



Introduction
The biological age of an organism can be estimated based on endocrine, immunological, or molecular observations (Jin, Reference Jin2010). Accelerated aging occurs when biological age is higher than chronological age and it has been observed in various conditions that are related to impairments in quality of life, morbidity, and mortality. On the psychological level, chronic and traumatic stressors have been identified as mediators of accelerated aging. Childhood maltreatment (CM) (i.e., experiences of physical, sexual, and/or emotional abuse as well as physical or emotional neglect during childhood) has been hypothesized to be a contributing factor to accelerated biological aging (Danese & McEwen, Reference Danese and McEwen2012) as it constitutes a major threat to psychological and physical health (Batten, Aslan, Macjejewski, & Mazure, Reference Batten, Aslan, Macjejewski and Mazure2004; Danese & Tan, Reference Danese and Tan2014; MacMillan et al., Reference MacMillan, Fleming, Streiner, Lin, Boyle, Jamieson and Beardslee2001; Springer, Sheridan, Kuo, & Carnes, Reference Springer, Sheridan, Kuo and Carnes2007). In particular, premature onset of age-related diseases has been observed after CM, including cardiovascular diseases, diabetes, and immune dysfunction (Batten et al., Reference Batten, Aslan, Macjejewski and Mazure2004; Castle, Reference Castle2000; Danese et al., Reference Danese, Moffitt, Pariante, Ambler, Poulton and Caspi2008; Danese et al., Reference Danese, Moffitt, Harrington, Milne, Polanczyk, Pariante and Caspi2009; Danese & McEwen, Reference Danese and McEwen2012). One possible explanation for this association is the allostatic load that impacts the immune system: chronic or repeated exposure to stress, especially during critical stress-sensitive developmental stages, could constantly trigger the activation of the immune system. This may ultimately result in elevated levels of inflammation as well as accelerated aging processes (Danese & McEwen, Reference Danese and McEwen2012. CM has been consistently linked to shorter telomere length, a well-established biomarker of cellular age (Boeck et al., Reference Boeck, Krause, Karabatsiakis, Schury, Gündel, Waller and Kolassa2018; Ridout et al., Reference Ridout, Levandowski, Ridout, Gantz, Goonan, Palermo and Tyrka2018; Tyrka et al., Reference Tyrka, Price, Kao, Porton, Marsella and Carpenter2010, Reference Tyrka, Carpenter, Kao, Porton, Philip, Ridout and Price2015). Moreover, alterations of mitochondrial biogenesis (Tyrka et al., Reference Tyrka, Price, Kao, Porton, Marsella and Carpenter2010, Reference Tyrka, Parade, Price, Kao, Porton, Philip and Carpenter2016), which are important in cellular aging, apoptosis, and higher levels of reactive oxygen species were found in association with CM (Boeck et al., Reference Boeck, Koenig, Schury, Geiger, Karabatsiakis, Wilker and Kolassa2016).
The biological aging of cells, tissues, and organs is characterized by diverse biomolecular changes, such as alterations in the proteome, the metabolome (reviewed by Hoffman, Lyu, Pletcher, & Promislow, Reference Hoffman, Lyu, Pletcher and Promislow2017), and the epigenome (Horvath, Reference Horvath2013; Weidner et al., Reference Weidner, Lin, Koch, Eisele, Beier, Ziegler and Wagner2014). Healthy aging has been associated with highly defined DNA methylation (DNAm) patterns (Drinkwater, Blake, Morley, & Turner, Reference Drinkwater, Blake, Morley and Turner1989; Fuke et al., Reference Fuke, Shimabukuro, Petronis, Sugimoto, Oda, Miura and Jinno2004; Jones, Goodman, & Kobor, Reference Jones, Goodman and Kobor2015; Wilson, Smith, Ma, & Cutler, Reference Wilson, Smith, Ma and Cutler1987), which have been proposed as biomarkers of chronological age (Horvath, Reference Horvath2013; Weidner et al., Reference Weidner, Lin, Koch, Eisele, Beier, Ziegler and Wagner2014). One promising candidate is the gene that codifies for the elongation of very long chain fatty acids protein 2 (ELOVL2), a gene located on chromosome 6, for which an age-related increase in DNAm has been repeatedly observed as a robust estimate for chronological age (Bysani et al., Reference Bysani, Perfilyev, de Mello, Rönn, Nilsson, Pihlajamäki and Ling2017; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012; Johansson, Enroth, & Gyllensten, Reference Johansson, Enroth and Gyllensten2012; Naue et al., Reference Naue, Sänger, Hoefsloot, Lutz-Bonengel, Kloosterman and Verschure2018; Park et al., Reference Park, Kim, Seo, Bae, Kim, Lee and Kim2016; Zbieć-Piekarska et al., Reference Zbieć-Piekarska, Spólnicka, Kupiec, Parys-Proszek, Makowska, Pałeczka and Branicki2015). ELOVL2 encodes for the fatty acid elongase 2, an enzyme involved in the synthesis of long polyunsaturated fatty acids (PUFAs; Leonard et al., Reference Leonard, Kelder, Bobik, Chuang, Lewis, Kopchik and Huand2002) in the endoplasmic reticulum, and it plays a key role in the structural organization of the cellular membrane (Jump, Reference Jump2002). In an epigenome-wide approach, Garagnani et al. (Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012) showed that ELOVL2 DNAm in whole blood displayed the strongest correlation with chronological age in a cohort of 501 individuals ranging from newborns to 99 years of age. The results were replicated in several different cell types, such as white blood cells and liver cells (Bysani et al., Reference Bysani, Perfilyev, de Mello, Rönn, Nilsson, Pihlajamäki and Ling2017; Johansson et al., Reference Johansson, Enroth and Gyllensten2012; Naue et al., Reference Naue, Sänger, Hoefsloot, Lutz-Bonengel, Kloosterman and Verschure2018; Park et al., Reference Park, Kim, Seo, Bae, Kim, Lee and Kim2016; Zbieć-Piekarska et al., Reference Zbieć-Piekarska, Spólnicka, Kupiec, Parys-Proszek, Makowska, Pałeczka and Branicki2015). In sum, ELOVL2 DNAm has emerged as a promising marker for age estimation, with a mean absolute deviation from chronological age ranging between 5 and 6.41 years (Bacalini et al., Reference Bacalini, Deelen, Pirazzini, De Cecco, Giuliani, Lanzarini and Garagnani2017; Johansson et al., Reference Johansson, Enroth and Gyllensten2012; Park et al., Reference Park, Kim, Seo, Bae, Kim, Lee and Kim2016; Spólnicka et al., Reference Spólnicka, Pośpiech, Pepłońska, Zbieć-Piekarska, Makowska, Pieta and Branicki2018). The remaining variance might be the result of other – so far unknown – environmental factors that potentially accelerate or slow down biological aging, like lifetime chronic or traumatic stress exposure.
Lifetime stress has been associated with accelerated epigenetic aging (Zannas et al., Reference Zannas, Arloth, Carrillo-Roa, Iurato, Röh, Ressler and Mehta2015). In a longitudinal study, Boks et al. (Reference Boks, van Mierlo, Rutten, Radstake, De Witte, Geuze and Vermetten2015) showed that war-associated trauma exposure during adulthood was related to an increased epigenetic age, as measured by an established so-called “epigenetic clock” based on Illumina 450 K DNAm analyses (a whole methylome approach; Horvath, Reference Horvath2013) in peripheral whole blood samples (Boks et al., Reference Boks, van Mierlo, Rutten, Radstake, De Witte, Geuze and Vermetten2015). Similarly, using this “epigenetic clock,” experiencing violence during childhood was also associated with increased epigenetic age in DNA extracted from saliva samples of school-age children (Jovanovic et al., Reference Jovanovic, Vance, Cross, Knight, Kilaru, Michopoulos and Smith2017).
