



Introduction
Thromboembolic events account for around one quarter of deaths worldwide, being the most frequent condition underlying myocardial infarction and ischaemic stroke. The incidence of thrombosis increases with age and its complications are among the major causes of long-term morbidity and poor quality of life, particularly in western countries (Ref. Reference Wendelboe and Raskob1). Understanding the pathogenetic mechanisms of thrombosis is a major challenge to set up appropriate prophylactic interventions.
In recent years, many studies have focused on the role of oxidative stress, that is, a condition in which a massive reactive oxygen species (ROS) production overwhelms antioxidant defences, in inducing thrombosis (Refs Reference Emmi2–Reference Becatti6). It is known that ROS can stimulate coagulation by increasing the expression of tissue factor in endothelial cells, monocytes and vascular smooth muscle cells, by directly interfering with platelet activation, as well as by inducing oxidative structural and functional modifications to key proteins involved in the coagulation cascade (including tissue factor pathway inhibitor, TFPI, protein C, thrombomodulin, fibrinogen, antithrombin). Moreover, ROS can mediate thrombo-inflammation, also via leucocyte (particularly neutrophil) hyperactivation and extracellular traps release (Ref. Reference Bettiol7). Interestingly, while erythrocytes have traditionally been considered as playing a bystander role in haemostasis and thrombosis (Ref. Reference Weisel and Litvinov8), growing evidence suggests a direct involvement of these cells in ROS-induced thrombogenesis (Ref. Reference Barshtein, Ben-Ami and Yedgar9).
Erythrocytes produce high amounts of intracellular ROS by NADPH oxidase activation and haemoglobin autoxidation. Moreover, erythrocytes can uptake extracellular ROS released by other cells in the blood flow. Accumulated ROS can induce structural changes to cell membrane, resulting in an impaired erythrocyte function and in the generation of a hypercoagulable milieu.
In this review, we aim to connect the dots linking erythrocytes, oxidative stress and thrombosis (Fig. 1), addressing their potential implication for the clinical and therapeutic management of thrombosis.
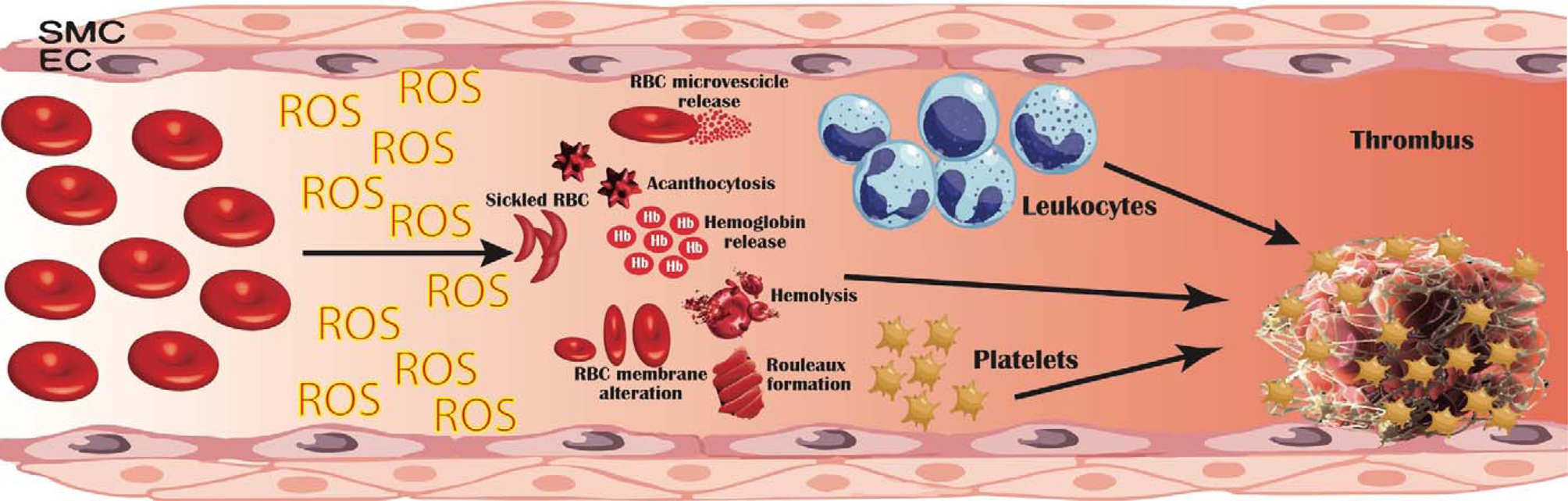
Fig. 1. Pathogenetic mechanisms linking erythrocyte oxidative modifications to thrombosis. Erythrocytes produce high quantities of intracellular reactive oxygen species (ROS), mostly by NADPH oxidase activation and haemoglobin autoxidation; furthermore, extracellular ROS released by other cells in the blood flow can be uptaken and accumulate within erythrocytes. This oxidative milieu can alter erythrocyte membrane, leading to an impaired erythrocyte function and promoting erythrocytes lysis, binding to endothelial cells (EC), activation of platelet, coagulation factors and leucocytes. Moreover, structurally altered erythrocytes are able to adhere to the vessel wall, contributing to thrombin generation within thrombus. This process results in an accelerated haemolysis and in a hypercoagulable state, in which structurally impaired erythrocytes contribute to increase thrombus size and to reduce its permeability and susceptibility to lysis. EC, endothelial cells; RBC, red blood cells; ROS, reactive oxygen species; SMC, smooth muscle cells.
