studies demonstrated the involvement of PARP activation in inflammatory reactions (for detailed review see in (Refs Reference Bai and Virag222, Reference Fehr223, Reference Kunze and Hottiger224)), as much as the protective effect of PARP inhibition in acute and chronic inflammation (Refs Reference Ahmad225, Reference Black226, Reference Czapski227, Reference Di Paola228, Reference Gonzalez-Rey229, Reference Hans230, Reference Hauser231, Reference Jijon232, Reference Jouan-Lanhouet233, Reference Kauppinen234, Reference Liu235, Reference Mazzon236, Reference Mazzon237, Reference Naura238, Reference Oumouna239, Reference Pacher and Szabo240, Reference Sahu, Narota and Naura241, Reference Sánchez-Fidalgo242, Reference Wardi243, Reference Virag244, Reference Mukhopadhyay245). As the majority of cellular PARP activity in mammals is attributed to PARP1, most of the available data in the context of inflammatory regulation focuses on the role of PARP1. However, PARP inhibitors target not only PARP1, but also PARP2-derived PAR synthesis (Ref. Reference Wahlberg246). Hence, for evaluating the anti-inflammatory effect of PARP inhibitors, it would be important to learn more about the tissue-specific targets of PARP2 activity and their relevance in pro- and anti-inflammatory pathways. It is also to be noted that in certain instances, the regulatory action of PARPs involves only the scaffolding function of the enzymes, and independent of their catalytic activity. One such example is the nuclear factor-κB (NF-κB) coactivator function of PARP1 (Ref. Reference Hassa247), but possibly there are cell type-dependent factors determining the nature of interaction between PARP1 and NF-κB that may be relevant in different inflammatory settings.
Nevertheless, in the past decade it turned out that PARP2 has multi-faceted regulatory roles in inflammatory mechanisms, which have been involved in various conditions, including neuroinflammation, hepatitis, colitis and psoriasis. In this chapter, we provide an overview on the role of PARP2 in immune modulation to explain why PARP2 deficiency tends to be protective in inflammatory pathologies.
PARP1/2 inhibition by PJ34 protects mice from concanavalin A (ConA)-induced liver injury (Refs Reference Jouan-Lanhouet233, Reference Arshad248), a murine model of immune cell-mediated hepatitis in humans (Ref. Reference Tiegs, Hentschel and Wendel249). It later turned out, that it is the deficiency of PARP2, and not of PARP1, which is protective against the hepatolysis caused by ConA (Ref. Reference Filliol176). Surprisingly though, neither PJ34, nor the genetic deletion of PARP2 suppressed the inflammation that is key in the promotion of hepatolysis induced by ConA.
The ConA-induced liver injury is dependent on Natural Killer T (NKT) lymphocytes (Refs Reference Kaneko250, Reference Takeda251, Reference Toyabe252). In the spleen and liver of PARP2−/− mice, a marked reduction was found in the percentage of type I or invariant NKT (iNKT) cells, the major NKT population in the liver. This resulted in reduced hepatocyte death after ConA induction, while the recruitment and activation of immune cells, such as conventional T lymphocytes, and their proinflammatory cytokine expression was unaffected (Ref. Reference Filliol176). The reduction of iNKT lymphocyte subset may be explained by the role PARP2 play in T cell receptor α-chain (TCRα) rearrangement process during thymopoiesis (Ref. Reference Yelamos33), as the iNKT cells are characterized by the expression of TCRα chain with a unique rearrangement (Vα14-Jα18).
There are some other points in this context that might worth considering. Although the most potent activator of iNKT cells is thought to be the synthetic glycolipid antigen α-galactosylceramide (Refs Reference Brossay253, Reference Kawano254), which was also used in the study by Filliol et al. (Ref. Reference Filliol176) to analyse iNKT cells in the PARP2−/− mice, numerous endogenous lipid antigens, both bacterial and self-lipids, capable of iNKT activation have been identified (Refs Reference Cameron255, Reference Kinjo256, Reference Kinjo257, Reference Mattner258, Reference van Eijkeren259, Reference Zhou260). As described above, studies performed on PARP2−/− mice in the past 15 years revealed characteristic changes in the lipidome of several tissues with local, as well as systemic consequences on animal physiology (Refs Reference Szanto83, Reference Marton85, Reference Szanto151). It seems feasible that lipidomic alterations may have far-reaching effects on the differentiation and activation of iNKT cells in PARP2−/− mice.