Considering these findings, it is still unknown whether acquired premature epigenetic aging due to environmental stressors (such as childhood or lifetime adversity) is transmitted to the next generation. Previous studies on the intergenerational transmission of the biological consequences of CM were limited in that they were not able to distinguish between epigenetic intergenerational transmission and postnatal environmental as well as psychosocial influences. To overcome this limitation, we assessed immune cells from umbilical cord blood. This approach allowed us to uniquely address the question of whether epigenetic alterations observed in mothers with CM have been transmitted to their newborns at the time of birth, before other environmental factors have had an influence on their epigenetic pattern.
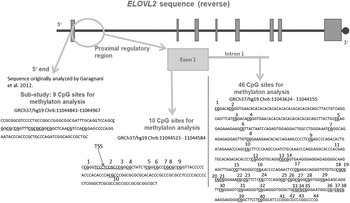
Based on previous findings, we assessed potential accelerated biological aging by analyzing the association between DNAm of the well-established biomarker for biological age (ELOVL2) and CM. In a first substudy, we targeted ELOVL2 5′ end (slightly upstream of the 5′ end of the gene) DNAm, a region that has been previously identified as a strong biomarker for age (Bysani et al., Reference Bysani, Perfilyev, de Mello, Rönn, Nilsson, Pihlajamäki and Ling2017; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012; Johansson et al., Reference Johansson, Enroth and Gyllensten2012; Naue et al., Reference Naue, Sänger, Hoefsloot, Lutz-Bonengel, Kloosterman and Verschure2018; Park et al., Reference Park, Kim, Seo, Bae, Kim, Lee and Kim2016; Zbieć-Piekarska et al., Reference Zbieć-Piekarska, Spólnicka, Kupiec, Parys-Proszek, Makowska, Pałeczka and Branicki2015). We then conducted explorative analyses of DNAm in an extended area of the ELOVL2 gene: we examined the DNAm within exon 1, which is located within the CpG (cytosine–phosphate–guanine dinucleotide) island of the ELOVL2 gene promoter and encompasses one transcription start site (TSS), and intron 1, which extends from the downstream region into the 3′ shore of the CpG island (Figure 1). We chose these regions for two reasons: (a) DNAm within CpG islands (DNA sequences rich in CpG sites) has special importance in the regulation of transcription activity (Deaton & Bird, Reference Deaton and Bird2011) and (b) DNAm within the CpG island located in the promoter of the ELOVL2 gene is especially dependent on chronological aging (Bysani et al., Reference Bysani, Perfilyev, de Mello, Rönn, Nilsson, Pihlajamäki and Ling2017; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012; Johansson et al., Reference Johansson, Enroth and Gyllensten2012; Naue et al., Reference Naue, Sänger, Hoefsloot, Lutz-Bonengel, Kloosterman and Verschure2018; Park et al., Reference Park, Kim, Seo, Bae, Kim, Lee and Kim2016; Zbieć-Piekarska et al., Reference Zbieć-Piekarska, Spólnicka, Kupiec, Parys-Proszek, Makowska, Pałeczka and Branicki2015). Furthermore, we investigated whether the associations between ELOVL2 DNAm patterns and CM experiences of mothers could also be observed in the cord blood cells of their newborns. For this purpose, this study assessed the DNAm of the ELOVL2 gene of immune cells from women with and without CM experiences, as well as from their newborns. Immune cells were isolated shortly after parturition to avoid the influence of psychosocial interaction as a possible factor for stress-associated methylation changes in the offspring. In order to assess potential phenotypic implications of the ELOVL2 DNAm, we additionally analyzed ELOVL2 gene expression in peripheral blood mononuclear cells (PBMC).
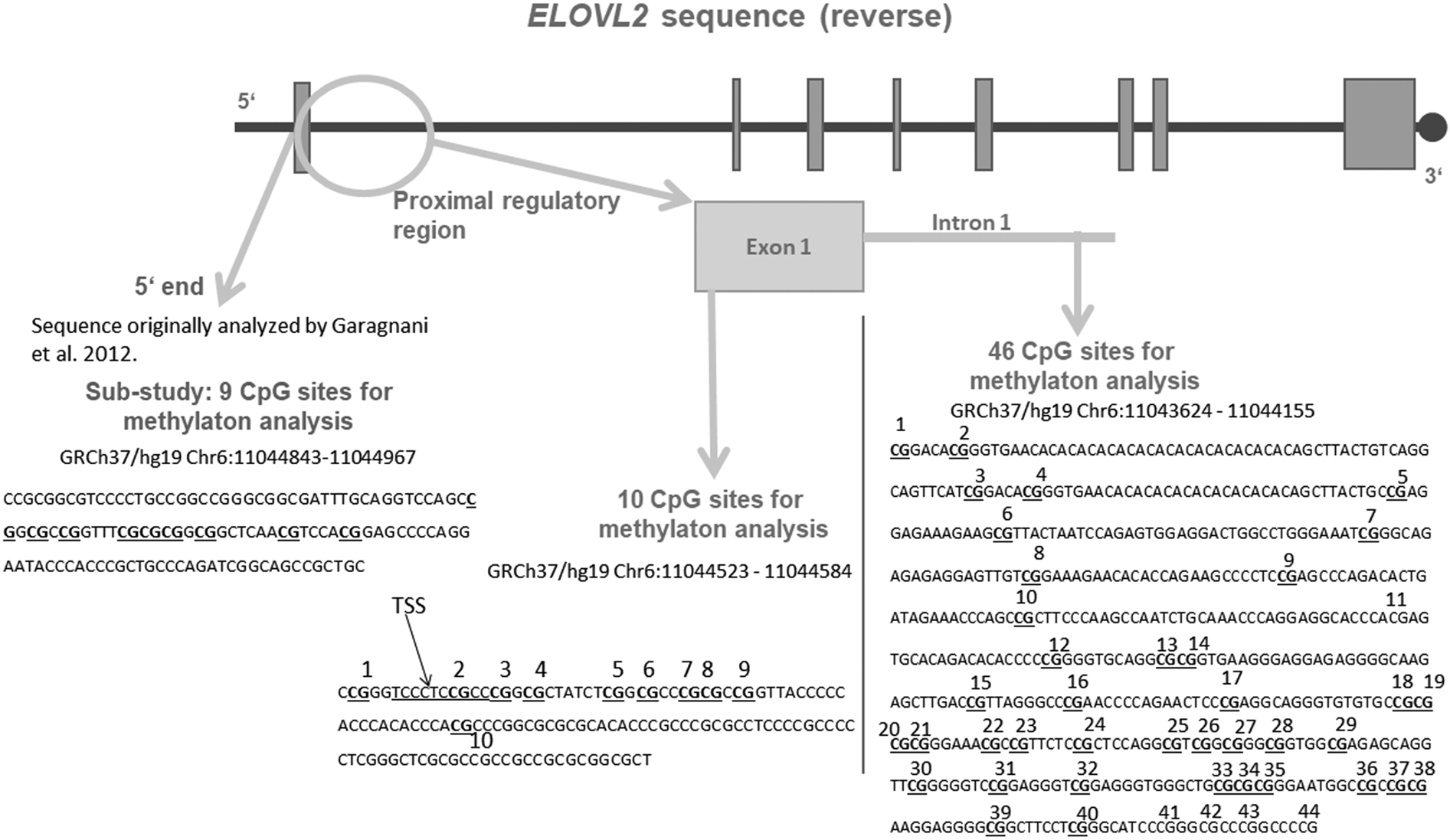
Figure 1. Schematic representation of the targeted sequence of the ELOVL2 gene. Bold and underlined are the CpG (cytosine–phosphate–guanine dinucleotide) sites that were included for analyses after data cleaning. The region analyzed in our substudy is shown in grey in the figure, at the 5′ end of the ELOVL2 gene. This 5′ end region lies approximately 250 bp upstream from the region targeted within the exon 1 and covered 9 CpG sites, all included in the analyses. The 10 CpG sites of the ELOVL2 exon 1 DNA methylation (DNAm) were analyzed. Regarding ELOVL2 intron 1, 44 CpG sites were covered by the mass spectrometry approach. After data processing, 39 CpG sites remained for analyses (underlined). TSS = transcription start site. Genomic and CpG islands annotations were based on the human UCSC Genome Browser assembly (February 2009, GRCh37/hg19).