Erythrocytes as leading actors in thrombosis
Haematocrit and thrombosis
The concept that erythrocytes contribute to haemostasis was formulated more than a hundred years ago, based on the evidence that bleeding time in anaemic patients was prolonged also in the presence of a normal platelet count (Ref. Reference Duke10), and that a negative correlation existed between haematocrit and bleeding time (Ref. Reference Hellem, Borchgrevink and Ames11). On the other hand, an abnormally high haematocrit, as observed in patients with polycythaemia vera or taking erythropoietin, has been associated with an increased risk of thrombosis (Ref. Reference Marchioli12). Erythrocytes primarily influence blood viscosity, which increases in a nonlinear manner with haematocrit. Increased viscosity decelerates blood flow and is a component of the Virchow's triad leading to a prothrombotic state (Ref. Reference Wolberg13). Indeed, haematocrit-related blood viscosity influences the interaction between platelets and blood vessel surfaces, with a remarkable rheological effect. Indeed, erythrocytes generally move down the centre of blood vessels, while platelets occupy marginal positions, to easily adhere at sites of vessel-wall injury (Ref. Reference Aarts14). In the presence of abnormally high haematocrit, platelets tend to accumulate near the vessel wall with arterial shear rates, increasing their interactions with the activated endothelium (Ref. Reference Goldsmith15). In vessels of small calibre, erythrocytes may aggregate and concentrate along the flow axis, thus further resulting in platelet margination. Moreover, as erythrocytes have a lower viscosity compared to platelets (Ref. Reference Dintenfass16), an increased haematocrit determines a reduced local viscosity (Ref. Reference Fahraeus17), which results in a decreased wall shear stress and a lower local nitric oxide (NO) release (Ref. Reference Baskurt18). As NO prevents the activation of endothelial cells and platelets, this leads to cellular activation in a pro-thrombotic sense.
Also, at low shear rates, the peculiar erythrocyte morphology allows electrostatic interactions and cell aggregation into piled-up ‘rouleaux’ structures, which cause an increased viscosity and hydrodynamic resistance (Ref. Reference Piety19). This phenomenon is more common in larger venous vessels at lower shear rates, such as in the lower limbs, which indeed are an elective site of venous thrombosis (Ref. Reference Yu20). Notably, fibrinogen is essential for the formation of rouleaux under low shear conditions (Ref. Reference Lominadze and Dean21) as it is able to bridge nearby cells, stimulating aggregates formation; the connection between fibrinogen and erythrocytes seems to be mediated by an integrin receptor on erythrocytes membrane, the β3 integrin (Ref. Reference Carvalho22) and/or the integrin-associated protein (CD47) (Ref. Reference De Oliveira23).
Erythrocyte structure and thrombosis
Even when haematocrit is within physiological ranges, erythrocytes can promote a pro-thrombotic state following structural and functional cell alterations. Erythrocytes are uniquely deformable cells with a characteristic biconcave shape capable of undergoing reversible shape changes into a bullet-like shape each time they pass inside microvessels. This morphology is essential to guarantee oxygen/carbon dioxide exchange between tissues and blood; indeed, by maximising the active contact area between erythrocytes and the vessel wall, as a result of erythrocyte deformation and high surface-to-volume ratio, gas exchange is optimised (Ref. Reference Danielczok24).
In some diseases, including sickle cell disease, β-thalassemia, haemolytic anaemias and hereditary stomatocytosis, as well as in chronic conditions such as diabetes, hypertension and coronary heart disease, erythrocytes show more rigid and less deformable structure (Refs Reference Vaya25, Reference Symeonidis26). This results in a lower ability to squeeze through capillaries and in an increased platelet margination, contributing to a prothrombotic state (Refs Reference Aarts27, Reference van Gelder, Nair and Dhall28).
