


Cannabinoids are highly lipophilic molecules that have been shown to alter the functional activities of immune cells in vitro and in vivo. The term ‘exogenous cannabinoids’ describes cannabinoids that are extracted from the marijuana plant Cannabis sativa or are synthesised in the laboratory. The marijuana cannabinoids delta-9-tetrahydrocannabinol (Δ9-THC), cannabinol (CBN) and cannabidiol (CBD) have been the most studied exogenous cannabinoids. Δ9-THC acts as a partial receptor agonist: on binding to cannabinoid receptors it elicits a response that never reaches the maximum efficacy. This compound is the major psychoactive and immunomodulatory component in marijuana and primarily exerts immunosuppressive effects on immune cells at peripheral sites and within the central nervous system (CNS). Synthetic exogenous cannabinoids that have been used widely in research include CP55940, WIN55212-2, SR141716A and SR144528 (Fig. 1). CP55940 and WIN55212-2 are full receptor agonists: on binding to cannabinoid receptors they elicit responses that garner maximum efficacy.
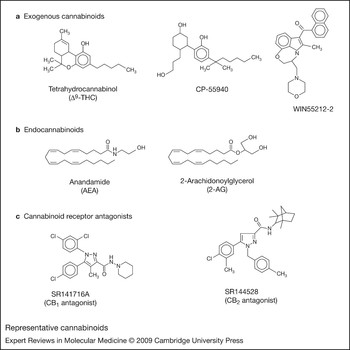
Figure 1. Representative cannabinoids. (a) Exogenous cannabinoids. Δ9-THC is a partial agonist for CB1 and CB2; CP55940 and WIN55212-2 are full agonists for CB1 and CB2. (b) Endogenous cannabinoids. AEA and 2-AG show agonist behaviour at CB1 and CB2. (c) Cannabinoid receptor antagonists. SR141716A is an antagonist for CB1; SR144528 is an antagonist for CB2. Abbreviations: AEA, arachidonoylethanolamide (anadamide); CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; CP55940, (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol; SR141716A, 5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide hydrochloride; SR144528, (1S-endo)-5-(4-Chloro-3-methylphenyl)-1-((4-methylphenyl)methyl)-N-(1,3,3-trimethylbicyclo(2.2.1)hept-2-yl)-1H-pyrazole-3-carboxamide; Δ9-THC, delta-9-tetrahydrocannabinol; WIN55212-2, (R)-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone.
Endocannabinoids (‘endogenous cannabinoids’) constitute a second group of cannabinoids; these are found natively in vertebrate systems and are produced by a variety of cell types, including endothelial cells (Ref. Reference Gauthier1), adipocytes (Ref. Reference Gonthier2), glial cells (Ref. Reference Walter3), macrophages (Ref. Reference Di Marzo4) and Purkinje cells (Ref. Reference Maejima5). These molecules are constituent elements of the ‘endocannabinoid system’, which also encompasses mediators responsible for their synthesis, metabolism and catabolism, as well as the cannabinoid receptors that serve as their molecular targets. Endocannabinoids are derivatives of integral components of cellular membranes and act as hydrophobic lipid messengers. As a result of their hydrophobicity, these molecules are not able to translocate in aqueous environments and, upon release, activate cannabinoid receptors locally or on nearby cells. Within the CNS, these bioactive lipids act as synaptic modulators (retrograde messengers); however, unlike other synaptic messengers such as the neurotransmitters acetylcholine and dopamine, endocannabinoids are not presynthesised and stored in vesicles but are produced ‘on demand’.
The first endocannabinoid to be identified was arachidonoylethanolamide (AEA; anandamide), which was isolated from porcine brain (Ref. Reference Devane6). AEA is the amide component of arachidonic acid and ethanolamine. The second endocannabinoid to be identified was 2-arachidonoylglycerol (2-AG), which was isolated from canine gut (Ref. Reference Mechoulam7). 2-AG is an ester derivative of arachidonic acid and glycerol, and is synthesised from the hydrolysis of 1,2-diacylglycerol (DAG) by a DAG lipase. In the brain, 2-AG is more bioactive and abundant than AEA. Both AEA and 2-AG are transported across the cell membrane before being degraded by fatty acid amide hydrolase (FAAH), although 2-AG can also be degraded by monoacylglycerol lipase (MGL), a serine hydrolase (Refs Reference Dinh8, Reference Dinh9, Reference Saario10). Several reports suggest that transport across cellular membranes is facilitated by carrier proteins, which have yet to be fully identified (Refs Reference Di Marzo11, Reference Beltramo12, Reference Rakhshan13, Reference Day14). In addition, while cannabinoid receptors serve as the molecular targets of AEA and 2-AG, it has been reported that these two compounds also can exert their effects in a receptor-independent fashion (Refs Reference Gardner15, Reference Hinz16, Reference Oz17, Reference De Morrow18).
Cannabinoid receptors
CB1
The initial evidence for the existence of a cannabinoid receptor came from pharmacological studies. Treatment of neuroblastoma cells with Δ9-THC, or with the synthetic compounds levonantradol and desacetyllevonantradol, resulted in inhibition of plasma membrane activity of adenylate cyclase, the enzyme that catalyses the conversion of ATP to cyclic AMP (cAMP) and pyrophosphate (Refs Reference Howlett and Fleming19, Reference Howlett20). However, in contrast to levonantradol, dextronantradol was shown to have no effect on this activity, indicating that the inhibition was stereoselective – a requisite condition for a receptor-mediated action. Additional studies demonstrated that the putative cannabinoid receptor was coupled to an inhibitory guanine-nucleotide-binding complex (Gi/o), because treatment with pertussis toxin reversed the inhibitory effect on adenylate cyclase (Ref. Reference Howlett, Qualy and Khachatrian21). Radioligand binding assays and in situ mRNA hybridisation demonstrated that the receptor was distributed throughout the brain and was localised predominantly to the cerebellum, cerebral cortex, hippocampus, basal ganglia and spinal cord (Refs Reference Devane6, Reference Matsuda22, Reference Herkenham23, Reference Westlake24). Subsequently, the gene encoding the receptor (CNR1) was isolated and cloned from a rat brain cDNA library (Ref. Reference Matsuda22), revealing the sequence for a 473 amino acid long, seven-transmembrane G-protein-coupled protein with an extracellular, glycosylated N-terminus and an intracellular C-terminus (Fig. 2a). This receptor was referred to initially as the ‘neuronal’ or ‘central’ cannabinoid receptor and has since been designated cannabinoid receptor 1 (CB1).

Figure 2. Human cannabinoid receptor amino acid sequences. (a) Amino acid sequence of the full-length human cannabinoid receptor 1 (CB1). The putative asparagine-linked glycosylation sites are shown as Ψ. The seven transmembrane domains are highlighted and noted TM1 to TM7. The Genebank accession numbers are NM_016083, NM_001840 and NP_057167.2. (b) Amino acid sequence of the full-length human cannabinoid receptor 2 (CB2). The putative asparagine-linked glycosylation site is shown as Ψ. The seven transmembrane domains are highlighted and noted TM1 to TM7. The Genebank accession numbers are NM_001841 and NP_001832.