Since microbial antigens modulate iNKT activation (Refs Reference Kinjo257, Reference Mattner258, Reference An261, Reference Ma262), gut microbiota composition may have significant effect on iNKT cell function not only in intestine but probably distant tissues, as well (Refs Reference Cameron255, Reference An261, Reference Ma262, Reference Olszak263). Interestingly, PARP1−/− mice display a more diverse gut microbiota composition compared to their wild-type littermates (Refs Reference Vida264, Reference Larmonier265, Reference Zhuang266), although these alterations were not linked to immune modifications in PARP1−/− mice so far. Future studies might determine if a functional association exists between gut microbiota and immune cell homoeostasis in PARP2−/− mice.
Even more so, that iNKT cells have been associated with the regulation of gut microbiota composition (Refs Reference de Aguiar267, Reference Selvanantham268, Reference Shen269), and thereby with the control of intestinal inflammation (Refs Reference Selvanantham268, Reference Shen269, Reference Lee270). Along this line, it was reported that the depletion of PARP2 by antisense oligonucleotide delivery reduced intestinal inflammation in the interleukin-10 (IL10)-deficient mouse model of colitis, as evidenced by the reduced lymphocyte infiltration, as well as attenuated TNFα and IFNγ secretion in the colon of mice treated with the PARP2-specific oligonucleotide compared to the control mice with colitis (Ref. Reference Popoff173). It was not established what may underlie the anti-inflammatory effect of PARP2 silencing in this model. The application of the antisense oligonucleotide resulted in only a small reduction in PARP2 expression in the colonic epithelial cells, hence it is unlikely to be responsible for the effect. More likely is the interference with innate immune responses caused by the suppression of PARP2 expression. In contrast to the colon, the oligonucleotide caused a significant decrease in PARP2 expression in the liver of mice (Ref. Reference Popoff173). It is possible that the lipid homoeostasis of liver is disturbed upon PARP2 depletion that affects iNKT cell activation. The gut also harbours iNKT cells, and it was suggested that a bi-directional interactive network, also called as the liver-gut axis, exists between iNKT cells of liver and gut and the gut microbiota, that together control inflammatory processes in the two organs (Ref. Reference Marrero271). Of note, there are discrepancies in the literature as to the role of iNKT cells in inflammatory regulation, as well as several gaps on the influence of PARP2 on innate immune processes that warrant further investigations.
Nevertheless, there is evidence that PARP2 mediates intestinal inflammation at the level of adaptive immunity, as well. The T cell-specific downregulation of PARP2 ameliorated LPS-induced acute colitis in mice (Ref. Reference Bencsics174). Lower IL17 production was measured in the colon of T cell-PARP2-KO mice than in the control mice after LPS treatment, suggesting that local activation of T helper 17 (Th17) cells, a key component in the pathogenesis of inflammatory bowel disease (Ref. Reference Caprioli, Pallone and Monteleone272), was impaired in mice with the T cell-specific conditional PARP2 deletion. This could be the result of suppression by regulatory T (Treg) cells, as Treg cell number was increased in the intestinal mucosa of LPS-treated T-cell-specific PARP2−/− mice compared to control animals after LPS treatment (Ref. Reference Bencsics174). To note, Th17 and Treg cells share a common precursor and require a common tumour growth factor (TGF)β signal for differentiation. In the presence of pro-inflammatory cytokines (e.g. IL6), TGFβ drives the differentiation of naїve CD4+ T cells into Th17 cells, however, in the absence of these cytokines the TGFβ signal directs towards Treg differentiation (Refs Reference Bettelli273, Reference Mangan274, Reference Veldhoen275). Hence, Th17 and Treg cells have opposite functions in immune regulation in autoimmunity, as Th17 cells can trigger, while Treg cells can suppress autoimmune diseases (Ref. Reference Lee276). Interestingly, PARP2 was shown to modulate TGFβ signalling in vitro, as PARP2, in complex with PARP1, seems to negatively regulate the Smad-dependent transcriptional responses induced by TGFβ in HaCaT keratinocyte cultures (Ref. Reference Dahl89). A similar mechanism may exist in T cells, and PARP1 and PARP2 may coordinately assist the differentiation of naїve CD4+ T cells in different conditions, as PARP1 regulates TGFβ signalling in T cells, as well (Ref. Reference Zhang277). The CD4+ T cells of PARP1−/− mice display higher expression of TGFβ receptors (Ref. Reference Zhang277), and PARP1−/− mice have higher number of Tregs in spleen, thymus and lymph nodes than in PARP1 + / + mice (Refs Reference Nasta278, Reference Zhang279).