Also, in sickle cell disease and β-thalassemia, the damaged erythrocyte membrane externalises phosphatidylserine, a negatively charged phospholipid which is physiologically located on the cytoplasmic side of the membrane. Phosphatidylserine exposure provides an active surface for prothrombin activation, determining a high thrombotic potential (Ref. Reference Ataga, Cappellini and Rachmilewitz29).
When exposed to high shear rates, inflammation, or in the above-mentioned diseases, erythrocytes can also generate microscopic extracellular membranous structures named microvesicles or microparticles, as a result of apoptosis activation or aging (Ref. Reference Heijnen30).
Microparticles enhance thrombin generation via the expression of phosphatidylserine and tissue factor, via the internalisation of free haeme and its transfer to vascular endothelium, as well as via the amplification of systemic inflammation through thrombin-dependent complement activation (Ref. Reference Zecher, Cumpelik and Schifferli31).
Erythrocytes and clot structure
Erythrocytes not only influence clot formation but also clot structure. Growing evidence shows that erythrocytes may be integrated into the thrombus, through unique liaisons with activated endothelial cells and/or exhibited subendothelial matrix (Ref. Reference Silvain32). Under normal circumstances, mature erythrocytes are not able to interface with endothelium; conversely, structurally and functionally altered erythrocytes (as observed in sickle cell disease, malaria or diabetes) show an increased stickiness and adhesion to the vascular endothelium, contributing to microvascular occlusions associated with thrombosis (Ref. Reference Smith33). Incorporation of erythrocytes in the thrombus influences fibrin network by increasing fibre diameter thus impacting on the viscoelastic clot properties (Ref. Reference Gersh, Nagaswami and Weisel34). In contracted clots and thrombi, erythrocytes have been shown to undergo a shape transformation from their native biconcave shape to a close-packed polyhedral structures covered by platelets and fibrin (polyhedrocytes) (Ref. Reference Cines35). Polyhedrocytes have been reported in coronary arterial thrombi from patients after myocardial infarction (Ref. Reference Cines35) and in pulmonary embolia (Ref. Reference Litvinov36). This structure decreases clot permeability to fibrinolytic agents, thereby increasing its resistance to lysis.
Interestingly, it has been suggested that erythrocytes can display also antithrombotic properties. In particular, haemoglobin deoxygenation is followed by an allosteric transition stimulating NO release from cysteine β93 of haemoglobin, with consequent capillary and postcapillary venules dilatation and inhibition of platelet reactivity (Ref. Reference Sun37). Moreover, ATP released from erythrocytes at low pH/reduced PO2 conditions or shear stress, can stimulate the activation of endothelial cell purinergic receptors, increasing NO production (Ref. Reference Kawai38). Also, it has been demonstrated that erythrocyte expression of ectoenzyme degrading ADP to AMP exerts antithrombotic properties by suppressing platelet aggregation (Ref. Reference Netsch39).
Therefore, erythrocytes structural and functional integrity displays critical roles in physiological haemostasis and thrombosis.
Erythrocyte and platelet interactions
Erythrocytes interact with platelets via different mechanisms. As previously described, erythrocytes exert a rheological effect, concentrating along the flow axis and causing platelet margination (Refs Reference Dintenfass16, Reference Fahraeus17). As a consequence, platelets are in close contact with the vessel wall, where they can interact with other clotting factors.
Moreover, as erythrocytes have a lower viscosity compared to platelets (Ref. Reference Dintenfass16), an increased haematocrit determines a reduced local viscosity (Fahraeus effect), except in capillaries that are smaller than erythrocytes, where the viscosity increases because of the presence of platelets (Ref. Reference Fahraeus17).
The reduced viscosity near the vessel wall determines a decreased wall shear stress and a reduced NO release (Ref. Reference Baskurt18), leading to cellular activation in a pro-thrombotic sense.
Erythrocytes can interact directly with platelets at venous shear rates, although erythrocyte-platelet binding has been described also in the so-called ‘white’ arterial thrombi mainly composed of activated platelets and fibrin (Ref. Reference Silvain40).