CB1 inhibits the phosphorylation of A-type potassium channels through the coupled inhibitory G protein (Gi/o), as explained in further detail below in the cannabinoid receptor signalling section. Inhibition of the phosphorylation of A-type potassium channels results in continuous potassium currents that may prevent neurotransmission (Refs Reference Howlett and Mukhopadhyay25, Reference Demuth and Molleman26). N-type calcium channels also are inhibited by CB1 through direct interaction with the inhibitory G protein (Gi/o). CB1-mediated restriction of neurotransmission via potassium and calcium channels accounts for cognitive impairment and sedative-like effects experienced by marijuana users (Refs Reference Demuth and Molleman26, Reference Gifford27).
CB2
Following the identification of CB1, a ‘peripheral’ or ‘non-neuronal’ cannabinoid receptor was cloned from a human promyelocytic cell line (HL60) cDNA library, and was designated cannabinoid receptor 2 (CB2) (Ref. Reference Munro, Thomas and Abu-Shaar28). The gene for this receptor (CNR2) was shown to encode a 360 amino acid long, seven-transmembrane G-protein-coupled receptor (Fig. 2b) with, like CB1, an extracellular, glycosylated N-terminus and an intracellular C-terminus. Unlike CB1, there is a considerable level of sequence variation for CB2 among human, mouse and rat species, particularly when comparing rat and human sequences. There is 81% amino acid identity between rat and human CB2, as compared with 93% amino acid identity between rat and mouse CB2 (Ref. Reference Griffin29). The C-terminus in particular differs in rat as a result of intronic DNA (Ref. Reference Brown30). Of note, it is the C-terminus of CB2 that plays a critical role in regulating receptor desensitisation and internalisation (Ref. Reference Bouaboula31); thus, sequence variation within this region should be taken into consideration when investigating physiological, pharmacological and immunological responses of CB2 in diverse species.
Another distinctive feature of CB2 in comparison with CB1 is that its distribution is predominantly in cells and tissues of the immune system, including the thymus, tonsils, B cells, T cells, macrophages, monocytes, natural killer (NK) cells and polymorphonuclear cells. B cells express the highest amounts of CB2, followed by NK cells, macrophages and T cells, in that order (Refs Reference Galiegue32, Reference Schatz33). Recent studies have demonstrated that CB2 is also expressed within the CNS during various states of inflammation (Refs Reference Nunez34, Reference Ramirez35, Reference Cabral and Marciano-Cabral36, Reference Fernandez-Ruiz37), as discussed in detail later in the article. This expression of CB2 has been localised primarily to microglia, the resident macrophages of the CNS. CB2 expression is detected in these cells upon activation by various insults and stimuli, but measurable levels of CB2 expression cannot be detected in resident, unstimulated microglia. In addition, during neuroinflammation, infiltrating immunocytes from peripheral non-neuronal sites that enter the brain as a result of breakdown of the blood–brain barrier contribute to the overall expression of CB2. Table 1 lists select references for reports of the distribution of CB1 and CB2 in various immune tissues and cell types.
Table 1. Distribution of cannabinoid receptors in the immune system

aCannabinoid receptor type not specified.
Other receptors
There is accumulating evidence that additional cannabinoid receptors exist, primarily from studies in which CB1 knockout or CB1–CB2 double-knockout mice have been used to investigate the pharmacology and pharmacokinetics of Δ9-THC, AEA and cannabinoid analogues. Recently, it has been suggested that the G-protein-coupled receptor GPR55, first cloned and identified in silico from an expressed sequence tags (ESTs) database (Refs Reference Sawzdargo38, Reference Baker39, Reference Pertwee40), may be a novel cannabinoid receptor. Similar to CB1 and CB2, GPR55 has seven conserved transmembrane sequences and is activated by plantonic and synthetic exogenous cannabinoids such as Δ9-THC, CBD, abnormal CBD, HU-210 and CP55940, and by the endogenous cannabinoids AEA, 2-AG and noladin ether (Ref. Reference Ryberg41). Unlike CB1 and CB2, GPR55 is not activated by the synthetic agonist WIN55212-2, but is coupled to a Gα12/13 protein instead of a Gi/o protein (Ref. Reference Ryberg41) and has been shown to increase intracellular calcium levels upon activation (Ref. Reference Lauckner42). GPR55 expression has been identified in a variety of tissues including spleen, gastrointestinal tissues and brain (Ref. Reference Baker39). However, the physiological and pharmacological functional relevance of GPR55 has yet to be elucidated.
Another receptor reported to be a candidate cannabinoid receptor is the transient receptor potential vanilloid 1 (TRVP1) receptor, a ligand-gated cation channel and a member of the transient receptor potential channel family (Ref. Reference Caterina43). TRVP1 receptors are inherently activated by naturally occurring compounds such as capsaicin, vanilloids and resiniferatoxin (Ref. Reference Szallasi44). Its implied role as a cannabinoid receptor is based on the ability of the endogenous cannabinoid AEA, shown to be structurally similar to capsaicin, to bind to and activate this receptor (Refs Reference Melck45, Reference Zygmunt46, Reference Smart47, Reference Ross48). Nevertheless, despite the various speculative reports of additional cannabinoid receptor subtypes, a novel cannabinoid receptor that meets rigid criteria pharmacologically and functionally has yet to be identified (Refs Reference Jarai49, Reference Di Marzo50, Reference Breivogel51, Reference Wiley and Martin52).
Cannabinoid receptor signalling
Both CB1 and CB2 are involved in activating signalling cascades that include adenylate cyclase and cAMP, mitogen-activated protein kinase (MAPK), and modulation of levels of intracellular calcium (Refs Reference Demuth and Molleman26, Reference Bayewitch53, Reference Slipetz54, Reference Bouaboula55, Reference Zoratti56). Upon cannabinoid receptor interaction with its cognate ligand, the receptor-coupled G protein exchanges the inactive guanine nucleotide GDP for its active form GTP, and the heterotrimeric G protein dissociates into α and βγ subunits (Fig. 3a). The βγ subunits are thought to take part in signalling pathways distinct from those of the α subunit, such as the regulation of phospholipase C (PLC) isoforms and activation of the MAPK signalling network (Ref. Reference Howlett and Mukhopadhyay25) (Fig. 3b). CB2 partly exerts its effects through initiation of phospholipase C (PLC) and inositol 1,4,5-trisphosphate (IP3) signalling pathways that result in increased levels of intracellular calcium (Ref. Reference Zoratti56). The α subunit binds to, and inhibits the activity of, adenylate cyclase, thereby preventing synthesis of the second messenger cAMP and negatively affecting downstream cAMP-dependent signalling events. As a decrease in cAMP production underlies a mechanism in which CB1 prevents neurotransmitter release and maintains the homeostatic integrity of the CNS, decreased cAMP production also may represent a mode by which CB2 signalling in response to endocannabinoids maintains immunological homeostasis or, alternatively, in response to exogenous cannabinoids such as Δ9-THC superimposes a perturbing immunosuppressive effect (Ref. Reference Cabral57).