Method
Recruitment of study participants and assessment of sociodemographic and psychological data
The study was part of the “My Childhood – Your Childhood” project conducted in Ulm, Germany. The study was approved by the ethics committee of Ulm University and was conducted in accordance with the Declaration of Helsinki. Umbilical cord blood from infants born in the maternity ward of Ulm University Gynecological Hospital (N = 5426) was collected between October 2013 and December 2015 and was transported directly to the Department of Clinical & Biological Psychology at Ulm University. During their stay on the maternity ward, women received information about the study within the first week after parturition. The following exclusion criteria were applied: mothers below 18 years of age, mothers with insufficient knowledge of the German language, mothers with severe complications during parturition, and mothers and/or infants with severe health problems. All women who agreed to participate in the study provided written informed consent (N = 533) and specific consent for processing umbilical cord blood from their newborns. Collected infant blood samples were immediately discarded if the mother met any of the exclusion criteria, declined participation in the study, or declined the use of their newborn's cord blood. The participant mothers completed a screening interview that included the German version of the Childhood Trauma Questionnaire (CTQ; Bader, Hänny, Schäfer, Neuckel, & Kuhl, Reference Bader, Hänny, Schäfer, Neuckel and Kuhl2009) conducted by trained study personnel. Basic sociodemographic data were also assessed.
The CTQ sum score (Bernstein & Fink, Reference Bernstein and Fink1998) was used to test the associations between DNAm and the severity of CM (or CM load). For description of demographic and clinical data of the study participants (Table 1), the mild cutoff criterion (Bernstein & Fink, Reference Bernstein and Fink1998) of the CTQ was used to classify mothers with (CM+) and without a CM history (CM−). Mothers with mild to severe traumatic childhood experiences in at least one subscale of the CTQ (emotional, physical or sexual abuse, and emotional or physical neglect) were categorized as CM+; the others were categorized as CM−. Demographic data based on the moderate CTQ cutoff are reported in section 1 of the Supplementary Material. Here, mothers with at least moderate childhood experiences in one or more subscales were categorized as CM+, otherwise as CM−.
Table 1. Demographic and biological characteristics
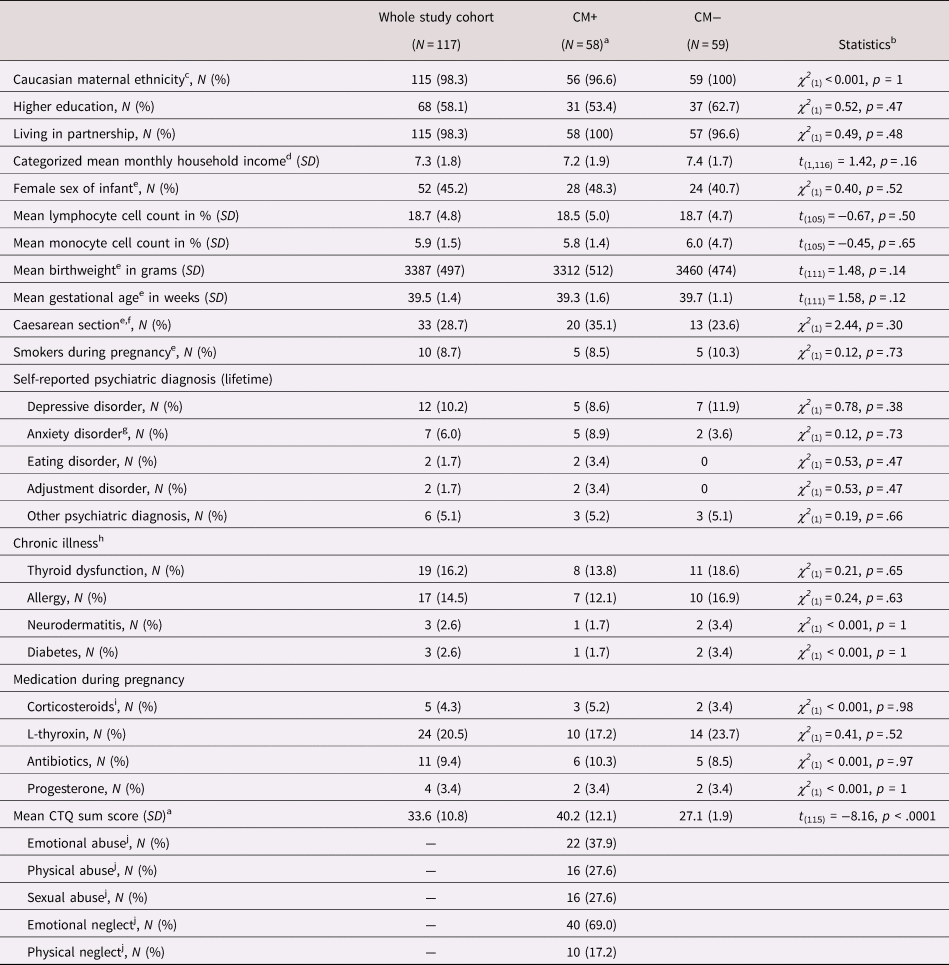
Note: Group differences calculated with chi-square tests for binomial and t tests for continuous variables. SD = standard deviation; CM = childhood maltreatment; CTQ = Childhood Trauma Questionnaire; CTQ sum score = childhood maltreatment load.
a The group comparisons presented in this table were assessed using the mild cutoff of the CM classification.
b Main effect of CTQ classification (t tests or chi-square tests).
c One study participant of Brazilian origin and one of North American origin.
d Two CM+ did not provide income data. Monthly household income (in €) was ranged between 1 and 9 as follows: 1 < 400; 2 = 400–1000; 3 = 1000–1500; 4 = 1500–2000; 5 = 2000–2500; 6 = 2500–3000; 7 = 3000–3500; 8 = 3500–4000; 9 > 4000.
e For gestational and neonatal characteristics, only mother–infant dyads were included: N CM− = 58; N CM+ = 55
f Included planned (N CM− = 16, N CM+ = 12) and emergency (N CM− = 4, N CM+ = 1) forms of caesarean section
g One woman from each CM group had a diagnosis of depression and anxiety disorder
h One CM+ women had asthma, neurodermatitis, and allergy; one CM+ had diabetes and thyroid dysfunction; one CM+ woman had an allergy and thyroid dysfunction.
i Only taken medication with more than one occurrence included
j Amount of women with at least mild experiences in this CTQ subscale.