Beside straight adhesive interactions (Refs Reference Turitto and Baumgartner41, Reference Santos42), erythrocytes can stimulate platelet degranulation and aggregation via chemical signalling, (i.e. through the release of ATP and ADP under low pO2 and low pH), as well as through the action of extracellular haemoglobin released from damaged erythrocytes (Ref. Reference Helms43). Indeed, haemoglobin is a strong NO scavenger, and the release of extracellular haemoglobin from damaged erythrocytes determines a reduction in NO bioavailability, thus preventing the suppressive effect of NO on platelet activation (Ref. Reference Helms43). Concomitantly, the release of arginase from damaged erythrocytes determines the cleavage of L-arginine, a substrate for NO production (Ref. Reference Helms43).
Oxidative stress and thrombosis
In the last years, a prominent role of oxidative stress in regulating both endothelial dysfunction and thrombus formation is emerging.
The importance of oxidative stress in thrombogenesis was first demonstrated in an experimental mice model of thrombosis (mice lacking functional eNOS), where NO deficiency was significantly associated to arterial thrombosis. These mice showed lower bleeding times if compared to wild-type animals (Ref. Reference Freedman44). Later on, it has been shown that a moderate iron overburden significantly stimulates thrombus formation, via a defective vasoreactivity as well as via an enhanced ROS production (Ref. Reference Barr45).
ROS interfere with pro- and anticoagulant molecules
ROS can interfere with the coagulation process via a plethora of multiple, interconnected mechanisms. ROS, mostly generated by NOX enzymes, can directly stimulate the coagulation cascade by increasing the expression of tissue factor in endothelial cells, monocytes and vascular smooth muscle cells (Refs Reference Golino46–Reference Herkert48). ROS can also promote a procoagulant state via oxidative modification of proteins involved in the coagulation pathway, such as the anticoagulant proteins protein C (Ref. Reference Nalian and Iakhiaev49), thrombomodulin (Ref. Reference Glaser50) and the TFPI, resulting in their inactivation (Ref. Reference Ohkura51). Indeed, in mice models lacking superoxide dismutase (SOD-/- mice), larger, rapidly growing venous thrombi were observed, due to an impaired SOD1-mediated protein C activation (Ref. Reference Dayal52). Also, the heparin-binding capacity of antithrombin is decreased following oxidation by hydrogen peroxide (Ref. Reference Van Patten53) or lipid peroxide (Ref. Reference Gray and Barrowcliffe54).
Furthermore, lipid oxidation can inactivate the anticoagulant function of protein Z-dependent protease inhibitor, a specific inhibitor of membrane-associated factor Xa (FXa) (Ref. Reference Huang55).
Similarly, it has been observed that leucocyte-produced ROS can oxidise fibrinogen, altering its secondary structure and the overall clot architecture, characterised by reduced porosity and by tight fibrin network with filaments of decreased average size. Also, these oxidative alterations result in an impaired fibrinogen function, both in terms of thrombin-catalysed fibrin polymerisation and fibrin susceptibility to plasmin-induced lysis. This mechanism has been linked to increased thrombosis risk in Behcet's syndrome (Refs Reference Becatti4, Reference Becatti56), cirrhosis (Ref. Reference Becatti5), and it has been also described in post-acute myocardial infarction (Ref. Reference Becatti6), pulmonary hypertension (Ref. Reference Miniati57) and pulmonary embolism (Refs Reference Cellai58, Reference Lami59).
ROS and platelets
Besides affecting the activity of pro- or anti-coagulant molecules through oxidative modification, ROS can also directly interfere with platelets and other cells involved in haemostasis and thrombosis.
Intraplatelet ROS can activate platelets, by oxidising arachidonic acid, generating isoprostanes (Ref. Reference Pignatelli60); this mechanism has been linked with an increased risk of deep venous thrombosis in patients with hypercholesterolaemia (Ref. Reference Davi61), diabetes mellitus (Ref. Reference Davi62), homozygous homocystinuria (Ref. Reference Davi63) and in obese women (Ref. Reference Davi64). Concomitantly, ROS can also indirectly promote platelet activation by negatively regulating mechanisms of platelet inhibition, such as NO scavenging (Ref. Reference Tajima and Sakagami65). In hyperhomocysteinemia superoxide formation by hyperactive platelets has been described as one of the key pathways contributing to arterial thrombosis in this condition (Ref. Reference Riba66).