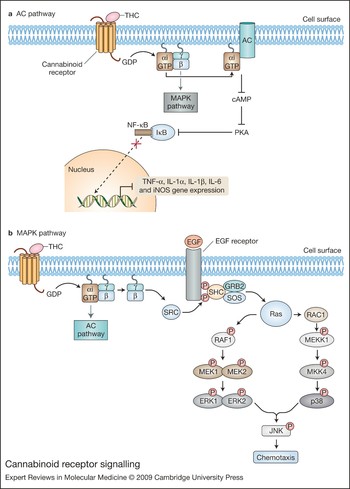
Figure 3. Cannabinoid receptor signalling. (a) The adenylate cyclase (AC) pathway. Giα signalling downregulates the pro-inflammatory immune response via adenylate cyclase. Upon cannabinoid receptor activation, the α subunit of the Gi protein interacts with adenylate cyclase to inhibit its activity. This results in a decrease of cAMP production, which leads to inactivation of protein kinase A (PKA). IκB-α remains unphosphorylated due to PKA inactivation, thus preventing activation, nuclear translocation and DNA binding of NF-κB and other transcription factors. These events ultimately lead to the downregulation of pro-inflammatory mediator gene expression. (b) The mitogen-activated protein kinase (MAPK) pathway. Following cannabinoid receptor activation, the βγ subunits of the Gi protein induce Ras-dependent MAP kinase signalling pathways, culminating in chemotaxis. The SRC kinase is activated upon interaction with Gi βγ subunits and phosphorylates tyrosine residues of the EGF receptor. The SHC–SOS–GRB2 trimer binds the EGF receptor via the SHC adaptor protein, while the guanine-nucleotide-exchange factor SOS activates Ras. Following Ras activation, two independent signalling pathways are initiated, leading to the activation of p38 (encoded by MAPK14) and ERK1 and 2 (encoded by the MAPK1 and MAPK2 genes, respectively). p38 as well as ERK1 and 2 can activate JNK, which induces chemotaxis of immune cells. Abbreviations: EGF, epidermal growth factor; ERK, extracellular-signal-regulated kinase; IL, interleukin; iNOS, inducible nitric oxide synthase; JNK, c-jun N-terminal kinase; MEK1 and MEK2, MAPK kinase encoded by MAP2K1 and MAP2K2 genes, respectively; MEKK1, MAPK kinase kinase encoded by MAP3K1 gene; MKK4, MAPK kinase 4 encoded by MAP2K4 gene; RAC1, Ras-related C3 botulinum toxin substrate 1; RAF1, v-Raf-1 murine leukaemia viral oncogene homologue; SHC, Src homology 2 domain-containing; SOS, son of sevenless; TNF, tumour necrosis factor.
Role of CB2 in immune modulation
Effect of exogenous cannabinoids on host resistance and immunity
Exogenous cannabinoids have been shown to decrease host resistance to a variety of infectious agents. Administration of Δ9-THC to mice lessened their ability to resist infection with the bacterial agent Listeria monocytogenes and herpes simplex virus 2 (Refs Reference Cabral and Staab58, Reference Morahan59). In mice and guinea pig models of genital herpes, treatment with Δ9-THC increased incidence of viral lesions and recurrence (Refs Reference Juel-Jensen60, Reference Cabral and Dove-Pettit61). In additon, cannabinoids compromised host resistance to Legionella pneumophila, Staphylococcus albus, Treponema pallidum, Friend leukaemia virus and Acanthamoeba (Refs Reference Cabral and Staab58, Reference Klein, Newton and Friedman62, Reference Arata63, Reference Arata64, Reference Marciano-Cabral65, Reference Newton, Klein and Friedman66). These observations are consistent with the notion that exogenous cannabinoids can affect the activities of immune cells.
Indeed, in vitro studies using cells of human and rodent origin have demonstrated that cannabinoids alter the functionality of a diverse array of immune cells. Δ9-THC and the synthetic cannabinoids CP55940 and HU-210 inhibited cell-contact-dependent cytolysis of tumour cells, which is mediated by macrophages and macrophage-like cells (Refs Reference Burnette-Curley and Cabral67, Reference Coffey68). Δ9-THC suppressed proliferation of B and T cells in response to cell-specific mitogens (Refs Reference Burnette-Curley and Cabral67, Reference Coffey68, Reference Klein69, Reference McCoy70), suppressed the cytolytic activity of NK cells (Ref. Reference Klein69), and inhibited cell-killing activity, proliferation and maturation of cytotoxic T lymphocytes (CTLs) (Ref. Reference Klein69). In addition, exogenous cannabinoids affected immune cell recruitment and chemotaxis to sites of infection and/or injury (Refs Reference Raborn71, Reference Sacerdote72). In murine models of granulomatous amoebic encephalitis (GAE) and atherosclerosis, macrophages and macrophage-like cells exposed to Δ9-THC displayed less migration to sites of infection (Refs Reference Ullrich73, Reference Cabral and Marciano-Cabral74, Reference Steffens75, Reference Kishimoto76). Thus, the collective data suggest that exogenous cannabinoids such as Δ9-THC inhibit the functional activities of a variety of immunocytes – an outcome that is consistent with the role of these compounds in decreased host resistance to infectious agents.
Role of CB2 in cell-mediated and humoral immunity
The recognition that select exogenous cannabinoids acted as anti-inflammatory agents and that immune cells also expressed cannabinoid receptors served as an impetus for studies aimed at defining a functional linkage between these two events. The preponderance of studies to date indicates that the cannabinoid receptor that is linked to modulation of the majority of immune functional responses is CB2.
Effects of exogenous cannbinoids
The partial agonist Δ9-THC, as well as the full cannabinoid agonists CP55940 and WIN55212-2, has been reported to enhance human tonsillar B cell growth when used at nanomolar concentrations (Ref. Reference Derocq77) by a CB2-mediated mechanism. Consistent with these results, the CB2-selective antagonist SR144528 reversed the stimulating effects of CP55940 on human tonsillar B cell activation (Ref. Reference Carayon78). By contrast, several reports have indicated that cannabinoids suppress the antibody response of humans and animals (Refs Reference Friedman, Ader, Felten and Cohen79, Reference Klein80), at least in part through inhibition of adenylate cyclase by a pertussis-toxin-sensitive G-protein-coupled mechanism (Ref. Reference Kaminski81). Furthermore, CB2 was shown to be downregulated at the mRNA and protein levels during B cell differentiation (Ref. Reference Carayon78). Together, these observations suggest that the CB2 plays a stimulatory role in B cell activation and growth, while exerting a suppressive role in B cell differentiation and antibody production.
Cannabinoids also have been reported to suppress various activities of T cells, apparently through CB2. For example, in vivo administration of Δ9-THC to mice resulted in significant inhibition of NK cytolytic activity without affecting ConA-induced splenocyte proliferation (Ref. Reference Massi82). Concomitant with this inhibition, levels of interferon γ (IFN-γ) were reduced significantly, and CB1 and CB2 antagonists reversed this effect. In view of these observations, it was suggested that both CB1 and CB2 were involved in the network that mediates NK cytolytic activity.