Due to sample availability, analyses of DNAm were conducted in a subset of study participants selected as follows: of the N = 533 women agreeing to participate, N = 153 provided both maternal blood and fetal cord blood samples. The project “My Childhood – Your Childhood,” from which the cohort for this study was selected, used the mild cutoff of the CTQ for the categorization of mothers according to their CM experiences. Accordingly, the mild CTQ cutoff was used for selection and characterization of the epigenetic cohort analyzed in this study as follows: all 58 pairs of mild CM+ women and their infants who provided both maternal and fetal cord blood samples were included in epigenetic analyses. With the focus on those mothers who were willing to further participate in the overall project (mother–infant dyads were invited to participate in two more psycho-diagnostic interviews, 3 months and 1 year after birth) and matching for maternal age, gestational week at the time of birth, birthweight, and sex of the infant, 59 CM− mother–infant dyads were selected for epigenetic analyses as controls. CM− mothers who were included in the epigenetic analyses did not differ from CM− mothers who were not included (N = 36) with respect to their age, CTQ sum score, sex of their newborns, ethnicity, smoking habits during pregnancy, chronic illnesses (thyroid dysfunction, allergies, neurodermatitis, diabetes, asthma, or coagulation disorders), lifetime psychological diagnosis, or medication during pregnancy (all p values > .05). From the 117 dyads included for epigenetic analyses, data from the offspring of three mothers who gave birth to twins were excluded and the blood cell isolation from one infant failed. Thus, the final cohort included 117 mothers (mild CTQ cutoff: N = 58 CM+ and N = 59 CM−; moderate CTQ cutoff: N CM+ = 33 and N CM− = 84) and 113 infants (mild CTQ cutoff: N CM+ = 55 and N CM− = 58; moderate CTQ cutoff: NCM+ = 32 and N CM− = 81). CM+ mothers did not differ from CM− mothers in terms of age, duration of the pregnancy, ethnicity, newborn's sex, chronic illnesses, or lifetime psychiatric disorders (Table 1). The demographic and clinical characteristics of the participants were obtained from clinical diagnostic interviews in which mothers were asked to report their chronic health outcomes and lifetime psychiatric diagnoses (Table 1). None of the self-reported lifetime diagnoses (depressive disorder, anxiety disorder, eating disorder, or adjustment disorder) had a main effect on methylation of ELOVL2 5′ end, exon 1 or intron 1 (all p values > .05).
Sample collection and DNA extraction
Venous blood from mothers and umbilical cord blood from newborns were drawn into collection tubes (Sarstedt S-Monovette, Nürmbrecht, Germany) buffered with CPDA (citrate-phosphate-dextrose solution with adenine). Maternal PBMC and umbilical cord blood mononuclear cells (UBMC) from infants were isolated by Ficoll-Hypaque density gradient centrifugation following the manufacturer's instructions (GE Healthcare, Chalfont St Giles, UK). Cells were suspended in cryopreservation solution (dimethyl sulfoxide, Sigma-Aldrich, St. Louis, MO, USA; heat-activated fetal calf serum, Sigma-Aldrich, dilution 1:10), placed in isopropanol-filled cryocontainers (Mr. Frosty, Nalgene, USA) and stored at −80°C until further analysis.
In addition, another small volume of whole blood from N = 108 mothers and N = 90 infants was collected into collection tubes (Sarstedt S-Monovette) buffered with EDTA (ethylene-diamine-tetraacetic acid). These samples were used for hemograms at the Department of Clinical Chemistry of Ulm University. Genomic DNA was extracted from freshly thawed PBMC and UBMC using a semiautomatic MagNaPure 96 System (Roche, Penzberg, Germany). DNA concentrations in the eluates were quantified using a Qubit spectrophotometer (Life Technologies, Darmstadt, Germany). DNA was lyophilized, resuspended in DNAse-free water (Life Technologies) to a final concentration of 40 ng/μl and stored at −20°C. Frozen DNA samples were transported on dry ice to the lab facilities from the Institute for Human Genetics at Ulm University for pyrosequencing analyses of the ELOVL2 5′ end and to Varionostic GmbH (Ulm, Germany) for ELOVL2 exon 1 and intron 1 methylation analyses.
Identification of epigenetic targeted regions
In order to replicate previous results on a region from the ELOVL2 that showed strong correlations with age, in a substudy (N = 116 mothers and 112 newborns) we targeted nine CpGs that are located approximately 250 bp upstream from one TSS, in the 5′ end of the ELOVL2 gene (GRCh37/hg19 chr6:11044843-11044967; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012; Bacalini et al., Reference Bacalini, Deelen, Pirazzini, De Cecco, Giuliani, Lanzarini and Garagnani2017). For details regarding the pyrosequencing analyses of the 5′ end, please refer to section 3 of the Supplementary Material.
We then performed explorative analyses on an extended area of the ELOVL2 gene: mass array spectrometry was used for the examination of the ELOVL2 intron 1 DNAm. Due to methodological reasons, mass array spectrometry could not be applied for the evaluation of DNAm of exon 1; pyrosequencing was thus performed (see section 4 of the Supplementary Material for more details). About 500–1000 ng of genomic DNA were bisulfite-treated and PCR-amplified before downstream processing.
Assessment of intron 1 DNAm using mass array spectrometry
The EpiTYPER assay (Sequenom Inc., San Diego, CA) was used to quantify the DNAm levels of individual CpG sites in ELOVL2 intron 1. Two primers were used to amplify the region GRCh37/hg19, chr6:11043624-11044155. PCR products were processed following the manufacturer's protocol. For each CpG unit, the percentages of methylated CpG sites over the sum of methylated and unmethylated CpG sites were obtained and used for statistical analyses (Ehrich et al., Reference Ehrich, Nelson, Stanssens, Zabeau, Liloglou, Xinarianos and van den Boom2005). Details regarding data processing of the mass array data are provided in section 5 of the Supplementary Material. The mean percentage of gene methylation over all remaining CpG sites after data processing was calculated and used for statistical analyses.
Assessment of exon 1 DNAm using bisulfite pyrosequencing
We further assessed DNAm levels at the TSS within exon 1 of ELOVL2 (GRCh37/hg19, chr6:11044523-11044584). Using the pyrosequencing technology, 10 CpG units were analyzed (Figure 1). PyroMark Q24 software (Qiagen) was used for sequencing analyses and quantification of the DNAm at each CpG site. All CpG units and samples fulfilled the data processing criteria (<50% of samples with missing data and 70% of coverage). Thus, all 10 CpG units within ELOVL2 exon 1 and all samples (N = 117 mothers and 113 children) were included in the analyses.
ELOVL2 gene expression analyses
A detailed description of the ELOVL2 gene expression analyses using the qPCR-based Taqman assay is given in section 6 of the Supplementary Material.