PARP1−/− CD4+ T cells also have an increased capacity to differentiate into Th17 cells in response to TGFβ and IL6 in vitro (Ref. Reference Zhang277). Although this supports the role of PARP1 in the TGFβ signalling of CD4+ T cells, it is unlikely to occur in vivo in the steady state since PARP1 deficiency in mice is associated with generally lower IL6 levels, and PARP1−/− CD4 + T cells secreted less IL6 in response to antigen stimulus in vitro than PARP1+ / + T cells (Ref. Reference Zhang277). However, pro-inflammatory stimulus can upregulate IL6 production in mice, and this may explain why PARP1−/− mice show augmented inflammation in mouse models of Th17-mediated inflammatory diseases, such as the experimental autoimmune encephalomyelitis (EAE), the murine model of multiple sclerosis (Refs Reference Kamboj280, Reference Selvaraj281), and the imiquimod-induced psoriasis-like dermatitis (Ref. Reference Kiss282).
Further studies are required to determine the potential role of PARP2 in TGFβ signalling in naїve CD4+ T cells, but we might as well assume that PARP2 cooperates with PARP1 in regulating the TGFβ signalling of T cells in a similar manner to what was found in HaCaT cultures. However, it would not explain why PARP2−/− mice, in contrast to PARP1−/− mice, are protected against Th17-mediated inflammation. In EAE, PARP2−/− mice displayed a delayed disease onset and a significantly reduced number of Th17 lymphocytes and hence inflammation in the central nervous system (Ref. Reference Kamboj280). In the imiquimod-induced psoriasis model PARP2−/− mice exhibited ameliorated dermatitis compared to PARP2 + / + mice (Ref. Reference Antal150). Immune cell subsets were not characterized in the infiltrate of the skin of PARP2+ / + and PARP2−/− mice with psoriasis-like dermatitis, but the smaller IL17 concentration measured in inflamed skin lysates of PARP2−/− mice suggests lower presence or suppressed activation of Th17 cells. These data suggest the existence of a PARP2-specific regulation of Th17 differentiation. It was shown in PARP2-depleted human keratinocytes and in the skin of PARP2−/− mice that phosphorylation of NF-κB p65 subunit is reduced in response to psoriasis-mimicking pro-inflammatory stimulus (Ref. Reference Antal150). The repressed activation of NF-κB and hence the amelioration of inflammatory response was a result of induced estradiol biosynthesis in PARP2-deficient keratinocytes (Ref. Reference Antal150). On one hand, decreased activation of NF-κB in keratinocytes can lead to decreased production of chemokines that recruit the Th17 subset to the site of inflammation. On the other hand, it was found in the imiquimod-treated skin of PARP2−/− mice that the expression of IL6, that is a NF-κB target gene, reduced compared to PARP2+ / + mice, which may have hampered the TGFβ-mediated differentiation of resident naїve T cells into Th17 cells. In addition, estradiol has a direct effect on Th17 cell differentiation. Although there are contradictory data in the literature (Refs Reference Fuseini283, Reference Newcomb284), estradiol and oestrogens in general are considered as negative regulators of Th17 cell differentiation (Refs Reference Bhadricha285, Reference Chen286, Reference Garnier287, Reference Lélu288, Reference Tyagi289, Reference Zhang290). Of note, only the skin has been studied in the imiquimod-treated PARP2−/− mice, but in the case of whole-body knock-out mice, the probability of the contribution of systemic effects must not be ruled out. A systemic shift towards oestrogen action in PARP2−/− mice could also explain, at least in part, the protective phenotype of PARP2−/− mice in EAE (Ref. Reference Kamboj280), as multiple studies demonstrated the protective role of oestrogens in EAE (Refs Reference Lélu288, Reference Benedek291, Reference Elloso292, Reference Haghmorad293, Reference Laffont294, Reference Offner295).