These and other studies have indicated that cannabinoids not only exert direct effects on immune cells but also have effects beyond the initial target cell in that they alter the expression of chemokines and cytokines– key mediators in a complex network of cross-signalling among immune cells for homeostatic balance between pro-inflammatory (T helper 1; Th1) and anti-inflammatory (T helper 2; Th2) activities. For example, Δ9-THC treatment of BALB/c mice resulted in a decrease in levels of IFN-γ, interleukin (IL)-12 and IL-12 receptor β2 in response to Legionella pneumophila infection (Ref. Reference Klein83). The use of cannabinoid receptor antagonists linked both CB1 and CB2 to decreased levels of IFN-γ and IL-12 and the suppression of Th1 immunity to Legionella. However, studies employing a tumour model implicated CB2 as the receptor responsible for Δ9-THC-mediated inhibition of immunity by a cytokine-dependent pathway (Ref. Reference Zhu84). In these studies, using a weakly immunogenic mouse lung cancer model, Δ9-THC decreased tumour immunogenicity. At both the tumour site and in the spleens of Δ9-THC-treated mice, levels of the immune-inhibitory Th2 cytokines IL-10 and transforming growth factor (TGF) were augmented, whereas levels of the immune-stimulatory Th1 cytokine IFN-γ were downregulated. In vivo administration of the CB2 antagonist SR144528 blocked the effects of Δ9-THC, suggesting that Δ9-THC promoted tumour growth by inhibiting antitumour immunity through a CB2-mediated, cytokine-dependent pathway. Collectively, the results from several studies suggest that exogenous cannabinoids elicit a shift from a Th1 pro-inflammatory cytokine expression profile to a Th2 anti-inflammatory profile, and that CB2 may be linked to this effect.
Effects of endogenous cannabinoids
Endocannabinoids also have been reported to affect immune function, mostly though CB2. The effects of AEA and palmitoylethanolamide, as well as Δ9-THC, on the production of tumour necrosis factor (TNF)-α, IL-4, IL-6, IL-8, IL-10, IFN-γ, and p55 and p75 TNF-α soluble receptors have been examined (Ref. Reference Berdyshev85). AEA diminished production of IL-6 and IL-8 at low nanomolar concentrations and inhibited that of TNF-α, IFN-γ, IL-4 and p75 TNF-α soluble receptor at micromolar concentrations. Palmitoylethanolamide, at concentrations similar to those of AEA, inhibited the synthesis of IL-4, IL-6 and IL-8, and the production of p75 TNF-α soluble receptor. However, palmitoylethanolamide did not affect TNF-α and IFN-γ production. Neither AEA nor palmitoylethanolamide had an effect on IL-10 synthesis. Based on these observations, it was suggested that various endogenous fatty acid ethanolamides participated in the regulation of the immune response, and that the inhibitory properties of AEA and palmitoylethanolamide were due to activation of CB2 since it has been shown that palmitoylethanolamide has a high affinity for the CB2 receptor and does not bind the CB1 receptor (Ref. Reference Facci86). AEA also has been shown to exert an inhibitory effect on chemokine-elicited lymphocyte migration (Ref. Reference Joseph87). The inhibition of stromal derived factor 1 (SDF-1)-induced migration of CD8+ T cells was found to be mediated through the CB2. However, there also are reports that AEA can exert potentiating effects. AEA acted as a synergistic growth factor for primary murine marrow cells and haematopoietic growth factor (HGF)-dependent cell lines (Ref. Reference Valk88). AEA also augmented production of IL-6 by astrocytes infected with Theiler's murine encephalomyelitis virus (Ref. Reference Molina-Holgado89). However, in these studies the enhancing effect of AEA was shown to be blocked by the CB1 antagonist SR141716A, suggesting involvement of CB1 rather than CB2 in the elevation of levels of this pleiotropic cytokine.
By contrast to AEA, 2-AG has been associated primarily with augmentation of immune responses. 2-AG stimulated the release of nitric oxide (NO) from human immune and vascular tissues and from invertebrate immunocytes via CB1 (Ref. Reference Stefano90), and haematopoietic cells expressing CB2 migrated in response to 2-AG (Ref. Reference Jorda91). Distinct profiles for CB2 expression in lymphoid tissues have been reported to be dependent on the state of receptor activation, and it has been proposed that cell migration constitutes a function of CB2 upon stimulation with 2-AG (Ref. Reference Rayman92). Furthermore, 2-AG induced the migration of human peripheral blood monocytes and promyelocytic leukaemia HL60 cells that have been differentiated into macrophage-like cells (Ref. Reference Kishimoto76), apparently through a CB2-dependent mechanism. Subsequent studies showed that 2-AG apparently accelerated production of chemokines by the HL-60 cells (Ref. Reference Kishimoto93). In addition, rat microglia have been reported to synthesise 2-AG in vitro, an event that has been linked to increased proliferation through a CB2-dependent mechanism (Ref. Reference Carrier94).
Role of CB2 in macrophages and macrophage-like cells
A major target of the action of exogenous and endogenous cannabinoids appears to be cells of macrophage lineage. Cannabinoids suppress macrophage functions such as phagocytosis, bactericidal activity, and spreading (Refs Reference Friedman, Ader, Felten and Cohen79, Reference Klein, Friedman and Watson95), interfere with macrophage cell-contact-dependent lysis of tumour cells, herpesvirus-infected cells and amoebae, and deplete macrophage-elicited soluble tumouricidal activity (Refs Reference Burnette-Curley and Cabral67, Reference Burnette-Curley96). These observations are consistent with reports that Δ9-THC inhibits the synthesis of proteins associated with primed and activated macrophages (Ref. Reference Cabral and Mishkin97), alters cytokine secretion by activated macrophages (Refs Reference Watzl, Friedman, Specter and Klein98, Reference Nakano99), and inhibits cytokine gene expression by microglia (Ref. Reference Puffenbarger100). Cannabinoids also have been found to affect the production of NO by macrophages and macrophage-like cells (Refs Reference Coffey68, Reference Jeon101). Although it is now evident that cannabinoids exert a variety of effects on the activities of macrophage and macrophage-like cells, a picture is emerging as to the role of CB2 in these processes and the state of cell activation under which it is functionally relevant.
Differential expression of CB2
Macrophages and macrophage-like cells such as microglia undergo a maturation, differentiation and activation process characterised by differential gene expression and the acquisition of correlative distinctive functional capabilities (Refs Reference Adams and Hamilton102, Reference Hamilton and Adams103, Reference Hamilton104). These cells can be driven sequentially in response to multiple signals from ‘resting’ to ‘responsive’, ‘responsive’ to ‘primed’, and ‘primed’ to ‘fully activated’ states, a process that mimics events in vivo. Using in vitro models, it has been shown that levels of CB2 receptor mRNA and protein are modulated differentially in relation to cell activation state (Ref. Reference Carlisle105). The CB2 is not detected in resting cells, is present at high levels in responsive and primed cells, and is identified at greatly diminished levels in fully activated cells (Fig. 4). These observations suggest that the CB2 is expressed ‘on demand’ and that the modulation of CB2 levels is a feature common to cells of macrophage lineage as they participate in the inflammatory response and undergo differential gene expression and acquisition of distinctive functional properties. Furthermore, the relatively high levels of CB2 recorded for macrophages when in responsive and primed states suggest that these cells, and possibly other immune cell types, exhibit a functionally relevant ‘window’ of CB2 expression during which they are most susceptible to cannabinoid-mediated action. Furthermore, since CB1 is expressed at low and constitutive levels in microglia as compared with CB2, the kinetics of expression of the two receptors as linked to immune functional activities may be distinct.