Data processing and statistical analyses
Data processing and statistical analyses were conducted with R version 3.2.3 (R Core Team, 2014). For demographic and clinical descriptive analyses, student t tests and χ2 tests were applied. The Shapiro–Wilk test was used to test the normal distribution of model residuals. Averaged DNAm data residuals from the substudy on ELOVL2 5′ end methylation were normally distributed within the mothers, but not within the newborns. Pearson correlations were used for correlation analyses of the maternal data. For analyses that included the newborns’ ELOVL2 5′ end data, the nonparametric Kendall's tau correlation was used. Regarding exploratory analyses, all methylation data from the ELOVL2 exon 1 and intron 1 data were skewed, and thus nonparametric Kendall's tau correlations were used for correlation analyses. We further included potential confounding factors as covariates for maternal analyses, analyzing intron 1 and exon 1 separately. For analyses of the effects of age, the covariates were CM load (operationalized by the CTQ sum score) and the relative counts of blood cell types (percentage of monocytes and percentage of lymphocytes in whole blood as separate covariates). For analyses of the effect of CM, the covariates were age and relative cell counts of monocytes and lymphocytes. For analyses of the effects of maternal CM and maternal age on infants’ ELOVL2 5′ end, exon 1, and intron 1 DNAm, the sex of the infant and gestational age (in weeks) were included as covariates. Since the criteria for the application of standard linear models were not fulfilled (e.g., not normally distributed residuals), nonparametric permutation tests (Freedman & Lane, Reference Freedman and Lane1983) were used to test for statistical significance when the effect of a covariate or interaction analysis was tested (standardized β coefficients are reported). The multiple testing adjustment procedure false discovery rate (FDR; Benjamini & Hochberg, Reference Benjamini and Hochberg1995) was used for the original p values in individual CpG analyses to counteract the risk of false positives. Samples were measured blinded to the experimenter. All tests were performed two-tailed, with α ≤ .05.
Results
DNAm of ELOVL2 sequence previously described as a biomarker for age (5′ end)
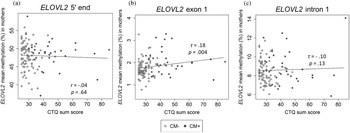
There was a positive correlation between age and the DNAm of the ELOVL2 5′ end (r = .79, p < .001, Figure 2a). This effect remained significant when the covariates CTQ sum score and cell counts of monocytes and lymphocytes were included in the analyses (r = .57, p < .001). All nine analyzed CpG units showed a positive correlation with age after FDR adjustment (Pearson's r ranged from .60 to .82; all p adj values < .001). There were, however, no associations between the mean DNAm of this region and the CTQ sum score (r = −.04, p = .65, as shown in Figure 3a). Single CpG unit analyses showed no correlational associations with the CTQ sum score (all p values > .05). Regarding intergenerational effects, we used nonparametric tests because the newborns’ ELOVL2 5′ end residuals were not normally distributed. The mean methylation of this ELOVL2 sequence was not correlated between mothers and their newborns (τ = .02, p = .77). There were no associations between maternal CTQ sum score and children's ELOVL2 methylation (τ = -0.09, p = .15). (See section 7 of the Supplementary Material for CM-associated group comparisons).
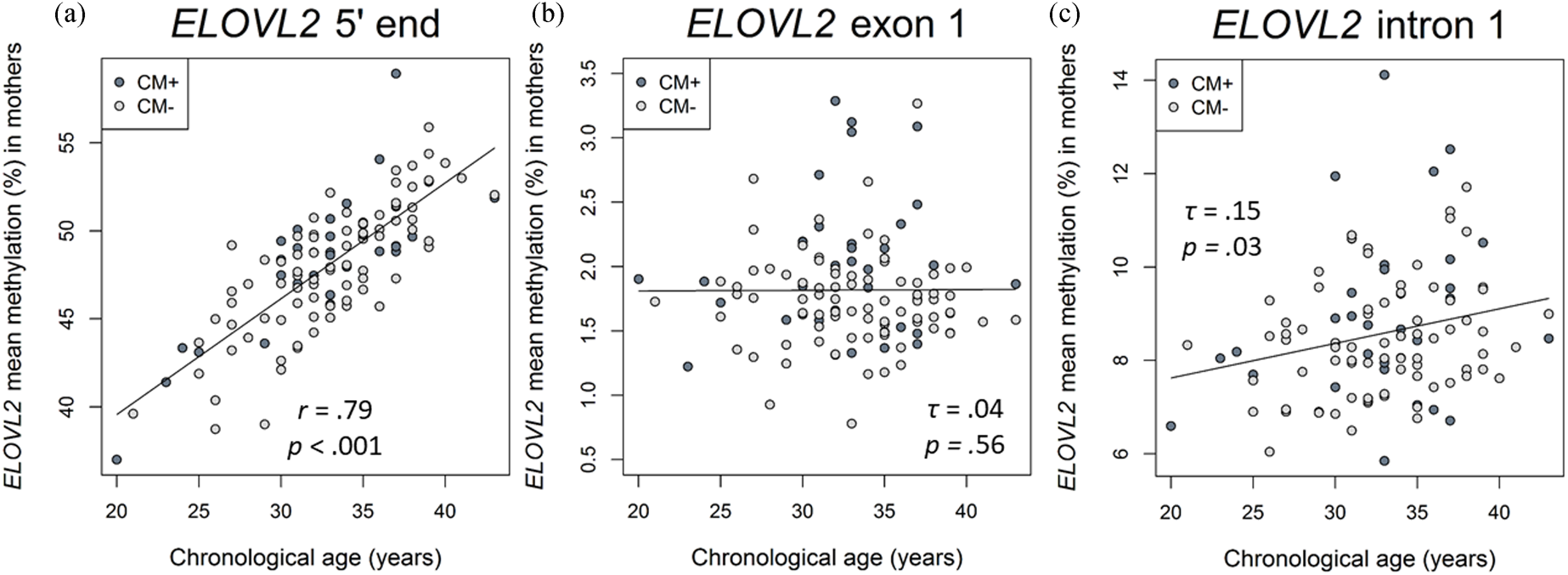
Figure 2. Associations of DNA methylation (DNAm) of ELOVL2 5′ end, intron 1, and exon 1 targeted regions with chronological age in mothers. (a) ELOVL2 5′ end mean DNAm in maternal peripheral blood mononuclear cells (PBMC) strongly correlated with chronological age (N = 116). (b) ELOVL2 exon 1 mean DNAm was not associated with chronological age (N = 117). (c) ELOVL2 intron 1 mean DNAm in maternal PBMC increased with chronological age (N = 110). Mothers with a childhood maltreatment (CM) history (CM+) were classified according to the moderate Childhood Trauma Questionnaire (CTQ) cutoff.
