



Introduction
Tick-borne encephalitis (TBE) is a vector-borne infection which is endemic in large parts of Eurasia. The etiologic agent is a virus of the Flaviviridae family. In the European Union and European Economic Area, there are between 2000–3500 yearly cases reported, and in recent years, the TBE virus has expanded to countries where the disease has previously not occurred. Thus, the epidemiology of TBE is monitored by the European Centre for Disease Control in European Surveillance System. The Czech Republic is located in the middle of the highly endemic area of Europe and has one of the highest incidences [Reference Beaute1].
The clinical spectrum of the TBE virus infection ranges from asymptomatic infections to serious cases with CNS involvement in the form of meningitis or meningoencephalitis. Up to 40% of patients with TBE can develop serious long-term or permanent neurologic sequelae and the fatality rate can reach up to 2% [Reference Taba2, Reference Bogovic3].
There is no specific therapy for TBE available and thus vaccination remains the only effective way to prevent this serious CNS infection [Reference Heinz4]. Vaccination against TBE is generally recommended for persons living in or travelling to endemic areas by a number of international and national public health authorities [Reference Steffen5–Reference Velay7].
There are currently two inactivated vaccines available that are approved by the European Medical Agency – Encepur® containing the antigen of the Karlsruhe 23 virus strain (K23), and FSME-Immun® based on the antigen of the Neudörfel strain [Reference Demicheli, Debalini and Rivetti8].
With respect to the onset of protection and its durability, two types of vaccination schemes are recommended by the manufacturers. Use of the rapid scheme provides both a high rate of protection in combination with early protection [Reference Schosser9]. For this reason, vaccination with this scheme is especially recommended for people travelling to endemic areas. There are some key differences between the two aforementioned vaccines when using the rapid vaccination scheme. Notably, Encepur® comprises of four doses in total. In the rapid scheme, Encepur® is administered with three shots at days 0, 7 and 21 with the first booster at day 365, whereas FSME-Immun® is administered with two shots at days 0 and 14 and the third shot at 5–12 months [Reference Jelinek10]. The standard scheme – with the first shot at day 0, the second within an interval of 30–90 days, and the third within an interval of 150–360 days has a slower onset of protection but provides longer durability [Reference Jelinek10].
Immunogenicity and durability of protection after TBE vaccination was proven by numerous studies on both adult and paediatric populations [Reference Zent11–Reference Harabacz15]. Both TBE vaccines in question are generally well tolerated with a very low occurrence of serious adverse events [Reference Loew-Baselli6, Reference Buhler16].
Vaccination against TBE has been recommended by some authors for HIV-1 infected people at risk [Reference Geretti17, Reference Crum-Cianflone and Sullivan18], but in fact only little is known about its immunogenicity and safety within this special population group [Reference Panasiuk, Prokopowicz and Panasiuk19, Reference Wolf20]. Moreover, this limited information relates to the TBE vaccination with the standard scheme; however, no data on the reliability of the frequently patient-preferred rapid scheme of TBE vaccination in HIV-1 infected individuals are available. Thus, we decided to investigate the immunogenicity and safety of the TBE vaccine following a rapid scheme administration in persons infected with HIV-1. The major aim of our study was to evaluate whether the rapid scheme of TBE vaccination is applicable to routine clinical practice – i.e. whether it provides a high level of immunogenicity and antibody persistence along with a favourable safety profile. Immunogenicity and safety data will be compared to historical data of HIV-negative individuals vaccinated by the rapid scheme of TBE vaccination. Some immunological markers and coinfections (CMV, Toxoplasma gondii and tuberculosis/TB) were analysed as factors impacting the antibody response following TBE vaccination.
Methods
The study was conducted at the HIV Clinic of the Na Bulovce Hospital as an open-label, prospective study. All enrolled persons were in follow-up care at the site due to their HIV-1 infection. Enrolment criteria consisted of the following: the patient's wish to be vaccinated against TBE by the rapid scheme, antiretroviral therapy with viral load <20 copies of HIV RNA/ml and CD4 + T lymphocytes (CD4)>400 cells/μl at least 3 months before enrolment in the study. Absolute counts of CD8 + T lymphocytes and CD4/CD8 ratio were included in the analysis. The age of all participants was between 18 and 50 years. The upper age limit was chosen to exclude older participants who might have altered immune responses [Reference Stiasny21]. None of the participants were vaccinated against TBE or had had this infection in the past, as confirmed by the negativity of specific TBE antibodies at the baseline [Reference Baldovin22, Reference Paulke-Korinek23]. There were also no contraindications to TBE vaccination in any participant.