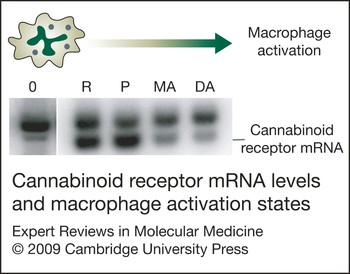
Figure 4. Cannabinoid receptor mRNA levels and macrophage activation state. Levels of cannabinoid receptor 2 (CB2) mRNA are modulated differentially in relation to macrophage activation state, as reported in Ref. Reference Carlisle105. CB2 is detected at low levels in ‘resting’ cells (0), is present at high levels in ‘responsive’ (R) and ‘primed’ (P) cells, and is identified at greatly diminished levels in cells that have been subjected to multistep activation (MA) or direct activation (DA). The resting state was achieved by growing cells on an agar substratum, whereas the responsive state was obtained by growing cells on a plastic surface. Cells were treated with interferon (IFN)-γ (100 U/ml), IFN-γ (100 U/ml) plus lipopolysaccharide (LPS) (100 ng/ml), or LPS (100 ng/ml) to obtain prime, multistep activated, or directly activated states, respectively. Southern blot analysis was performed on mutagenic reverse transcriptase PCR (MRT-PCR) products of total RNA from mouse peritoneal macrophages (Ref. Reference Rayman92). The upper band of each doublet represents amplified genomic DNA used as an internal quantitative standard. The lower band of each doublet represents amplified product from mRNA.
Chemotaxis as a signature activity of ‘responsive’ macrophages
Chemotaxis and antigen presentation are signature activities of macrophages and macrophage-like cells when in responsive and primed states of activation – states that are associated with early stages of the inflammatory response. Chemotaxis describes the ability of cells to migrate towards an increasing concentration gradient of stimulating agent (Refs Reference Harris106, Reference Harris107, Reference Jin and Hereld108, Reference Kehrl109) and is distinct from chemokinesis, which represents stimulus-dependent random cellular motion (Refs Reference Becker110, Reference Keller111, Reference Lauffenburger and Horwitz112, Reference Mitchison and Cramer113). During chemotaxis, macrophage interaction with a chemoattractant results in the initiation of a rapid and directed movement that is associated with a complex array of cellular events that includes changes in ion fluxes, alterations in integrin avidity, production of superoxide anions, and secretion of lysosomal enzymes (Ref. Reference Murdoch and Finn114). Classical chemoattractants include bacterial-derived N-formyl peptides, the complement fragment peptides C5a and C3a, and lipids such as leukotriene B4 and platelet-activating factor (Refs Reference Schiffmann115, Reference Goldman and Goetzl116, Reference Hanahan117, Reference Gerard and Gerard118). Chemokines – cytokines of 8 to 17 kDa molecular mass that are selective for leukocytes in vitro and elicit accumulation of inflammatory cells in vivo – represent a second group of chemoattractants (Refs Reference Baggiolini119, Reference Baggiolini120, Reference Kim121, Reference Le122). As in the case of cannabinoid binding to cannabinoid receptors, the specific effects of chemokines on target cells are mediated by G-protein-coupled receptors (Refs Reference Murdoch and Finn114, Reference Charo and Ransohoff123). Ligation of chemokines to their cognate receptors initiates a series of signal transduction events that results in leukocyte trafficking during infection, inflammation, tissue injury and tumour development (Ref. Reference Charo and Ransohoff123).
The current data indicate that cannabinoids act through CB2 to alter macrophage migration, with exogenous cannabinoids such as Δ9-THC exerting an inhibitory effect and, conversely, endocannabinoids such as 2-AG eliciting a stimulatory effect. For example, in vivo and in vitro treatment of rat peritoneal macrophages with CP55940 resulted in decreased migration in vitro to the peptide formal-methionyl-leucine-phenylalanine (fMLP) in a mode that is linked primarily to CB2 (Ref. Reference Sacerdote72). Also, the chemotactic response of mouse macrophages to fMLP was decreased by CBD (Ref. Reference Sacerdote124), which binds weakly to CB2. CB2 was implicated in this response since the CB2-selective antagonist SR144528 prevented the decrease in migration. In contrast to events observed for Δ9-THC, 2-AG triggered migration of microglia, and CB2 was involved in this effect (Ref. Reference Walter3). Recently, a combined pharmacological and genetic approach (employing macrophages from knockout mice) demonstrated that Δ9-THC and CP55940 inhibited mouse peritoneal macrophage chemotaxis to CCL5 (RANTES) through CB2 (Ref. Reference Raborn71). The Δ9-THC and CP55940 deactivation of migratory responsiveness to the chemokine CCL5, an event that is mediated through activation of the cognate G-protein-coupled chemokine receptor CCR5, suggested that signalling through CB2 leads to ‘cross-talk’ between that receptor and CCR5. These and other studies implicate the CB2 in a network of G-protein-coupled receptor signal transduction systems, inclusive of chemokine receptors, that act co-ordinately to modulate macrophage migration.
Antigen processing as a signature activity of ‘primed’ macrophages
It has also been shown that CB2 is involved in cannabinoid-mediated inhibition of processing of antigens by macrophages. In studies examining the effect of Δ9-THC on the processing of intact lysozyme by macrophages, Δ9-THC impaired the ability of a macrophage hybridoma to function as an antigen-presenting cell based on its ability to secrete IL-2 upon stimulation of a soluble-protein-antigen-specific helper T cell hybridoma (Ref. Reference McCoy70). Δ9-THC exposure resulted in a significant reduction in the T cell response to the native form of lysozyme after pretreatment of the macrophages with nanomolar drug concentrations. However, Δ9-THC did not affect IL-2 production when the macrophages presented a synthetic peptide of the antigen to T cells, suggesting that the drug interfered with antigen processing, not peptide presentation. The cannabinoid inhibition of the T cell response to native lysozyme was stereoselective, consistent with the involvement of a cannabinoid receptor: bioactive CP55940 diminished T cell activation whereas the relatively inactive stereoisomer CP56667 did not. The macrophage hybridoma expressed mRNA for CB2 but not for CB1. Furthermore, the CB1-selective antagonist SR141716A did not reverse the suppression caused by Δ9-THC, whereas the CB2-selective antagonist SR144528 completely blocked the Δ9-THC suppression of the T cell response. Collectively, these results implicated macrophages as the target of cannabinoid inhibition of antigen processing through CB2.
CB2 and neuroinflammation
CB2 localisation in the CNS
Early studies to define the functional relevance of CB1 and CB2 suggested that CB1 was compartmentalised to the CNS while the expression of CB2 was limited to cells and tissues of the immune system. The development of phenotypically normal CB2-knockout mice (Ref. Reference Buckley125) was a major breakthrough that contributed to elucidation of the role of CB2 in immune modulation, including that in the CNS. In addition to the CB2-knockout mouse strain developed by Buckley and colleagues (Ref. Reference Buckley125), Deltagen (San Mateo, CA, USA) developed a CB2-knockout mouse strain that is commercially available through Jackson Laboratories (Bar Harbor, ME, USA) (Ref. Reference Buckley126); these CB2-knockout mice strains have mutations in the C-terminus and the N-terminus of CB2, respectively. The tissues from these mice have been employed extensively in studying CB2 function and CB2-mediated responses. Macrophage function in helper T cell activation in the knockout mouse developed by Buckley and colleagues is not sensitive to the inhibitory effects of Δ9-THC, in contrast with macrophage function in wild-type counterparts (Ref. Reference Buckley125).