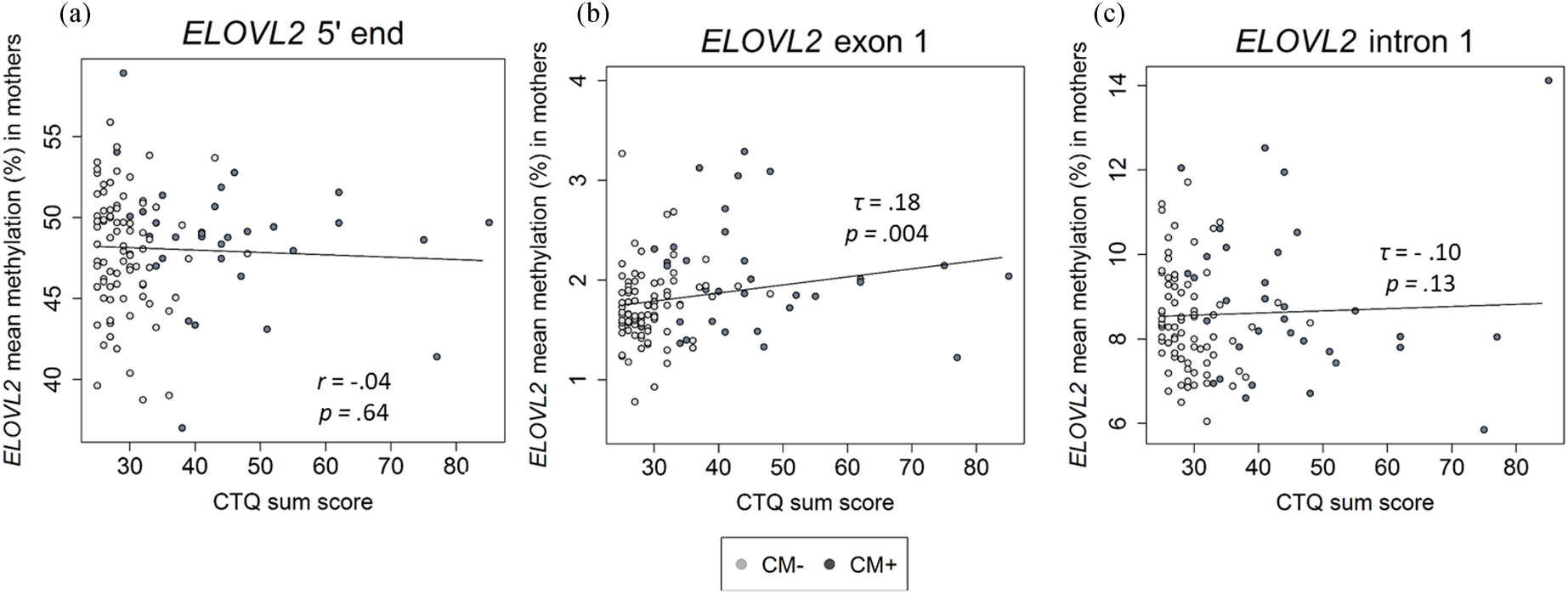
Figure 3. Association between childhood maltreatment (CM) and mean DNA methylation (DNAm) of ELOVL2 5′ end (N = 116), exon 1 (N = 117), and intron 1 (N = 110) in mothers. (a)–(c) Correlational analyses showed that an increased CM load (CTQ sum score) was associated with higher DNAm of the ELOVL2 exon 1, but not of ELOVL2 5′ end or intron 1. CTQ = Childhood Trauma Questionnaire. CTQ sum score = Childhood maltreatment load. CM+ mothers classified according to the moderate CTQ cutoff.
Associations between ELOVL2 exon 1 and intron 1 DNAm and chronological age in mothers
Correlation analyses showed that the mean DNAm of ELOVL2 intron 1 increased with chronological age in mothers (mean of DNAm (%) ± SD = 8.5 ± 1.4%, τ = .15, p = .03; Figure 2c). This effect remained significant (β age = .22, p = .02) when CTQ sum score and cell counts of monocytes and lymphocytes were included as covariates in the analysis. The individual analysis of each single CpG site showed that the methylation levels of the units CpG 2.4 (τ = .16, p = .03), CpG 16 (τ = .14, p = .04), and CpG 39.40 (τ = .14, p = .04) were positively associated with chronological age. However, the associations of age with these CpG single units did not remain after the FDR correction for multiple testing (p FDR > .05)
In contrast, the mean DNAm of exon 1 (mean of DNAm (%) ± SD = 1.8 ± 0.5%) was not significantly associated with maternal chronological age (τ = −.04, p = .56; Figure 2b) and none of the single CpG units analyzed within ELOVL2 exon 1 correlated with maternal chronological age (all p values > .05).
Association between ELOVL2 methylation in exon 1 and intron 1 and CM in mothers
The mean DNAm across ELOVL2 intron 1 did not correlate with the severity of CM experiences (τ = −.10, p = .13; Figure 3c). No significant associations between CTQ sum score and ELOVL2 intron 1 methylation levels were found at any individual CpG unit (original p values > .05). Adjustment for the covariates age and monocyte and lymphocyte cell counts did not alter the results.
In contrast to intron 1, ELOVL2 exon 1 mean methylation (mean ± SD = 1.8 ± 0.4%) was positively associated with the severity of CM experiences assessed as CTQ sum score (τ = .18, p = .004; Figure 3b). The inclusion of age, monocyte cell count, and lymphocyte cell count as covariates in one model did not alter the significance of the results (CM severity: β CTQ sum score = .19, p = .05). CM+ mothers showed significantly increased ELOVL2 exon 1 mean methylation compared with CM− mothers (see section 7 of the Supplementary Material). Regarding the associations between individual CpG sites within ELOVL2 exon 1 and the severity of CM experiences, the methylation levels of CpG 2, CpG 3, CpG 4, CpG 6, and CpG 8 were positively correlated with the CTQ sum score (Kendall's τ ranged from .13 to .21; p = .02 to .001). After inclusion of the covariates (age, monocyte cell count, and lymphocyte cell count), CpG 3 and CpG 8 remained positively associated with the severity of CM experiences (see Table S4 of the Supplementary Material), but these associations were reduced to statistical trends (CpG 3) or became non-significant (CpG 8) after FDR correction (Table S4).
ELOVL2 gene expression in PBMC
ELOVL2 gene expression was not detectable, either in the maternal PBMC or in the newborns’ UBMC (see section 6 of the Supplementary Material).
ELOVL2 DNAm of exon 1 and intron 1 in newborns from CM-exposed mothers
The infants’ mean DNAm of ELOVL2 intron 1 and exon 1 were 6.6 ± 1.2% and 2.8 ± 1.2%, respectively. Mean DNAm levels of ELOVL2 within intron 1 as well as exon 1 were not associated with the severity of maternal CM experiences (intron 1: β CTQ sum score = −.10, p = .29; exon 1: β CTQ sum score = .07, p = .42) or with maternal age (intron 1: β age = −.002, p = .98; exon 1: β age = −.008, p = .93). Neither mean methylation of ELOVL2 intron 1 (τ = −.01, p = .85) nor ELOVL2 exon 1 (τ = −.08, p = .23) were correlated between infants and their mothers.
Discussion
This is the first study to investigate the potential effect of a history of CM experiences on the ELOVL2 gene DNAm pattern, a gene associated with biological aging. The results of DNAm on the 5′ end of the ELOVL2 gene replicated previously described associations between chronological age and ELOVL2 methylation at a sequence considered as a biomarker for age (Bacalini et al., Reference Bacalini, Deelen, Pirazzini, De Cecco, Giuliani, Lanzarini and Garagnani2017 Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012). However, there were no effects of CM on the DNAm of the ELOVL2 5′ end. Our explorative testing of DNAm of intron 1 and exon 1 ELOVL2 regions showed higher mean and site-specific methylation levels of ELOVL2 exon 1 in CM+ compared with CM− women. However, these differences might not be biologically relevant because the overall DNAm changes were very small and the effects of CM were rather minimal in comparison with the effects of age on the ELOVL2 5′ end – the core biomarker region of ELOVL2 for biological aging. Moreover, mean DNAm levels of ELOVL2 intron 1 were not associated with CM experiences, but were significantly positively associated with chronological age. Importantly, there was no dose-dependent effect of maternal CM experiences on neonatal ELOVL2 5′ end, intron 1, and exon 1 methylation levels.