ROS and leucocytes
ROS also modulate platelet-leucocyte interactions: ROS produced by NOX2 can affect the expression of P-selectin (CD62) and CD40L, that are transferred to the platelet surface upon activation. P-selectin and CD40L promote leucocyte recruitment and activation (Refs Reference Turkoz67, Reference Martinez68) and their levels are associated with an increased risk of venous thromboembolism in various conditions (Ref. Reference Ay69), such as in Behçet's syndrome (Refs Reference Turkoz67, Reference Martinez68). Concomitantly, ROS can induce leucocyte recruitment via different complementary mechanisms: they can directly act as a chemoattractant for neutrophils and monocytes, mostly via upregulation of IL-8 (Ref. Reference Miyoshi70) and of monocyte chemotactic protein-1 (MCP-1) production, respectively (Ref. Reference Lee, Lee and Kim71). Moreover, they can increase the expression of leucocyte adhesion molecule expression (such as platelet-endothelial cell adhesion molecules-1, PECAM-1) and promote leucocyte endothelial adhesion (Ref. Reference Rattan72).
Also, ROS can activate mast cells, which on their turn produce ROS, mostly via NOX2, with consequent redox-sensitive calcium channels activation, increase in cytoplasmic calcium concentrations required for the induction of mast cell degranulation (Ref. Reference Chelombitko73) and leucocyte recruitment and activation (Ref. Reference Krystel-Whittemore, Dileepan and Wood74). The leucocyte-ROS axis is particularly relevant in the process of thrombo-inflammation, which sustains thrombotic events in various immune-mediated conditions such as thrombosis in Behçet's syndrome (Ref. Reference Becatti4).
In Behçet's syndrome, ROS have been shown to stimulate neutrophils to release extracellular traps (NETs) (Ref. Reference Bettiol7). NETs are structures composed of cell-free DNA, histones, microbicidal proteins and proteases, that are extruded by dead neutrophils, mostly by low-density granulocytes (LDGs), following infective or inflammatory stimuli (Ref. Reference Brinkmann75). NETs can directly induce thrombogenesis (Ref. Reference Folco76), by activating the intrinsic and extrinsic coagulation pathways, and by enhancing thrombin production in plasma, probably via histone/polyphosphate triggering (Ref. Reference Folco76). Concomitantly, NETs can stimulate neutrophils to further produce ROS, in a self-sustaining process.
Also, in Behçet's syndrome, leucocyte ROS levels have been correlated with a peculiar profile of circulating miRNAs (i.e. small non-coding RNAs that act as post-transcriptional regulators of gene expression) affecting pathways related to cell-matrix interaction, oxidative stress and blood coagulation (Refs Reference Bagni77, Reference Emmi78), suggesting a contribution of epigenetic mechanisms in ROS-induced thrombo-inflammation.
Connecting the dots: the erythrocyte-ROS axis in thrombosis
As described in the previous paragraphs, erythrocyte can contribute to thrombogenesis via different mechanisms and growing studies suggest a key role of oxidative stress in linking erythrocytes to thrombosis (Supplementary Table 1).
Erythrocytes have a plethora of enzymatic (e.g. superoxide dismutase, catalase, glutathione peroxidase and peroxiredoxin-2 (PRDX-2)) and non-enzymatic antioxidant defences. Among the latter, reduced glutathione (GSH) is a ubiquitous intracellular antioxidant which inhibits free radical formation and more generally acts as a redox buffer, detoxifier and chemokine scavenger. Erythrocytes can export GSH at a constant rate of ~ 21 nmol/h/ml erythrocytes, contributing to the extracellular GSH reservoir (Ref. Reference Giustarini79). GSH is synthesised de novo from cysteine, glycine and glutamate by the enzymes, γ-L-glutamate L-cysteine ligase and glutathione synthetase (Ref. Reference Giustarini79). Reduced GSH concentration has been reported in various conditions characterised by an increased cardiovascular risk, such as diabetes mellitus (Ref. Reference Murakami80), hypertension (Ref. Reference Pouvreau81), haemodialysis and peritoneal dialysis (Ref. Reference Ross, Koo and Moberly82), and is considered as an indicator of an impaired oxidative stress.
Within erythrocytes, oxidative stress can be sustained by ROS released from neutrophils and macrophages into the plasma and taken up by erythrocytes, particularly in microcirculation, where the erythrocytes are in close contact with the vasculature (Ref. Reference Mohanty, Nagababu and Rifkind83). Also, erythrocyte also contains NADPH oxidases, which can generate endogenous ROS (Ref. Reference George84). Endogenous and exogenous ROS induce oxidation of iron contained in haemoglobin, from Fe2+ containing haemoglobin to Fe3+-containing methaemoglobin.
Fe3+ induces iron-dependent free radical generation (Fenton reaction) which causes lipid peroxidation, haemolysis and endothelial perturbation. This triggers a haemolysis/oxidative cycle, which promotes vascular injury, thrombus formation and atherothrombotic events (Ref. Reference Woollard85) as observed in severe haemolytic syndromes (Ref. Reference Woollard85).