In addition, CB2-knockout mice have been utilised to study the specificity of various CB2 antibodies. However, CB2 localisation within the CNS has proven to be an elusive target. While some laboratories have reported detection of CB2 in the brain (Refs Reference Buckley126, Reference Van Sickle127, Reference Gong128), other laboratories have not been able to identify this protein, raising concern about the reliability and specificity of the CB2 antibodies used. In the studies performed that identified the CB2 protein in brainstem neurons, a polyclonal antibody against the C-terminus was used to detect the receptor, and the CB2-knockout strain developed by Buckley and colleagues and wild-type mice were used as the knockout and positive controls, respectively, to confirm the specificity of the polyclonal CB2 antibody (Refs Reference Buckley126, Reference Van Sickle127). The knockout control was appropriate for those experiments since this knockout strain has a deletion in the C-terminus of the CB2 protein. In other studies, on brain of wild-type mice, an antibody that was raised against the C-terminus of the CB2 protein was used to demonstrate receptor expression (Refs Reference Buckley126, Reference Gong128, Reference Onaivi129, Reference Onaivi130). The investigators identified the CB2 protein in various brain regions using an antibody specific for the N-terminus of the CB2 protein; however, a knockout control using tissues from CB2-knockout mice was not used to confirm the specificity of this antibody (Refs Reference Buckley126, Reference Gong128). Similar potentially confounding issues have been raised for CB1 antibodies. Grimsey and colleagues demonstrated that various CB1-specific antibodies used in immunostaining and western blot analyses displayed a multitude of variability in expression profiles – an outcome that was attributed to possible conformational changes, dimerisation with other G-protein-coupled receptors, or post-translational modifications (Ref. Reference Grimsey131). It was postulated that such factors, separately or combined, could result in epitope masking or insufficient binding of antibody (Ref. Reference Grimsey131). Collectively, these studies highlight the importance of employing specific cannabinoid receptor antibodies whose specificity can be confirmed using appropriate knockout controls, particularly when investigating the complex arena of the CNS. While localisation of the CB2 protein in the CNS may have been elusive, in vitro studies have shown that in addition to expression in microglia (Ref. Reference Carlisle105), CB2 has since been identified in neurons, oligodendrocytes and other glial cells in vitro and in intact tissue (Refs Reference Nunez34, Reference Ramirez35, Reference Cabral and Marciano-Cabral36). This receptor can be induced on demand during early inflammatory events (Refs Reference Ramirez35, Reference Fernandez-Ruiz37) and is linked to attenuation of pro-inflammatory cytokine production by microglia, as discussed earlier (Refs Reference Shohami132, Reference Cabral133, Reference Molina-Holgado134, Reference Klegeris135, Reference Sheng136).
Neuroinflammatory disorders: CB2 as a therapeutic target?
Like macrophages at peripheral sites, microglia are able to phagocytose and process antigens, and upon activation produce pro-inflammatory factors including the cytokines IL-1, IL-6 and TNF-α (Refs Reference Kreutzberg137, Reference Benveniste138, Reference Baker139). Pro-inflammatory mediators released from microglia are cytotoxic and also can secondarily activate astrocytes, leading to a further induction of the expression of inflammatory factors. The resultant ‘storm’ of pro-inflammatory mediators contributes to breakdown of the blood–brain barrier and plays a critical role in promoting influx into the CNS of immunocytes from peripheral non-neuronal sites that also express CB2. Microglia are believed to play a major role in many neuropathogenic diseases and disorders such as Alzheimer disease, multiple sclerosis, amyotrophic lateral sclerosis, and HIV (human immunodeficiency virus) encephalitis.
Alzheimer disease
Alzheimer disease is the most common neurodegenerative disorder that causes senile dementia. The defining neuropathological features of the disease are the presence of extracellular neuritic amyloid plaques and intracellular neurofibrillary tangles in the brain; as neurodegeneration progresses, there is accelerated neurofibrillary tangle formation, neuroinflammation and neuronal loss.
It has been reported that cannabinoids can be neuroprotective in Alzheimer disease by inhibiting the activation of microglia (Ref. Reference Ramirez35) induced by amyloid plaques consisting of extracellular aggregates of amyloid β (Aβ) peptides (Refs Reference Dickson140, Reference Selkoe141). Recently, the CB1/CB2 agonist CP55940 and the CB2 agonist JWH-015 were found to protect and rescue peripheral blood lymphocytes from Aβ- and H2O2-induced apoptosis by two alternative mechanisms (Ref. Reference Velez-Pardo and Del Rio142). A receptor-independent pathway was implicated through the demonstration that the reactive oxygen species dihydrorhodamine was not oxidised into fluorescent rhodamine 123 as a result of cannabinoid inhibition of Aβ-generated H2O2, while a receptor-dependent pathway was implicated through the activation of the transcription factor NF-κB and downregulation of the transcription factor p53, which can initiate apoptosis. The downregulation of p53 involved phosphoinositide 3-kinase, as demonstrated through the use of the phosphoinositide 3-kinase inhibitor LY294002 (Ref. Reference Velez-Pardo and Del Rio142). Collectively, these results suggested that cannabinoids have potential as neuroprotective compounds in Alzheimer disease.
Multiple sclerosis
Multiple sclerosis is a chronic, inflammatory demyelinating disease of the human CNS that primarily affects adults (Refs Reference Raine and Wu143, Reference Racke144, Reference Calderon145). The disease is characterised by T-cell-mediated degeneration of the myelin sheath that covers axons, resulting in an inflammatory process that stimulates other immune cells to secrete pro-inflammatory mediators and antibodies, breakdown of the blood–brain barrier, activation of macrophages, and production of ‘cytotoxic’ proteins such as metalloproteinases (Refs Reference Kieseier146, Reference Anthony147).
A significantly greater density of CB2-immunoreactive microglia/macrophages has been identified in affected regions of human multiple sclerosis post-mortem spinal cord (Ref. Reference Yiangou148). However, most studies aimed at assessment of effects of cannabinoids on multiple sclerosis, and the role of CB2 in this process have involved the use of mouse models, particularly the experimental autoimmune encephalomyelitis (EAE) model, which exhibits a CD4+-T-cell-mediated autoimmune disease (Ref. Reference Racke144). Δ9-THC markedly inhibited neurodegeneration in the EAE model and reduced the associated induced elevated level of glutamate in cerebrospinal fluid (Ref. Reference Fujiwara and Egashira149). CB2 mRNA expression and protein internalisation were upregulated significantly in activated microglia of mice experiencing EAE, implicating CB2 in this disease (Ref. Reference Ehrhart150). The cannabinoid WIN55212-2 ameliorated EAE and diminished cell infiltration of the spinal cord. WIN55212-2 was found to induce encephalitogenic T cell apoptosis, partly through CB2 (Ref. Reference Sanchez151). More recently, it has been proposed that CB2 plays a protective role in EAE pathology by targeting myeloid progenitor trafficking, which may contribute to microglial activation in the CNS (Ref. Reference Palazuelos152). In Theiler's virus infection of murine CNS, another mouse model for human multiple sclerosis, improved neurological deficits, concomitant with reduced microglial activation, major histocompatibility complex class II expression and T cell infiltration, were observed following treatment of mice with the synthetic cannabinoids WIN55212-2, ACEA (a CB1-selective agonist) and JWH-015 (a CB2-selective agonist) (Refs Reference Arevalo-Martin153, Reference Croxford and Miller154). In the Theiler's model of multiple sclerosis, clinical signs and axonal damage in the spinal cord were reduced by the AMPA (amino-3-hydroxy-5-methyl-4-isoxazolepropionate) glutamatergic receptor antagonist NBQX (Ref. Reference Docagne155). The cannabinoid HU-210 ameliorated symptoms, accompanied by a reduction of axonal damage. Furthermore, the HU-210-mediated reduction in AMPA-induced excitotoxicity in vivo and in vitro was linked to CB1 and CB2.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis is characterised pathologically by progressive degeneration of cortical motor neurons (upper motor neurons) and clinically by muscle wasting, weakness and spasticity that progresses to complete paralysis (Ref. Reference Kuncl, Asbury, McKhann and McDonald156). A pathological hallmark of amyotrophic lateral sclerosis is neuroinflammmation, a process that is mediated by pro-inflammatory cytokines, prostaglandins and NO (Ref. Reference Babu157).