The results suggest associations between ELOVL2 DNAm patterns and both age and CM: while mean DNAm of the ELOVL2 5′ end and intron 1 increased with chronological age, DNAm of ELOVL2 exon 1 and the severity of CM experiences were positively correlated. Our results of 5′ end replicated the strong evidence of age-related increased DNAm of the ELOVL2 gene (Bacalini et al., Reference Bacalini, Deelen, Pirazzini, De Cecco, Giuliani, Lanzarini and Garagnani2017; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012; Spólnicka et al., Reference Spólnicka, Pośpiech, Pepłońska, Zbieć-Piekarska, Makowska, Pieta and Branicki2018; Zbieć-Piekarska et al., Reference Zbieć-Piekarska, Spólnicka, Kupiec, Parys-Proszek, Makowska, Pałeczka and Branicki2015). Interestingly, since the 5′ end DNAm did not show differences associated with CM status or the severity of CM experiences, our results suggest that exposure to CM does not exert accelerated epigenetic aging in PBMC in this genomic region. Moreover, we found the association between age and DNAm changed within ELOVL2 in other noncoding regions, namely the intron 1 region. Our results thus extend the number of CpG sites of ELOVL2 that are influenced by chronological age and contribute to the understanding of the interplay of DNAm, aging, and environment. In contrast to ELOVL2 5′ end and intron 1, ELOVL2 exon 1 – which encompasses the TSS – was significantly higher methylated with increasing severity of maternal CM experiences. While our results on exon 1 DNAm are preliminary and need replication, they broaden the perspective of ELOVL2 regulation by factors that are considered to accelerate biological age, such as CM. ELOVL2 exon 1 DNAm might be more sensitive to environmental factors (e.g., CM) than to chronological age. CpG 2 and CpG 3 seem to be of particular interest since they are respectively located at the TSS sequence and immediately after it. CpG 3 showed a statistical trend to increase with higher severity of CM experiences. To summarize, different loci are associated with chronological age and CM in different ways, suggesting that the position of the CpG within the gene sequence is critical.
The most direct implication of changes in DNAm in intron 1 and exon 1 of the ELOVL2 gene is that, when added to a TSS like the one within exon 1, the methyl group (CH3) can physically impede the transcription (Suzuki & Bird, Reference Suzuki and Bird2008). Here, in tissues other than PBMC, an ELOVL2 methylation-mediated downregulation may decrease physiological levels of n-3 derived PUFAs, which are essential for perinatal brain development, maturation, and normal brain functions (Schuchardt, Huss, Stauss-Grabo, & Hahn, Reference Schuchardt, Huss, Stauss-Grabo and Hahn2010). In contrast, the molecular associations of age on ELOVL2 methylation occur in the non-expressed areas of the 5′ end and intron 1. While methylation of introns can also interact with gene expression and with regulatory elements such as enhancers and promoters (Wiench et al., Reference Wiench, John, Baek, Johnson, Sung, Escobar and Hager2011), this interaction is not as clear because it sometimes requires complex tridimensional conformations of the DNA strand (Binder, Reference Binder2009). These tags might be a consequence of other upstream molecular mechanisms associated with age and might regulate alternative splicing (Maunakea, Chepelec, Cui, & Zhao, Reference Maunakea, Chepelec, Cui and Zhao2013) as an adaptation of the immune system to increasing age. We were not able to detect gene expression of ELOVL2 in PBMC, confirming previous results (Sibbons et al., Reference Sibbons, Irvine, Pérez-Mojica, Calder, Lillycrop, Fielding and Burdge2018). We speculate that the small differences in DNAm found for ELOVL2 in human PBMC can also be a reflection of more significant changes in other cell types and tissues, especially in those where ELOVL2 activity is essential for fatty acid metabolism and therefore for physiological activity (e.g. energy metabolism in the liver). Several studies suggest that DNAm of age-related genes in PBMC represents regulation in the liver (Bysani et al., Reference Bysani, Perfilyev, de Mello, Rönn, Nilsson, Pihlajamäki and Ling2017; Naue et al., Reference Naue, Sänger, Hoefsloot, Lutz-Bonengel, Kloosterman and Verschure2018), where ELOVL2 mostly exerts its function.
An epigenetically mediated dysregulation of the ELOVL2 enzyme might have implications for cellular and physiological functions. ELOVL2 is responsible for the elongation of omega n-3 (anti-inflammatory; first unsaturated bond at the third carbon relative to the terminal methyl end) and omega n-6 (pro-inflammatory; first unsaturated bond at the sixth carbon) PUFAs (Bandeira-Melo, Bozza, & Weller, Reference Bandeira-Melo, Bozza and Weller2002; Bozza & Bandeira-Melo, Reference Bozza and Bandeira-Melo2005; Schmitz & Ecker, Reference Schmitz and Ecker2008) in the liver. An ELOVL2-mediated dysregulation of n-3 and n-6 PUFAs metabolism could explain the link between CM experiences and the observed higher risk for conditions related to fatty acid metabolism such as obesity (Hemmingsson, Johansson, & Reynisdottir, Reference Hemmingsson, Johansson and Reynisdottir2014), diabetes (Basu, McLaughlin, Misra, & Koenen, Reference Basu, McLaughlin, Misra and Koenen2017; Huang et al., Reference Huang, Yan, Shan, Chen, Li, Luo and Liu2015), and cardiovascular outcomes (Basu et al., Reference Basu, McLaughlin, Misra and Koenen2017), and could increase the risk for Alzheimer's disease (Snowden et al., Reference Snowden, Ebshiana, Hye, An, Pletnikova, O'Brien and Thambisetty2017) and major depression (Coryell et al., Reference Coryell, Langbehn, Norris, Yao, Dindo and Calarge2017; Kiecolt-Glaser et al., Reference Kiecolt-Glaser, Belury, Porter, Beversdorf, Lemeshow and Glaser2007). Thus, the higher methylation of exon 1 ELOVL2 observed with increasing CM severity might be part of a pathway towards adult physical and psychological pathology in combination with other environmental factors. Our cohort consisted of a more or less representative group of individuals of the population from Ulm and were rather clinically healthy. Therefore, future studies should address the role of ELOVL2 epigenetic regulation on clinical disease outcomes in the context of severe CM.
Even though DNAm patterns are tissue-specific (Sielker, Relton, Gaunt, Slagboom, & Heijmans, Reference Sielker, Relton, Gaunt, Slagboom and Heijmans2018), recent studies showed that age-associated ELOVL2 methylation increases exceptionally in a systemic manner, including observations in immune cells (Bader et al., Reference Bader, Hänny, Schäfer, Neuckel and Kuhl2009; Sielker et al., Reference Sielker, Relton, Gaunt, Slagboom and Heijmans2018). Accelerated immunological aging in CM+ individuals has been suggested due to several findings: shorter telomere length (Boeck et al., Reference Boeck, Krause, Karabatsiakis, Schury, Gündel, Waller and Kolassa2018; Ridout et al., Reference Ridout, Levandowski, Ridout, Gantz, Goonan, Palermo and Tyrka2018; Tyrka et al., Reference Tyrka, Carpenter, Kao, Porton, Philip, Ridout and Price2015), higher levels of reactive oxygen species and oxidative stress (Boeck et al., Reference Boeck, Koenig, Schury, Geiger, Karabatsiakis, Wilker and Kolassa2016), and an increase in the ratio of memory/naïve lymphocytes with age and stress exposure (Chakravarti & Abraham, Reference Chakravarti and Abraham1999; Weng, Reference Weng2006). One could thus speculate that the observed CM-associated higher DNAm of ELOVL2 exon 1 in our study could rather be an effect of a skewed distribution of the subcellular populations of PBMC (naïve vs. memory cells). While we did not observe any changes in the distribution of lymphocytes and monocytes in CM+ women, future studies should focus on compositional shift and differential ELOVL2 DNAm in subcellular populations of those cell types.