Serostatus of CMV (CLIA, LIAISONTM, DiaSorin, Italy), T. gondii (ELISA, TestLineTM, TestLine, Czech Republic) and a history of TB was checked in all enrolees.
The research project was approved by the Ethics Committee of Na Bulovce Hospital (01.09.2017/8578/EK-Z) and all enrolled individuals signed informed consent.
The vaccination was performed using the inactivated vaccine Encepur Adult® containing 1.5 μg of K23 antigen in the standard dose of 0.5 ml administered intramuscularly. This vaccine was chosen because it is based on the antigen of the identical virus strain K23 found in the diagnostic test used to measure antibody levels.
All participants were vaccinated in the rapid scheme as recommended by the manufacturer – the first dose at day 0, second dose at day 7, third dose at day 21 and at day 365 the first booster dose was administered [Reference Jelinek10].
The immunogenicity of the vaccination was assessed by the production of TBE-specific IgG antibodies [Reference Garner-Spitzer24]. The TBE IgG were measured in serum by using an ELISA assay based on the K23 antigen (MASTAZYMETM FSME IgG), a commercial test marketed by Mast Diagnostica GmbH, and the concentrations were calculated in Vienna units – VIEU/ml [Reference Velay7, Reference Baldovin22, Reference Holzmann25]. The antibody concentrations were measured at baseline, 7 days after application of the third dose (day 28), on day 60 and 180. The timings of measurements on day 28 and 60 were chosen to be comparable with historical data of HIV-negative individuals. Day 180 was added to monitor the kinetics of antibodies between the third dose and the booster dose. Furthermore, on day 365, a blood sample was taken for antibody analysis just before a booster vaccination was applied. The next sampling took place on day 395 to assess the effect of the booster dose. The final TBE IgG measurement was performed on day 730 to measure the proportion of individuals that were still protected 2 years on from vaccination.
At baseline, the antibody concentrations were interpreted as negative if lower than 50 VIEU/ml [Reference Litzba26]. The concentration of specific TBE IgG antibodies higher than 126 VIEU/ml was considered protective [Reference Holzmann25, Reference Litzba26].
All participants were asked to record all adverse events following application of every single dose of vaccine.
Statistical analysis was performed using the software Stata, release 14.2 (Stata Corp LP, College Station, TX, USA). Continuous data were characterised by means and standard deviations and by medians and interquartile ranges. At each time point, geometric mean concentrations (GMC) of TBE-specific IgG and corresponding 95% confidence intervals were calculated from the data of all participants regardless of whether they reached protective levels – and compared using censored regression to consider values above the upper limit of quantification. Changes in seroprotection rates were evaluated using McNemar's test. Between-group comparison was performed by the Student's t-test. All statistical tests were evaluated at a significance level of 0.05.
Results
The study included 28 participants – 25 males (89.3%) and three females (10.7%). The mean values (± standard deviations) of their basic demographic, clinical and laboratory parameters were as follows: age 36.4 years (range 25–46), time since diagnosis of HIV 5.7 ± 3.6 years, CD4 count at baseline 844 cells/μl (range 438–1741 cells/μl), nadir CD4 count 434 cells/μl (range 19–1218 cells/μl), CD8 count at baseline 1128 cells/μl (range 479–2348 cells/μl), CD4/CD8 ratio 0.82 ± 0.32, time from CD4 nadir 4.5 ± 3.4 years and time since ARV initiation was 4.1 ± 3.5 years. The mean time prior to vaccination with a viral load of HIV RNA consistently lower than 20 copies/ml was 2 years (range 0.3–7.3years) (Table 1).
Table 1. Basic characteristics of study cohort

s.d., standard deviation; IQR, interquartile range; ARV, antiretroviral therapy.
a Time from HIV diagnosis to vaccination.
b Time from nadir CD4 + to vaccination.
Positivity of CMV IgG antibodies (semiquantitative method) was found in 28/28 participants (100%). Positivity of T. gondii IgG antibodies (≥12 IU/ml) was found in 7/28 participants (25%). Difference between responders and non-responders in terms of T. gondii seropositivity has not reached a statistical significance at any time point. No case of TB was found within the history of study cohort.