CBN delayed the onset of symptoms in mice suffering from experimentally induced amyotrophic lateral sclerosis without affecting survival (Ref. Reference Weydt158), and treatment of mice with WIN55212-2 after onset of symptoms delayed overall disease progression (Ref. Reference Bisland159). Also, the CB2 agonist AM-1241 prolonged survival in a G93A-SOD1 mutant transgenic mouse model of amyotrophic lateral sclerosis when administered at onset of disease symptoms (Ref. Reference Shoemaker160). mRNA and receptor binding of CB2 were selectively upregulated in spinal cords of these mice in a fashion that paralleled disease progression. Daily injections of AM-1241 initiated at onset of symptoms increased the survival interval after disease onset by 56%. Collectively, the results suggested that the CB2 agonist extended the interval for motor neuron degeneration and prolonged function in these affected mice.
HIV encephalitis
HIV encephalitis, also known as acquired immune deficiency syndrome (AIDS)–dementia complex, results in progressive memory loss, intellectual deterioration, behavioural changes, and motor deficits (Ref. Reference Spencer and Price161). The neuropathology of HIVE is characterised by neuronal loss, glial activation, presence of multinucleated giant cells, perivascular mononuclear infiltration, and in some cases vacuolar myelopathy and myelin pallor (Ref. Reference Spencer and Price161). The production of pro-inflammatory cytokines such as TNF-α by activated monocytes and microglia, and neurotoxins such as glutamate and NO, is the primary cause of brain damage associated with this disorder. In addition, HIV-specific gene products such as the transactivator Tat and the envelope glycoprotein gp120 that are released from infected monocytes and microglia contribute to neuropathology.
The simian immunodeficiency model comes closest to replicating events that are associated with HIV infection of the human CNS. Examination of brains of macaques with simian immunodeficiency virus (SIV)-induced encephalitis has led to the suggestion that the endocannabinoid system participates in the development of HIV-induced encephalitis (Ref. Reference Benito162). In this infectivity model, expression of CB2 was induced in perivascular macrophages, microglial nodules, and T cells. It was proposed that activation of CB2, expressed by perivascular macrophages, which play a critical role in viral entry into the CNS (Refs Reference Williams163, Reference Williams164), likely led to reduction of their antiviral response, thus favouring the entry of infected monocytes into the CNS (Ref. Reference Benito162). In addition, the endogenous cannabinoid-degrading enzyme FAAH was reported as overexpressed in perivascular astrocytes as well as in astrocytic processes reaching cellular infiltrates (Ref. Reference Benito162). Furthermore, activation of CB2 resulted in inhibition of the transendothelial migration of Jurkat T cells and primary human T cells by interfering with the CXCL12–CXCR4 chemokine receptor system (Ref. Reference Ghosh165). These observations suggest that activation of CB2 can alter the activation of other G-protein-coupled receptors such as CXCR4, which functions as a coreceptor for T-lymphotropic HIV. A similar observation in terms of a linkage to CB2 has been made for the chemokine receptor CCR5, which acts as the coreceptor for monotropic HIV (Ref. Reference Raborn71). Activation of CB2 with Δ9-THC, CP55940 or with the CB2-selective compound O-2137 inhibited activation of CCR5 by its native chemokine ligand CCL5. Collectively, these results indicate that CB2 as a Gi/o-protein-coupled receptor ‘crosstalks’ with several other G-protein-coupled receptors, especially chemokine receptors, to alter heterologous signal transduction pathways. Furthermore, these interactions might have implications for HIV infection, particularly for those receptors such as CXCR4 and CCR5 that act in a coreceptor capacity for HIV. Additionally, possible therapeutic implications of ‘crosstalk’ between cannabinoid receptors and other cellular receptors has come from studies suggesting that CB2, along with CB1, could play a role in linking the endocannabinoid sytem with the modulation of neural stem cell proliferation through bidirectional ‘crosstalk’ with TNF receptors (Ref. Reference Rubio-Araiz166).
Summary of cannabinoid receptors in neuropathological disorders
In summary, cannabinoid receptors appear to play an important role in neuropathological diseases. CB1 has been reported to be critical for the overall homeostatic balance and regulation of the CNS, while CB2 has been implicated as playing a functionally relevant role during neuroinflammation. Microglia, as resident macrophages in the CNS, not only play a role in host defence and tissue repair but also have been implicated as contributive to, if not causative of, a variety of inflammatory neuropathological processes. In these cells CB1 appears to be present at constitutive and relatively low levels while CB2 is expressed inducibly during the inflammatory process and at relatively high levels. Immune responses during the early phase of neuropathological processes appear to involve preponderantly CB2, and levels and functional relevance of this receptor may be amplified as disease progresses to later stages of inflammation. The recognition that immunocytes resident within the brain express CB2, during the inflammatory process suggests the existence of a temporal window during which these cells may be susceptible to therapeutic manipulation through the use of CB2-selective agonists. That is, selective targeting of the CB2 could result in dampening of untoward immune responses such as elicitation of a chemokine/cytokine ‘storm’ within the CNS that would result in breakdown of the blood–brain barrier, influx of immunocytes from peripheral non-neuronal sites, and further inflammation.
Clinical implications/applications
Cannabinoids, as ligands that signal through cannabinoid receptors, may be particularly useful as agents for therapeutic manipulation of hyperinflammatory immune responses within the CNS. These compounds are highly lipophilic and thus readily penetrate the blood–brain barrier, a challenge that is posed to a variety of agents that have therapeutic potential. Furthermore, through the application of appropriately engineered molecules, it may be possible to specifically target CB2, a condition that would obviate generation of untoward psychotropic effects that could be engendered if the CB1 were activated also.