In contrast to the results in maternal PBMC, we did not see any significant differences in ELOVL2 mean DNAm levels, neither in exon 1 nor intron 1 in UBMC, between the children of CM+ and CM− mothers. The use of fetal immune cells obtained from umbilical cord blood allowed us to compare, in a unique way, whether there is an intergenerational transmission of epigenetic signatures from mothers to their offspring (Ramo-Fernández et al., Reference Ramo-Fernández, Boeck, Koenig, Schury, Binder, Gündel and Kolassa2019). This is important as it suggests that the ELOVL2 DNAm of stem cells might not be affected by and does not directly transmit the maternal epigenetic effects of aging and CM to the next generation. In contrast, our results within the mothers suggest that own CM experiences and age-associated wear and tear seem to influence exon 1 and intron 1 DNAm of the ELOVL gene, starting from birth. Previous results showed an increase of more than 80% in ELOVL2 DNAm in a cohort including newborns to nonagenarians (Bacalini et al., Reference Bacalini, Deelen, Pirazzini, De Cecco, Giuliani, Lanzarini and Garagnani2017; Garagnani et al., Reference Garagnani, Bacalini, Pirazzini, Gori, Giuliani, Mari and Franceschi2012). Although our results need to be replicated in larger cohorts and in epigenome-wide studies, they indicate that newborns from mothers with and without CM experiences initially have the same epigenetic age with respect to the ELOVL2 gene DNAm, regardless of maternal age or CM status. How childhood rearing conditions influence ELOVL2 methylation and affect vulnerability for changes in fatty acid metabolism in these children needs to be investigated in future studies.
Some limitations of this study need to be kept in mind: First, the results cannot be generalized to other ethnicities and need further replication in an independent cohort. Second, even though statistically significant, the differences in DNAm within the exon 1 of the ELOVL2 gene between CM+ and CM− mothers (reported in the Supplementary Material) were relatively small. These results need future replication. The biological relevance of such small differences for physiological effects of ELOVL2 activity is questionable because of the small effect observed. Moreover, it cannot be excluded that other cell types or tissues show effects of CM that cannot be seen in PBMC and that might reflect relevant changes in those cell types (e.g., in the liver). The observed changes in DNAm might reflect a small percentage of a specific cell type within the PBMC fractions that show clear biological effect of CM on their methylation patterns, but that might be diluted with the overall PBMC cell population. To account for this, we included the relative amounts of monocytes and lymphocytes as covariates in our analyses.
Third, we used PBMC isolated from maternal peripheral blood and fetal blood collected from the umbilical cord. These blood types differ in the amount of memory immune cells, hematopoietic cells, and progenitor cells. Future studies should investigate how different immune cell compositions might influence ELOVL2 DNAm by investigating cell-type specific differences in DNAm in isolated immune cell subsets.
Another limitation of this study was the relatively low range of CTQ sum score reported by our cohort, which might not allow us to statistically detect site-specific effects of high maltreatment load on CpG mean methylation of the ELOVL2 gene. The increased risk for psychiatric and other physical diseases in CM-affected individuals is well established, but presumably increases with aging (i.e., from middle adulthood onwards, when aging processes in the body become visible). The project “My Childhood – Your Childhood” is a study of risk and resilience factors in the intergenerational transmission of CM and the participants range from having no experiences of CM to high CM load in rare cases. As a consequence, our cohort is more representative of the general population and does not consist of a high-risk population for psychological and physical diseases. Our cohort showed no associations between CM and disease outcomes, possibly because (a) our study design included only healthy women and (b) because of the skewed distribution of CM load (many participants with low CTQ sum scores, relatively few with very high scores) experienced by the participants. Another limitation from our cohort is that it included only women of childbearing age, which limited assessments of CM on psychiatric and physical health outcomes throughout the life span. Exposure to further stressors such as maternity and parturition, as well as increased chronological age, might trigger the development of health outcomes later in life. Finally, the physiological relevance of CM-associated changes in ELOVL2 methylation of exon 1 needs further investigation. Especially, the physiological role of omega-3 and omega-6 fatty acids is of interest in the context of CM. Previous studies suggested a buffering role of dietary omega-3 intake on the inflammatory response to acute stress (Hantsoo et al., Reference Hantsoo, Jasarevic, Criniti, McGeehan, Tanes, Sammel and Epperson2019), and individuals with CM have altered serum levels of unsaturated fatty acids (Koenig et al., Reference Koenig, Karabatsiakis, Stoll, Wilker, Hennessey, Hill and Kolassa2018). Nevertheless, the specific metabolic effects of changes in ELOVL2 exon 1 methylation with CM need detailed future investigations.
Conclusion
Our results replicate previous evidence of a strong correlation between chronological age and the DNAm of the ELOVL2 5′ end – a region that has been previously identified as a robust marker for biological age. Moreover, we found that, in PBMC, the ELOVL2 5′ end did not show increased methylation in association with CM. We extended these associations between age and DNAm to the intron 1 region of the ELOVL2 gene. Furthermore, we found preliminary evidence for a CM-associated increase of ELOVL2 exon 1 methylation in PBMC. Our results indicate region-specific effects of aging and CM on the DNAm of ELOVL2 intron 1 and 5′ end, and exon 1, respectively. Most importantly, for the children of mothers with varying degrees of CM experiences, the CM-associated alterations in ELOVL2 exon 1 DNAm seem not to be transmitted to the next generation. Therefore, no indication was found that a mother's CM affects the biological regulation of ELOVL2 in her infant. The functional relevance of the effects observed in our study needs to be elucidated in future studies, which have to clarify whether CM changes DNAm directly or whether the observed changes are secondary effects of CM-associated stress (e.g., changes in PBMC composition and/or physiological adaptations due to differences in energy metabolism and oxidative stress).
Supplementary Material
The Supplementary Material for this article can be found at https://doi.org/10.1017/S0954579420001972
Acknowledgments
We thank Traudl Hiller for her contribution to blood processing and PBMC isolation. We also acknowledge Nico Preising (UPEP, Ulm University) for providing access to technical resources required for lyophilization of biomaterial. We acknowledge the general support of Dr Frank Reister, the whole maternity ward staff at Ulm University Hospital and all the women who participated in our study. Finally, we also thank the whole “My Childhood – Your Childhood” team for their cooperative teamwork. AK is now at the Department of Clinical Psychology at the University of Innsbruck, Austria.
Financial Statement
This work was funded by a grant from the Federal Ministry of Education and Research of Germany (funding number: 01KR1304A). AMB and AMG were supported by a scholarship of the Konrad Adenauer Foundation and CB was supported by a scholarship of the Carl Zeiss Foundation.
Conflicts of Interest
None.
Authors’ Contributions
AK and ITK conceptualized the epigenetic study in the project “My Childhood – Your Childhood.” LR performed the selection of ELOVL2 regions to be epigenetically analyzed, and supervised and performed the biological sample processing with support from CB and AK. RNM performed biological sample processing of the substudy on ELOVL2 5′ end methylation with support from AMG. OA performed the pyrosequencing testing of the ELOVL2 5′ end methylation. AMB organized recruitment of the participants, performed screening interviews, and preprocessed clinical data. LR performed all data analysis. LR interpretated the results, together with AK, AMB, CB, AMG, and ITK. LR wrote the manuscript with substantial input from all coauthors. All authors read, critically revised, and approved the final version of this manuscript.





 Open access
Open access