Taken together, PARP2 may have more complex roles in inflammatory regulation than we are currently aware of, and more studies are needed to explore PARP2-specific immune modulatory effects that may facilitate efforts towards the development of PARP2-specific inhibitors and may open new avenues for the applicability of the clinically available PARP inhibitors outside tumour therapy.
PARP2 in cancer
Cancer development is a multistep process involving the acquisition of capabilities (cancer hallmarks) for maintaining proliferative signalling, deregulating cellular metabolism, cell death resistance, increased genomic instability, inducing angiogenesis, activating tumour invasion, promoting inflammation, enabling replicative immortality, evading growth suppressors and immune evasion (Ref. Reference Hanahan296). As aforementioned, PARP2 has been shown to play important roles in many of these processes, both at the level of the tumour cell itself and its environment, suggesting its involvement in tumour progression (Table 6). Accordingly, increasing evidence suggests though that PARP2 plays specific roles in different types of cancers through different mechanisms including transcription regulation, immunomodulatory functions and DDR regulation (Table 6).
Table 6. PARP2 in cancer

PCa, Prostate cancer; AR, Androgen receptor; T-ALL, T-cell acute lymphoblastic leukaemia.
PARP2 in breast cancer
In a mouse model of PyMT-driven spontaneous breast cancer (MMTV-PyMT) (Ref. Reference Guy, Cardiff and Muller300), it was observed that PARP2 deficiency in the whole mouse or PARP2 deficiency only in the mammary gland delayed tumour onset, without affecting tumour growth rate, while selective PARP2 deficiency in the myeloid lineage did not affect tumour initiation (Ref. Reference Zuo297). Interestingly, lung metastasis was significantly reduced in PARP2-deficient MMTV-PyMT mice as well as in hosted wild-type mice implanted with PARP2-deficient cells compared to control. Conversely, in another syngeneic mouse model of bone metastasis-prone breast cancer, PARP2 deficiency has been shown to increase bone metastasis mediated by converting the bone microenvironment to immunosuppressive by altering the balance of Treg and T helper 1 (Th1) cells (Ref. Reference Zuo297).
In a different syngeneic breast cancer mouse model in which the PARP2 proficient AT-3 breast cancer cell line was implanted in host-wild-type mice and in mice with a PARP2-deficiency only in T cells, we observed a significant reduction in AT-3-induced tumour growth in host mice with a deficiency of PARP-2 only in T cells compared to control host mice, suggesting a critical role of PARP2 in modulating the T cell response against breast tumour (Ref. Reference Moreno-Lama217). More recently, in a triple negative breast cancer model, it was demonstrated that selective degradation of PARP2 by proteolysis targeting chimera (PROTAC) has a therapeutic effect, both in vitro and in vivo (Ref. Reference Pu301).
In addition to these pre-clinical studies, PARP2 has also been suggested to play a role in the response to chemotherapy in breast cancer; multiple studies have shown associations with SNPs in PARP2 and breast cancer survival or risk. In a study using a cohort of postmenopausal breast cancer patients, it was shown that the intronic rs878156 SNP in PARP2 can modulate cancer specific survival in breast cancer patients as a function of chemotherapy (Ref. Reference Seibold302). Another study by Popanda et al. (Ref. Reference Popanda303) identified association between a PARP2 polymorphism and breast cancer risk. Finally, in ER-positive/HER2-negative breast tumours, the expression of PARP2 was associated with poor prognosis (Ref. Reference Santarpia304).