The oxidised Fe3+ methaemoglobin can be converted back into the reduced form by a cytochrome b5 reductase. However, if the reducing equivalents for this enzyme are scarce, haeme is further degraded to quaternary compounds with consequent ROS formation (Ref. Reference Mahdi86).
ROS damage erythrocyte membrane (Ref. Reference Barodka87), reduce cell deformability and induce cell lysis, by triggering a molecular signalling cascade with the activation of Ca2+ permeable cation channel (Ref. Reference Foller88). The influx of Ca2+ activates Ca2+-sensitive K+ channels, leading to phosphatidylserine exposure on the erythrocyte membrane (Ref. Reference Foller88). This provides an active surface for prothrombin activation: it has been postulated that even a small fraction of erythrocytes exposing phosphatidylserine can lead to thrombin generation, accounting for up to 30–40% of the thrombin-generating potential of whole blood (Ref. Reference Whelihan89). Notably, in a mouse model of sickle cell disease, reducing erythrocyte ROS production with manganese porphyrins, which suppress erythrocyte NOX activity (Ref. Reference MacKinney90) was found to result in a reduced phosphatidylserine exposure and improved eryptosis (Ref. Reference Thamilarasan91).
Beside directly stimulating thrombin generation, phosphatidylserine exposure on the erythrocyte membrane stimulates the release of microvesicles (Ref. Reference Morel92) with a high thrombotic potential (Ref. Reference Ataga, Cappellini and Rachmilewitz29), as previously described and considered a promising target for the treatment of thrombotic disorders (Ref. Reference Van Der Meijden93). Oxidation-induced damage on erythrocyte membrane further induces haemolysis. Under physiological conditions, the release of free haemoglobin and haeme can be inactivated by plasma haptoglobin and hemopexin (Refs Reference Smith and McCulloh94, Reference Cooper95) leading to their phagocytosis (Ref. Reference Thomsen96). Conversely, oxidised haemoglobin has a low affinity for haptoglobin, resulting in an impaired plasma clearance and in an increased release of haeme and iron (Ref. Reference Nagy97). Free redox-active haeme translocate into endothelial cells, triggering H2O2-mediated endothelial damage and overwhelming intracellular antioxidant defences.
Moreover, extracellular haeme derived from lysed erythrocytes mediates additional pro-thrombotic mechanisms: it stimulates neutrophil recruitment and NETosis (Ref. Reference Chen98), as observed in sicke cell disease (Ref. Reference Chen98) and promotes NLRP3 inflammasome activation and cytokine and lipid mediator production in macrophages (Ref. Reference Dutra99) which have been shown to potentiate venous thrombosis (Ref. Reference Gupta100). Specifically, free haemoglobin and haeme can stimulate the nuclear factor κB (NF-κB) under the control of a Toll-like receptor (TLR)-signalling pathway (Refs Reference Belcher101, Reference Ogasawara102) leading to the activation of hypoxia-inducible factor (HIF)-1α and HIF-2α (Ref. Reference Lisk103) which further induce inflammation, vasoconstriction and increase endothelial permeability (Ref. Reference Lisk103).
Furthermore, free haemoglobin can upregulate the expression of functional tissue factors in macrophages and desensitises tissue factor to the effects of antioxidants, such as glutathione or serum (Ref. Reference Bahl104). Also, it can scavenge NO, thereby impairing its regulatory effects on vasocostriction, endothelial adhesion molecule expression and platelet activation and aggregation, in a pro-thrombotic sense (Ref. Reference Rother105). Free haeme can induce platelet activation also by binding to glycoprotein-1b alpha (GPIbα) on platelets (Ref. Reference Singhal106), as well as through C-type lectin-like receptor-2 (CLEC-2) (Ref. Reference Bourne107).
A direct role of erythrocyte oxidative stress has been described in retinal vein occlusion, a condition characterised by vision loss resulting from hypoperfusion and hypoxia of the retina. Increased erythrocyte oxidative stress levels were found in patients with retinal vein occlusion; also, erythrocyte-derived ROS and erythrocyte lipid peroxidation were found to positively correlate with erythrocyte membrane viscosity and deformability (Ref. Reference Becatti108).
Similarly, in patients with cochlear vascular occlusion leading to sudden sensorineural hearing loss, a significant structural and functional involvement of erythrocyte membrane alterations was found, associated with enhanced levels of membrane lipid peroxidation and intracellular ROS production. Notably, in vitro experiments demonstrated that ROS display a critical role in impairing erythrocyte membrane fluidity (Ref. Reference Becatti109).