The seronegativity at baseline was confirmed in all participants since their baseline TBE IgG concentrations were ≤42.9 VIEU/ml. On day 28, the protective concentrations of TBE IgG were found in 35.7% of the 28 participants. Concentrations range in these 10 responders between 135.5–1472 and 3.7–101.6 VIEU/ml in the remaining 18 non-responders. On day 60, protective TBE IgG concentrations were found in 39.3% participants, on day 180 in 25.0%, and on day 365 – when blood was taken before the booster was administered – the proportion of persons with TBE IgG protective levels was as low as 21.4%. On day 395, 1 month after the booster administration, protective concentrations of TBE IgG were found in all but one of the vaccinees (96.4%). The proportion of protected vaccinees then decreased significantly (P = 0.008) to 71.4% on day 730. GMCs calculated from the data of all 28 participants were 78.7 VIEU/ml (95% CI 48.3–128.1) on day 28, 94.0 VIEU/ml (95% CI 63.4–139.4) on day 60, 65.5 VIEU/ml (95% CI 43.5–98.7) on day 180 and 55.8 VIEU/ml (95% CI 37.6–82.9) on day 365. The latter GMC was significantly lower compared to day 60 (P = 0.048). On day 395, after the booster, GMC significantly (P < 0.001) increased to 1303.9 VIEU/ml (95% CI 709.7–2395.5). Then the concentration levels dropped again, resulting in a GMC of 263.1 VIEU/ml (95% CI 131.9–525.1) on day 730, which was significantly lower than that on day 395 (P = 0.016) and the proportion of protected vaccinees decreased significantly to 71.4% (P = 0.008) (Fig. 1).
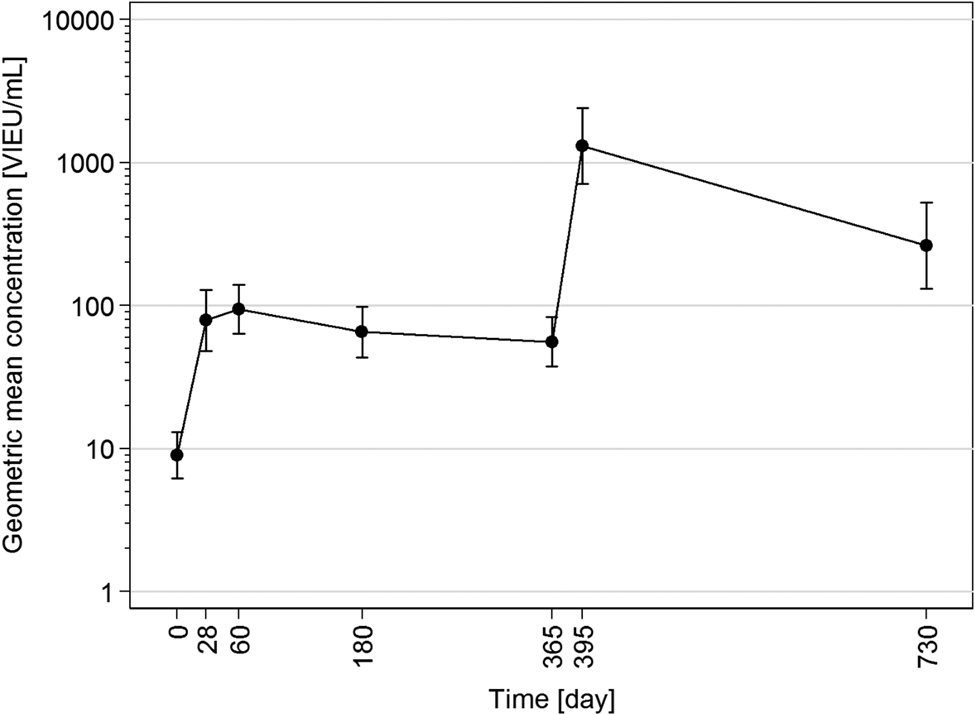
Fig. 1. Time course (day 0–730) of geometric mean concentrations with 95% confidence intervals calculated from data of all 28 participants.