PARP2 in prostate cancer
Genetic depletion or pharmacological inhibition of PARP2 with the selective inhibitor UPF-1069, inhibited androgen receptor (AR)-positive, but not AR-negative, prostate cancer cell growth in vitro (Ref. Reference Gui153). In addition, prostate tumour growth inhibition was also observed in vivo after selective targeting of PARP2 in an immunodeficient mouse model with subcutaneous implantation of AR-positive cancer cells (Ref. Reference Gui153). The transcription factor AR plays a critical role in prostate cancer progression. This effect of PARP2 in prostate cancer progression has been suggested to be due to the interaction of PARP2 with FOXA1, a critical factor facilitating AR recruitment to genome-wide prostate-specific enhancer regions. Moreover, analysis of PARP2 expression in prostate tumour samples has shown that overexpression of PARP2 is associated with prostate cancer aggressiveness (Ref. Reference Gui153).
PARP2 in haematological malignancies
The role of PARP2 has been explored in different haematological malignancies. Interestingly, in a mouse model of c-Myc-driven B cell lymphoma, PARP2 deficiency delays tumour progression by exacerbating replication stress in c-Myc-overexpressing B cells, resulting in accumulation of DNA damage and concomitant cell death that restricts the c-Myc–driven expansion of B cells in a p53-dependent manner (Ref. Reference Galindo-Campos298). However, in a mouse model of spontaneous p53-deficiency-driven T cell lymphoma, PARP2 deficiency accelerates the progression of T cell lymphomas (Refs Reference Nicolas129, Reference Navarro305). In both models, PARP2 deficiency triggers, in response to DNA damage accumulation, a p53-dependent DDR, resulting in cell cycle arrest and impaired survival of c-Myc-overexpressing pre-B cells or thymocytes respectively as the major intrinsic tumour-suppressor mechanism. Indeed, when p53 is removed, the protective effect associated with PARP2 deficiency disappears in the model of B cell lymphoma (Ref. Reference Galindo-Campos298) or even accelerated tumour progression in the case of T cell lymphoma (Refs Reference Nicolas129, Reference Navarro305). Interestingly, PARP2 deficiency does not influence the survival in a mouse model of Notch-1-driven T cell acute lymphoblastic leukaemia (Ref. Reference Galindo-Campos298). Altogether, these data suggest that the effect of PARP2-deficiency may be specific to c-Myc-driven haematological malignancies.
In an alkylator-induced mouse model of acute myeloid leukaemia, induced in multiple in-bred mouse strains, Cahan and Graubert identified network of cancer-inducing and supporting genes, among which PARP2 had a prominent place (Ref. Reference Cahan and Graubert306).
PARP2 in tumours of the digestive tract
Recently, PARP2 has been involved in different types of tumours of the digestive tract. For instance, the pro-tumour effect of circular RNA-RAD23B in the invasion of oesophageal cancer cells has been suggested to be mediated by promoting the expression of PARP2 (Ref. Reference Lan21). In gastric cancer, PARP2 deletion restrained cell proliferation, migration and invasion of gastric cancer cell lines (Ref. Reference Ma, Jiang and Wu23). Moreover, PARP2 have also been involved in pathways associated with hepatocellular carcinoma. For instance, PTTG3P acted as an oncogenic lncRNA to promote hepatocellular carcinoma development through upregulating CCND1 and PARP2 (Ref. Reference Zhou24). In addition, high expression of PARP2 and low expression of miR-149 in hepatocellular tumour tissues correlated with larger tumour mass size, capsular and vascular invasion, lymph node metastasis and high histological grade. Accordingly, high PARP2 expression is a poor prognosis factor and correlated with low survival rate (Ref. Reference Lin19).