Of major note, ROS-induced erythrocyte modifications are particularly relevant during aging. An age-dependent increase in erythrocyte oxidative stress markers paralleled by an age-dependent decline in the total plasma antioxidant capacity has been reported (Refs Reference Rizvi and Maurya110–Reference Li112). In rat models, an increase in plasma membrane redox system activity, lipid peroxidation and erythrocyte malondialdehyde has been reported in senescent erythrocytes, paired by a reduced L-cysteine influx and a consequent decrease in intracellular GSH (Ref. Reference Kumar and Rizvi113).
Beside erythrocytes, also platelets exhibit a progressive impairment in redox status during aging, with a marked increase in oxidative stress, hyperactivation and apoptotic markers, although this trend is reverted in old subjects (80–100 years) (Ref. Reference Jain114). Accordingly, erythrocyte and platelet oxidative stress has been suggested as one of the major mechanisms sustaining the pathogenesis of thrombotic events during aging, with potentially relevant implications in terms of thrombotic prophylaxis and treatment (Refs Reference Wang and Zennadi115, Reference Fuentes and Palomo116). In aging rat models, rapamycin, particularly when combined with metformin, was found to be a promising age-delaying agent, able to restore altered levels of redox biomarkers in erythrocytes (Refs Reference Singh117, Reference Singh118).
Therapeutic implications
Understanding the role of the erythrocyte-oxidative stress axis in inducing thrombosis offers the possibility of setting up new prophylactic strategies for cardiovascular preventions (Table 1).
Table 1. Therapeutic implications

Ang, angiotensin; LDL, low-density lipoprotein; LPS, Lipopolysaccharide; NO, nitric oxide; NOX, NADPH oxidase; ROS, reactive oxygen specifies; TLR, toll-like receptor.
Pharmacological therapies
Angiotensin-converting enzyme (ACE) inhibitors are among guideline-recommended first-line therapies in patients with hypertension to reduce the related risk of atherosclerotic disease and cardiovascular events. Growing evidence suggests that these agents exert cardiovascular effects that go beyond blood pressure reduction (Refs Reference Borghi119–Reference Borghi and Omboni121).
ACE inhibitors block the conversion of Ang I to Ang II, which induces endothelial dysfunction via promoting leucocytes recruitment and ROS production, with consequent enhanced LDL oxidation and NO degradation (Ref. Reference Borghi and Levy120).
Similarly, statins are lipid-lowering agents recommended in patients with hypercholesterolemia. In vitro and in vivo studies showed that statins can modulate the atherosclerotic process, through mechanisms additive to blood cholesterol reduction, that include anti-inflammatory and antioxidant actions (Refs Reference Pedersen122–Reference Oesterle, Laufs and Liao124).
Indeed, statins can interfere with leucocyte migration, proliferation and leucocyte/endothelial interactions (Ref. Reference Takemoto and Liao125). Also, statins (particularly atorvastatin) can block Rho and Rac activation, thus reducing endothelial activation, while increasing the expression of eNOS and the endothelial production of the vasorelaxant NO. As the activation of Rho family members is a major source of ROS production in the vasculature, statins can counteract oxidative stress mechanisms which contribute to an increased risk of thrombotic events (Refs Reference Dechend126–Reference Pignatelli128). Statins were found to contribute also to the resolution of venous thrombi, although the mechanism has not fully clarified (Ref. Reference Kessinger129).
Similar effects have been reported for antiplatelets (aspirin), anticoagulants (Xa inhibitor, rivaroxaban), thioredoxin inhibitors (Refs Reference Basili130–Reference Cammisotto132) and the oral anti-diabetic drug alogliptin (Ref. Reference Yisireyili133).
Vitamins
Among non-pharmacological agents, vitamins, particularly A, C and E, are known to reduce the risk of atherosclerosis and related complications.
In vitro and in vivo studies report that vitamin E exerts pleiotropic antithrombotic effects by reducing the expression and release of endothelial adhesion molecules, preventing leucocyte/endothelial cell interactions. Also, it counteracts cholesterol-induced atherosclerotic lesions progression by inhibiting smooth muscle cells proliferation and it can inhibit the formation of platelet-leucocytes aggregates and the activation of the clotting system (Refs Reference Crutchley and Que134–Reference Myung136). Notably, natural vitamin E consists of a family of eight compounds, four tocopherols and four tocotrienols. All tocopherols and tocotrienols are potent antioxidants with lipoperoxyl radical-scavenging actions able to counteract oxidative stress. In patients with type 2 diabetes mellitus and the haptoglobin 2-2 genotype presenting increased oxidative stress levels, vitamin E was found to reduce the risk of cardiovascular events (Ref. Reference Milman137); however, the cardioprotective effect of vitamin E supplementation in the general population as well as in other high-risk setting was disappointing (Refs Reference Stephens138–140).