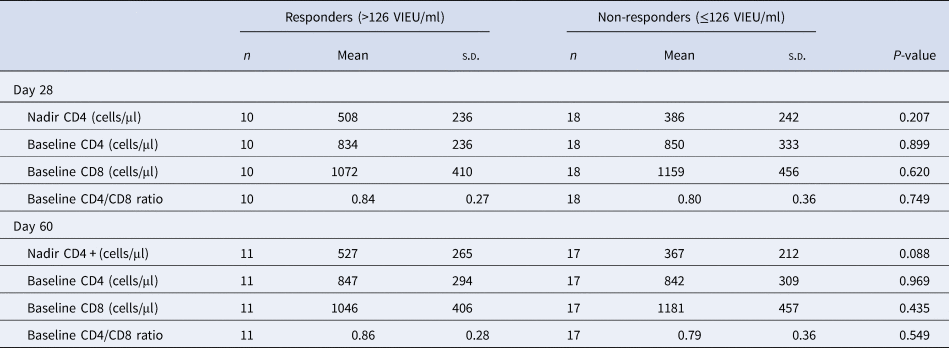
No differences were found between the group of vaccinees with confirmed early production of protective TBE IgG concentrations and non-responders in terms of the CD4 counts, CD8 counts and CD4/CD8 ratio. When measuring TBE IgG concentrations on day 28, the mean baseline CD4 values in both groups were practically identical – in responders 834 cells/μl and in non-responders 850/μl. Regarding the mean values of CD4 nadir, the participants with protective TBE IgG concentrations on day 28 had slightly higher values – 508/μl vs. 386/μl in non-responders, but this difference did not reach statistical significance (P = 0.207). Similarly, in the group of participants with protective TBE IgG concentrations at day 60, the mean baseline CD4 and CD8 counts of 847/μl and 1046/μl, respectively, did not differ significantly from those in non-responders with 842/μl and 1181/μl, respectively.
A larger difference between these groups was observed in the mean values of CD4 nadir, where responders had a mean CD4 nadir of 160 cells/μl higher (P = 0.088). This may suggest a significant difference, but it is not statistically proven.
No difference between responders and non-responders was found in terms of duration of ARV therapy.
The proportion of seropositivity of T. gondii was higher in the group of non-responders, but this difference has no statistical significance (Table 2).
Table 2. Comparison of CD4, CD8 cell counts and CD4/CD8 ratio in responders and non-responders on days 28 and 60
Throughout the entire course of our study (day 0–730), there was no virological failure or progression of HIV infection observed. Single virological blips (range 21–152 copies/ml) were observed in nine participants; however, viral loads returned below 20 copies/ml in all these vaccinees at follow-up 4 weeks later.
Eleven participants (39.3%) reported mild, local pain with a duration of less than 48 h after administration of one or more doses of the vaccine. In addition, two of them (7.1%) also reported local erythema with the duration of less than 48 h after the first dose of the vaccine.
Discussion
The early seroprotection rate of 35.7% achieved in our cohort 4 weeks after the start of vaccination is generally very low. In studies that looked at the immunogenicity of TBE vaccination in a rapid scheme in the general population, the rate of early seroprotection (1–3 weeks after the third dose) ranged from 95 to 100% of vaccinees [Reference Zent11, Reference Zent27, Reference Galgani28]. Standard methodology using ELISA for the analysis of specific antibodies does not offer an explanation for the low seroprotection rate. In the available literature, we did not find any publication evaluating the immunogenicity of TBE vaccination in the rapid scheme in individuals with presumably altered immune response. There are only publications available in which the immunogenicity of TBE vaccination in such subjects has been evaluated in a conventional scheme.
There are only two studies in the literature evaluating the immunogenicity of TBE vaccination in HIV-infected individuals. Panasiuk et al. vaccinated 29 HIV-1 individuals in a special scheme (0, 1, 2, 9 months), and 1 month after the last dose, the seroprotectivity rate was 44.8% [Reference Panasiuk, Prokopowicz and Panasiuk19]. In a small British study on a group of 16 haemophiliacs, four of whom were asymptomatic HIV-infected subjects, all HIV-negative haemophiliacs developed satisfactory levels of antibodies with geometric mean reciprocal titres of 193 after completion of the standard three-dose schedule, while vaccination failed in all four HIV-infected haemophiliacs with achieved geometric mean reciprocal titres of only 13 [Reference Wolf20].