The principal potential cellular target in the CNS for these compounds – given its involvement in early stages of the inflammatory response in generation of a cascade of inflammatory factors and its expression of CB2 – is the microglial cell. During activation, microglia, as macrophage-like cells, also upregulate an array of cell-surface receptors that may be critical in regeneration and/or degeneration of the CNS. Included among these are immunoglobulin (Ig) superfamily receptors, complement receptors, Toll-like receptors, cytokine/chemokine receptors, and opioid receptors. These cells, in addition to expressing both the CB1 and the CB2 in vitro (Refs Reference Carlisle105, Reference Waksman167), also produce the endocannabinoids 2-AG as well as AEA, although the latter is generated in lesser quantities (Ref. Reference Carrier94). Thus, microglia appear to harbour a fully constituted system of endogenous cannabinoid ligands and cognate receptors. Activation of CB2 on these cells appears to promote migration and proliferation. 2-AG induces migration of microglia and this occurs through the CB2 and abnormal-CBD-sensitive receptors, which subsequently leads to activation of the extracellular-signal-regulated kinase (ERK)1 and 2 (encoded by the MAPK1 and MAPK2 genes, respectively) signal transduction pathway (Ref. Reference Walter3). Furthermore, it has been shown that microglia express CB2 at the leading edge of lamellipodia, consistent with an involvement in cell migration. There is accumulating evidence that CB2 also is expressed in the CNS in vivo. The expression of CB2 in microglial, astrocyte and neuronal subpopulations has been identified in a variety of neurodegenerative disease models (Ref. Reference Fernandez-Ruiz37). This expression of CB2 in vivo has been attributed, in large measure, to microglia. In several neurodegenerative diseases, upregulation of microglial CB2 has been observed (Refs Reference Yiangou148, Reference Benito162, Reference Zhang168, Reference Benito169, Reference Maresz170, Reference Ashton171). In studies investigating the expression profile of FAAH and the CB2 in postmortem brain tissues from Alzheimer disease patients, it was observed that congregated microglia associated with neuritic plaques selectively overexpressed CB2 (Ref. Reference Benito172). In addition, CB2-positive microglia have been identified dispersed within active multiple sclerosis plaques and in the periphery of chronic active plaques (Ref. Reference Benito169).
The collective findings support the concept that, in addition to CB1, CB2 has a functionally relevant role in the CNS. This role appears to play out during the inflammatory process associated with a variety of neuropathies. In this context, it has been proposed that the role of the CB2 in immunity in the CNS is primarily one that is anti-inflammatory (Ref. Reference Carrier173). Since microglia exhibit phenotypic and functional properties of macrophages and inducibly express CB2 at maximal levels when in responsive and primed states, there may be a window of functional relevance for this receptor, comparable to that for macrophages at peripheral sites. That is, antigen processing and/or chemotaxis by these cells may also be susceptible to cannabinoids acting via CB2.
Indeed, studies using a mouse model of GAE, a chronic progressive human infection of the CNS that is caused by the opportunistic pathogen Acanthamoeba, revealed a paucity of cells expressing the Mac-1 (CD11b) protein marker for macrophages and macrophage-like cells at focal sites containing Acanthamoeba in the brains of infected mice treated with Δ9-THC as compared with vehicle-treated Acanthamoeba-infected controls (Ref. Reference Marciano-Cabral65). These observations indicated that microglia (and possibly macrophages introduced from peripheral sites) either did not migrate to infected areas or were selectively targeted by the Acanthamoeba and destroyed. Treatment of neonatal rat cerebral cortex microglial cultures with Δ9-THC resulted in inhibition of the migratory response to Acanthamoeba-conditioned medium, which harbours proteases and other factors released from amoebae that serve as chemotactic stimuli (Ref. Reference Cabral57). In addition, treatment with the potent CB1/CB2 agonist CP55940 resulted in a significant concentration-related decrease in microglial migration in response to conditioned medium. The highly selective CB2 ligand O-2137 exerted a profound and significant inhibition in the microglial migratory response to conditioned medium while treatment with the CB1-selective ligand ACEA had a minimal effect. Finally, treatment of microglia with the CB1 antagonist SR141716A did not block the inhibitory effect of CP55940 while treatment with the CB2-specific antagonist SR144528 resulted in a reversal of the inhibitory effect of CP55940. These collective results indicated that the cannabinoid-mediated inhibition of the conditioned-medium-stimulated microglial response to A. culbertsoni in mouse brain was linked, at least in part, to CB2. The mode by which Δ9-THC and other exogenous cannabinoids such as CP55940 signal through CB2 to inhibit the chemotactic response of microglia to Acanthamoeba remains to be defined. However, it is known that Acanthamoeba produces proteases, phospholipases and other factors (Ref. Reference Marciano-Cabral and Cabral174) that may act on phospholipids in microglial membranes, generating cleavage products (Ref. Reference Cabral175). It is postulated that bioactive lipid mediators thus generated include the endocannabinoid 2-AG that serves to drive chemotaxis by autocrine and/or paracrine activation of CB2. The exogenous cannabinoid Δ9-THC may alter this chemotactic response, as well as chemotactic responses to other stimuli, by superimposing an inhibitory effect through activation of CB2. That is, Δ9-THC could inhibit the synthesis and/or, release of 2-AG or, by virtue of its relative long half-life as compared with that of 2-AG, pre-empt this endocannabinoid from ligating to CB2.
Summary, research in progress, and outstanding research questions
There is currently a large body of data indicating that the CB2 plays a functionally relevant role during inflammation. This role is especially evident for cells of myeloid lineage, including macrophages and macrophage-like cells, as well as microglia that are resident in the CNS. These latter cells are morphologically, phenotypically, and functionally related to macrophages. The CB2 is differentially expressed by macrophages and macrophage-like cells, with highest levels detected when these cells are in responsive and primed states, suggesting the existence of a window of functional relevance during which activation of the CB2 modulates macrophage activities. Signature activities of responsive and primed macrophages are chemotaxis and antigen processing, respectively. The endocannabinoid 2-AG, elicited from macrophages and microglia during the activation process, stimulates a chemotactic response from these cells through the CB2. By contrast, exogenous cannabinoids such as Δ9-THC and CP55940 inhibit the chemotactic response as well as antigen processing of antigens, through activation of the CB2. Furthermore, exogenous cannabinoids such as Δ9-THC may superimpose an inhibitory effect on pro-chemotactic endocannabinoids.
Although in recent years major advances have been made regarding the functional relevance of the CB2, a number of outstanding research questions remains. Principal among these is definition of the mechanism through which exogenous cannabinoids such as Δ9-THC superimpose an inhibitory effect on endocannabinoid-mediated immune functional activities. In this context, are differential signal transductional pathways involved following CB2 activation by Δ9-THC versus endocannabinoids? Do exogenous cannabinoids by virtue of their relatively long half-life as compared with endocannabinoids persist in cells so as to affect receptor-mediated endocytosis and recycling of receptor–ligand complexes? In addition, what is the extent of the ability of the CB2 to ‘crosstalk’ with other G-protein-coupled receptors, especially chemokine receptors such as CXCR4 and CCR5, which also serve as coreceptors for HIV? Do the endocannabinoids AEA and 2-AG exert differential effects on immune function, thereby acting in an immune homeostatic role? That is, does AEA act in an anti-inflammatory capacity while 2-AG acts as a pro-inflammatory agent as is typical for other bioactive lipids such as select prostaglandins that exert pro-inflammatory versus anti-inflammatory activities? These are but a few of the salient questions that await resolution.
Acknowledgements and funding
Supported in part by NIDA/NIH award R01 DA005832.