Development and aging
The involvement of PARP1 in development and aging was known since the late ‘90s (Refs Reference Muiras307, Reference Wang308, Reference de Murcia309). The role for PARP1 in aging was mostly associated with its role in DNA repair, although subsequently PARP1 was associated with most hallmarks of aging (Refs Reference Vida310, Reference Muiras and Burkle311, Reference Mangerich312). Recent data suggest that PARP2 is likely to be involved in development and aging, furthermore, the role of PARP2 in development and aging is evolutionarily conserved, as such features were identified in plants as well (Ref. Reference Liu313).
Although PARP2 knockout mice are conceived, develop and are viably born, the double knockout of PARP2 and other DNA repair enzymes as PARP1 (Ref. Reference Menissier-de Murcia43) or ATM (Ref. Reference Huber72) are non-viable and double knockout embryos are lost in the first half of the pregnancy. Although, the exact cause of the underdevelopment and loss of the double knockout embryos is not explored in detail and not defined. Nevertheless, PARP2 is involved in decidualization (Ref. Reference Kelleher314) and in developing endometrial receptivity and supporting blastocyst implantation (Ref. Reference Soni48) that represent likely explanations for early embryonic lethality alongside deficient DNA repair. PARP2 interacts with transcription factors that influence the development of organs, as TTF-1 (Ref. Reference Maeda154) that impact on lung development. PARP2 regulates cell death processes (Refs Reference Dantzer30, Reference Yelamos33, Reference Cohausz and Althaus186).
At the other end of the life cycle of an individual, PARP2 is associated with aging. As discussed earlier, PARP2 is involved in processes that are considered as the hallmarks of aging as systemic inflammation (Ref. Reference Meier315), metabolic features (Ref. Reference Janssens316), cardiovascular health (Ref. Reference Hickman317), microbiome health (Refs Reference Shintani318, Reference Miko, Vida and Bai319). Chevanne et al. (Chevanne et al. (Ref. Reference Chevanne180)) showed that the expression of PARP1 and PARP2 in immortalized B lymphocytes from aged and centenarians, as compared to cells from young individuals, decrease supporting the previous observations that successful aging is associated with sustained PARP1 (and PARP2?) expression (Refs Reference Vida310, Reference Muiras and Burkle311, Reference Mangerich312). This observation can be linked to features we have discussed, such as the reduced thymopoiesis in the PARP2 knockout mice that resembles to the processes associated with thymic aging (Ref. Reference Yelamos33). The currently available data suggest that while PARP2 is associated with aging, it is likely not an individual aging factor, but PARP2 is likely a member of a web that acts in a coordinated fashion during aging.
Future prospects
Hereby, we reviewed the known biological properties and roles of PARP2, including inflammation, cancer, hematopoiesis, metabolism, oxidative stress-related diseases (Fig. 5). It is likely that there are other, yet uncovered biological functions of PARP2, such as its involvement in the life cycle of viruses (Refs Reference Fehr223, Reference Curtin320, Reference Papp321). Uncovering and understanding these pathways warrant further research.

Figure 5. The schematic representation of the PARP2-mediated physiological and pathophysiological functions. AR, androgen receptor; BER, base excision repair; CAF, cancer-associated fibroblast; DDR, DNA damage response; DSB, double-strand break; NRF2, nuclear factor erythroid 2–related factor 2; PARylation, poly-ADP-ribosylation; PPAR, peroxisome proliferator activated receptor; SREBP, Sterol regulatory element-binding protein; SSB, single-strand break; TCR, T cell receptor; TTF1, Thyroid transcription factor-1; topo, topoisomerase; WAT, white adipose tissue.