In a Czech study on vaccinees older than 60 years vaccinated by the standard scheme, a seroprotection rate of 68% was achieved 1 month after the second dose in those vaccinated with ENCEPUR® [Reference Jilkova29]. Low immunogenicity of TBE vaccination ranging between 35% and 39% was observed in heart transplant patients, during chemotherapy for breast cancer and in patients on immunosuppressive therapy for rheumatoid arthritis [Reference Dengler30–Reference Hertzell32].
In addition, the persistence of protective antibody concentrations in our group was considerably shorter than in the general population. Before the application of the booster 1 year after the start of vaccination, the proportion of vaccinees with a protective concentration of TBE IgG in our cohort was only 21.4%, which is significantly lower than in the HIV-negative population [Reference Schondorf13]. Thirty days after administration of the booster, the proportion of vaccinees with protective IgG concentrations rose to 96.4%, but during the following year, this proportion significantly decreased to 71.4%. This is substantially less than the observed immunogenicity of booster administration 1 year after the start of vaccination with the rapid scheme in the general population, where 99% of vaccinees still had protective TBE IgG concentrations a year after booster administration [Reference Beran14, Reference Rendi-Wagner33]. Also in the study of Zent et al., the proportion of vaccinees with protective antibody levels was 99% 1 year after the booster and still 98% after 2 years [Reference Zent34].
In our study, no differences in baseline CD4 counts were found between responders and non-responders to TBE vaccination. Polish authors have observed that vaccinees with CD4 > 500 exhibited better responsiveness achieving 55% seroprotectivity, while only 44.8% achieved seroprotection in those with CD4 < 500; however, this difference was not significant [Reference Panasiuk, Prokopowicz and Panasiuk19]. Also, in the British study, all four HIV-positive haemophiliacs had normal CD4 counts (1.47 ± 0.51.109 cells/l), but none of them produced protective levels of TBE antibodies [Reference Wolf20].
In our cohort, individuals with higher CD4 nadir had a better response to TBE vaccination, although this difference was only close to statistical significance. A similar observation was made in the study of Lange et al., where effectively treated HIV-infected individuals vaccinated by tetanus and diphtheria toxoid had a normal actual CD4 count, but their antibody response was worse in individuals with lower CD4 nadir compared to those with higher nadir CD4 count who responded better to vaccination [Reference Lange35].
Possible reasons for the significantly worse response to TBE vaccination in HIV vaccinees may be the phenomenon of premature ageing and immunosenescence, which is expressed more in untreated HIV-infected individuals – however, residual defects remain even in those on antiretroviral therapy [Reference Deeks36]. Associated cytokine abnormalities caused by dysregulation of cellular immunity undoubtedly also play an important role here. In studies focused on TBE vaccination failure in HIV-negative individuals, significant differences in the levels of some cytokines were observed – responders had higher levels of IL-2 and INF-γ, while non-responders had significantly higher levels of IL-10 and lower levels of IL-2 and IFN-γ [Reference Garner-Spitzer24, Reference Aberle37, Reference Litjens38].
The main limitations of our study are as follows: a relatively small number of study individuals, a small proportion of female individuals and an absence of CMV seronegative individuals.
According to the results of our study, TBE vaccination in a rapid schedule does not ensure a satisfactory onset of early protection in most HIV individuals, even though they are on treatment and have high CD4 levels and complete viral suppression. For this reason, in our opinion, this method of protecting HIV-infected people from TBE infection cannot be considered reliable in routine practice.
However, the need for rapid immunisation against TBE is still relevant and the question of how to increase the immunogenicity of vaccination needs to be answered. In conventional TBE vaccination schedules, the inclusion of an additional dose is usually recommended for vaccinees with presumable altered immune response [Reference Geretti17, Reference Hansson39].
We assume that in the short time interval during which the early protection must be provided, there is no longer space for an additional dose of vaccine and based on previous experience with TBE vaccination, an increase in immunogenicity may not be expected when the vaccine is administered subcutaneously [Reference Hopf40].
Therefore, we speculate that a possible solution could be dose escalation in TBE vaccination in the rapid scheme, but this would require validation in new studies.
Data availability statement
The data that support the findings of this study are available from the corresponding author [DJ] upon reasonable request.
Acknowledgements
Klára Labská, MD. Ph.D., National Institute of Public Health, Šrobárova 48, 100 42, Prague 10, Czech Republic.
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflict of interest
None.




 Open access
Open access