Vitamin C was found to enhance endothelium-dependent vasodilation, both in normotensive and hypertensive subjects (Ref. Reference Taddei141), thanks to its effects on NO availability (Ref. Reference Bendich142). However, contrasting findings were reported on the benefits of vitamin C supplementation for cardiovascular prevention.
In another study, it was shown that vitamin C (0.5–5 mM) increased the procoagulant activity of freshly isolated human erythrocytes, particularly those from cancer patients, via the externalisation of phosphatidylserine and the formation of phosphatidylserine -bearing microvesicles. Also, in rat models, the intravenous injection of vitamin C (0.5–1.0 g/kg) significantly increased thrombosis. (Ref. Reference Kim143).
Dietary regimens
Diets, especially high-fat or high-carbohydrate diets, can increase oxidative stress by elevating the levels of protein carbonylation and lipid peroxidation while impairing antioxidant defences (Ref. Reference Apel and Hirt144). In obese patients, insulin resistance greatly increases oxidative stress, thus contributing to the increased risk of hypertension, dyslipidaemia, type 2 diabetes, atherosclerosis and non-alcoholic fatty liver disease associated with this condition (Ref. Reference Jiang, Liu and Li145).
The cardioprotective role of specific nutritional regimens has been widely investigated. In a prospective cohort study on more than seven hundred patients with atrial fibrillation, the cardioprotective role of Mediterranean diet was investigated. Results indicated that adherence to Mediterranean diet could be associated with a reduction of cardiovascular events, through an antioxidant effect, as shown by a downregulation of sNOX2-dp (soluble NOX2-derived peptide) and F2-isoprostanes during this dietary regimen (Ref. Reference Pastori146).
Moreover, xanthohumol contained in beer, was found to prevent arterial and venous thrombosis in mice, by decreasing ROS accumulation and inhibiting platelet activation (Ref. Reference Xin147). Similar effects were suggested for antioxidants contained in red wine (Refs Reference Wollny148, Reference Gresele149) and olive oil (Ref. Reference Carnevale150). Also, nattokinase, a serine protease from the traditional Japanese food Natto, displays anti-inflammatory and anti-oxidative stress activities by inhibiting LPS-mediated TLR-4 and NOX2 signalling in macrophages, thereby exerting a protective effect against inflammation-induced thrombosis (Ref. Reference Wu151).
More recently, tailored nutritional interventions have been investigated to counteract thrombo-inflammation in peculiar chronic immune-mediated diseases, such as Behçet syndrome. Behçet syndrome displays a peculiar gut microbiota fingerprint, with an impaired production of short-chain fatty acids, especially butyrate (Ref. Reference Consolandi152), which can exert protective effects against cardiovascular diseases (Ref. Reference Brown and Hazen153). Butyrate-enriched dietary interventions were recently found to reduce ROS production and ROS-mediated fibrinogen structural and functional alternations in these patients (Ref. Reference Emmi154) paving the way for new cardioprotective therapies in this condition.
Concluding remarks
The erythrocyte/ROS axis is involved in the regulation of various processes that promote thrombosis. An impaired redox state induces erythrocyte membrane damage, leading to membrane fluidity alterations and decreased deformability. These changes impair erythrocyte function in the haemostatic process, promoting thrombosis via haemolysis, phosphatidylserine exposure, microvescicle release, induction of platelet activation and aggregation and vascular injury. Oxidised erythrocytes not only promote thrombus formation but also contribute to increase its size and to reduce its permeability and susceptibility to lysis and studies have suggested that the role of erythrocytes is particular once the thrombogenetic process has started and erythrocytes are entrapped within the growing thrombus (Ref. Reference Gutmann155). However, the wide plethora of mechanisms by which oxidised erythrocytes contribute to thrombosis is not completely elucidated.
Deeping current knowledge on the mechanisms linking ROS and erythrocytes and their crosstalk with leucocytes, platelets and pro- and anti-coagulant molecules will pave the way to new therapeutic strategies for reducing thrombosis risk, particularly in conditions characterised by a sustained thrombo-inflammatory milieu.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/erm.2022.25
Acknowledgements
All people who contributed to this work are listed as co-authors.
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflict of interest
All authors declare none.
Ethical standards
Not applicable.





 Open access
Open access