Early in vivo studies demonstrated a sensitivity of PARP1 and PARP2-deficient mice to genotoxic agents, indicating a role for these proteins in the DDR, although possibly with spatio-temporal differences. Accordingly, considering the critical role of the DDR in cancer, PARP1/2 inhibitors (PARPi) have emerged as an important new class of therapeutics in cancer. However, in spite of increasing evidence suggesting that PARP1 and PARP2 have discrete biological roles, PARPi currently approved for clinical use, targeting the highly conserved catalytic site of PARP1/2, do not discriminate between PARP1 and PARP2 (Ref. Reference Brooks, Anand and Cohen322). Taking into account the specific functions of these two proteins in various biological processes, including cancer models, a new generation of selective inhibitors has been initiated, aim to optimized anti-tumour immune response, reduce off-target effects and optimize the side-effect profile of the inhibitors. A clear example of this development is the generation of a selective PARP1 inhibitor by AstraZeneca that is already in clinical trials (Ref. Reference Illuzzi323). The development of selective PARP2 inhibitors is at an earlier stage. UPF-1069 has very limited selectivity which limits its clinical use (Ref. Reference Gui153). In addition to classical small molecules, PROTAC has emerged as a new strategy for selective PARP2 inhibition that has demonstrated efficacy in a pre-clinical breast cancer model (Refs Reference Pu301, Reference Kelleher314). Undoubtedly, the arrival of these PARP1/PARP2-selective inhibitors in the pre-clinical and clinical phase will allow validation of the results of genetic depletion of PARP2 in different tumours and may represent a very important therapeutic advance in the fight against certain types of cancer.
Acknowledgement
The project was funded by grants from the NKFIH (K142141, FK134588), from the Hungarian Academy of Sciences (POST-COVID2021-33, NKM2022-30). Project no. TKP2021-EGA-19 and TKP2021-EGA-20 have been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the TKP2021-EGA funding scheme. This project has received funding from the HUN-REN Hungarian Research Network. The Yélamos lab is funded by the Spanish Ministerio de Ciencia e Innovación (grant PID2020-112526RB-I00; funded by MCIN/AEI/10.13039/501100011033). The project is realized support from the Science Funding Initiative at the University of Debrecen. Figures 3 and 5 were created with BioRender.com.
Abbreviations
ACOX1, acyl coenzyme A oxidase I; AMPK, AMP-activated protein kinase; aP2, adipocytes protein 2; AR, androgen receptor; ART, ADP-ribosyl transferase enzymes; BER, base excision repair; CAF, cancer-associated fibroblast; CENPA, centromere protein A; CENPB, centromere protein B; CHD1L, Chromodomain-helicase-DNA-binding protein 1-like; CNS, central nerve system; ConA, concanavalin A; cyt c, cytochrome c; DDR, DNA damage response; DN, double-negative (CD4- CD8-); DP, double-positive thymocytes (CD4+ CD8+ ); DSB, double strand break; EAE, experimental autoimmune encephalomyelitis; ER, estrogen receptor; FAS fatty acid synthase; FEN1, Flap endonuclease 1; HR, homologous recombination; IFNγ, interferon γ; iNKT, invariant NKT; IL, interleukin; LPL, lipoprotein lipase; LPS, lipopolysaccharide; MARylation, mono-ADP-ribosylation; MCAD, medium-chain specific acyl-CoA dehydrogenase; MCD, malonyl-CoA decarboxylase; mCPT1b, muscle isoform of carnitine O-palmitoyltransferase 1; mTOR, mechanistic/mammalian target of rapamycin; NFE2L2, nuclear factor erythroid 2-related factor 2; NF-κB, nuclear factor-κB; NHEJ, non-homologous end joining; NKT, Natural Killer T; NRF2, nuclear factor erythroid 2–related factor 2; PARPi, PARP1/2 inhibitors; PARylation, poly-ADP-ribosylation; PGC1α, peroxisome proliferator activated receptor cofactor 1α; PPAR, peroxisome proliferator activated receptor; PROTAC, proteolysis targeting chimera; ROS, Reactive oxygen species; RXR, retinoid X receptor; SNP, single nucleotide polymorphism; SREBP, Sterol regulatory element-binding protein; T-ALL, T cell acute lymphoblastic leukaemia; TCRα, T cell receptor α-chain; Th1, T helper 1; Th17, T helper 17; topo, topoismerase; TTF1, Thyroid transcription factor-1; Treg, regulatory T; TGF, tumour growth factor; UCP-2, uncoupling protein-2; WAT, white adipose tissue; XRCC1, X-ray repair cross-complementing protein 1; 5′-dRP, 5′-deoxyribose phosphate.












 Open access
Open access